
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKey difference between transition state stabilization and ground state destabilization: increasing atomic charge densities before or during enzyme–substrate binding†
Deliang
Chen
 *a,
Yibao
Li
a,
Xun
Li
a,
Xuechuan
Hong
b,
Xiaolin
Fan
a and
Tor
Savidge
*cd
*a,
Yibao
Li
a,
Xun
Li
a,
Xuechuan
Hong
b,
Xiaolin
Fan
a and
Tor
Savidge
*cd
aJiangxi Key Laboratory of Organo-Pharmaceutical Chemistry, Chemistry and Chemical Engineering College, Gannan Normal University, Ganzhou, Jiangxi 341000, China. E-mail: deliang2211@hotmail.com
bState Key Laboratory of Virology, Key Laboratory of Combinatorial Biosynthesis and Drug Discovery and Hubei Province, Wuhan University School of Pharmaceutical Sciences, Wuhan, 430071, China
cDepartment of Pathology & Immunology, Baylor College of Medicine, Houston, TX 77030, USA. E-mail: tor.savidge@bcm.edu
dTexas Children's Microbiome Center, Texas Childrens Hospital, Houston, TX 77030, USA
First published on 21st June 2022
Abstract
The origin of the enormous catalytic power of enzymes has been extensively studied through experimental and computational approaches. Although precise mechanisms are still subject to much debate, enzymes are thought to catalyze reactions by stabilizing transition states (TSs) or destabilizing ground states (GSs). By exploring the catalysis of various types of enzyme–substrate noncovalent interactions, we found that catalysis by TS stabilization and the catalysis by GS destabilization share common features by reducing the free energy barriers (ΔG‡s) of reactions, but are different in attaining the requirement for ΔG‡ reduction. Irrespective of whether enzymes catalyze reactions by TS stabilization or GS destabilization, they reduce ΔG‡s by enhancing the charge densities of catalytic atoms that experience a reduction in charge density between GSs and TSs. Notably, in TS stabilization, the charge density of catalytic atoms is enhanced prior to enzyme–substrate binding; whereas in GS destabilization, the charge density of catalytic atoms is enhanced during the enzyme–substrate binding. Results show that TS stabilization and GS destabilization are not contradictory to each other and are consistent in reducing the ΔG‡s of reactions. The full mechanism of enzyme catalysis includes the mechanism of reducing ΔG‡ and the mechanism of enhancing atomic charge densities. Our findings may help resolve the debate between TS stabilization and GS destabilization and assist our understanding of catalysis and the design of artificial enzymes.
1. Introduction
Enzymatic reactions are of universal importance in biology where they control diverse life processes. Elucidating the origin of the enormous catalytic power of enzymes is of fundamental and practical importance to many fields, such as elucidating the mechanism of biological systems and designing artificial enzymes.1–4 Enzymes can catalyze reactions via noncovalent interactions or covalent bonds with substrates. However, the catalysis by forming covalent bonds with substrates alters the reaction paths and the origin of enzyme catalytic power is obvious.5 Thus, in exploring the origin of the enormous catalytic power of enzymes, it is important to elucidate why enzymes can catalyze reactions significantly via enzyme–substrate noncovalent interactions. The origin of enzyme catalytic power contributed by enzyme–substrate noncovalent interactions has been extensively studied and several mechanisms are proposed to explain the origin of the enormous catalytic power of enzymes. The most widely accepted mechanism proposes that enzymes catalyze reactions through stabilizing the transition states (TSs) of the reactions5–10 and stabilization of TS has emerged as a strategy for designing artificial enzymes.11–14 The most established finding in support of the TS stabilization mechanism is that the TSs of enzymatic reactions are more stable than the TSs of their reference reactions.15 The free energy barrier (ΔG‡) of an enzymatic reaction (ΔG‡cat) is lower than that of the reference solution reaction (ΔG‡uncat) and the TS of the enzyme reaction is more stable than that in the reference reaction (Fig. 1). The TS stabilization mechanism is also supported by catalysis powered by electrostatic interactions.16–19 For example, the enzyme–substrate electrostatic interactions of the substrate oxygen atom in the isomerization of 5-androstene-3,17-dione (5-AND) catalyzed by ketosteroid isomerase (KSI) (Fig. S1†) reduce the ΔG‡ of the reaction, and are extensively reported as supporting the electrostatic TS stabilization mechanism.18,20–22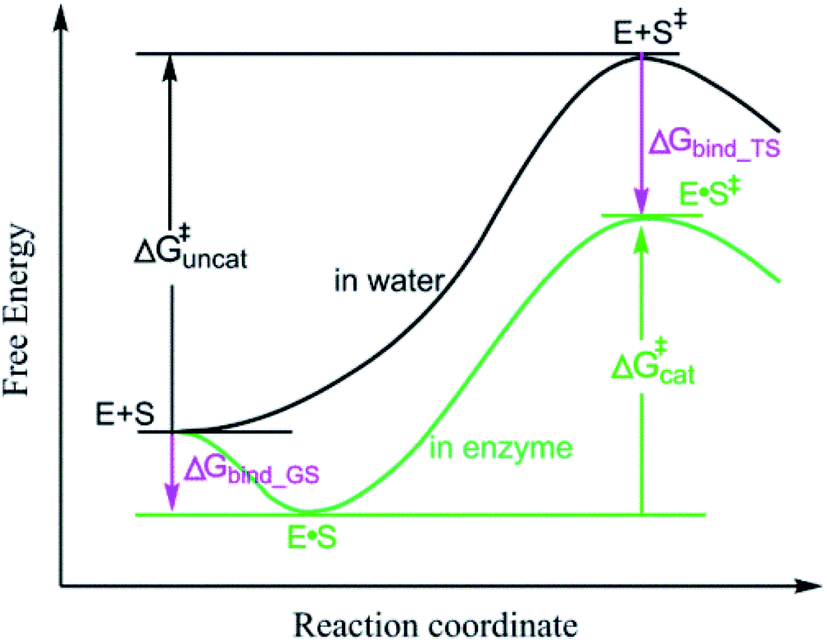 | ||
| Fig. 1 General reaction coordinate diagram comparing the free energy of an enzymatic reaction (green) with that of the corresponding reference reaction (black). S: substrate; E: enzyme. The enzymatic TS (E·S‡) is more stable than the reference TS (E + S‡). | ||
By contrast, many studies propose that enzymes can catalyze reactions through destabilizing the GSs of reactions.23–25 Ruben, et al. reported that, in the KSI-catalyzed isomerization of 5-AND, desolvation of the COO− group of Asp40 partially catalyzes the reaction via GS destabilization (Fig. S1†).24 By comparing the binding affinity of ground state analogues for KSI with a wild-type anionic Asp general base or with uncharged Asn and Ala in the general base position, this study showed that desolvation of the wild-type anionic Asp general base decreased the binding affinity of ground state analogues and thus destabilized the reaction's GS. In this example, the reduction in binding free energy caused by the desolvation of the wild-type anionic Asp general base was estimated using an experimental approach. Although the binding affinity of ground state analogues is different from the binding affinity of ground state substrates, this estimation is sufficient to demonstrate GS destabilization. Whether an enzyme–substrate interaction increases or decreases the binding free energy of the substrate for the enzyme can be determined without accurate quantification. It is well known that moving charged atoms from water to the nonpolar active sites of enzymes is unfavorable in free energy and the desolvation of charged atoms definitely destabilizes the GSs of enzymatic reactions. The catalysis by the desolvation of charged atoms, which has been reported in many studies,26–28 supports the GS destabilization mechanism.
Even though the TS stabilization and GS destabilization mechanisms are both supported by experimental and computational studies, whether enzymes catalyze reactions via TS stabilization or via GS destabilization is still under intense debate.15,29–32 Some studies are not generally supportive of GS destabilization as a catalytic mechanism because the GSs of the enzymatic reactions are more stable than the GSs of the corresponding reference reactions.15 Other studies support the GS destabilization mechanism based on the observation that the destabilization of specific atoms can reduce the ΔG‡s of reactions.24 The studies denying GS destabilization and the studies supporting GS destabilization are similar in defining GS destabilization based on the effects that enzyme–substrate interactions have on the binding affinity of GS substrates for enzymes. However, the studies denying the GS destabilization mechanism tend to consider all enzyme–substrate interactions in an enzymatic reaction; while the studies supporting the GS destabilization mechanism focus merely on specific enzyme–substrate interactions. Thus, the interactions supporting the GS destabilization mechanism are different from the interactions denying the GS destabilization mechanism, which is an important reason for the debate. In addition, the catalysis of specific enzyme–substrate electrostatic interactions via TS stabilization does not imply that other noncovalent interactions (e.g. nonpolar–polar interactions, electrostatic stress) cannot catalyze reactions. It is thus possible that TS stabilization and GS destabilization are not opposite to each other. However, it is not easy to resolve the debate by using experimental and traditional computational approaches because the debate cannot be resolved without considering the facts supporting both TS stabilization and GS destabilization simultaneously. To better understand how these contrasting mechanisms catalyze reactions, we investigated similarities and differences that are evident between the catalysis via TS stabilization versus GS destabilization. We found that TS stabilization and GS destabilization utilize the same molecular mechanism when lowering the ΔG‡s of reactions, but differ in achieving the requirement for ΔG‡ reduction. These findings may contribute significantly to our mechanistic understanding of how TS stabilization and GS destabilization augment the power of enzymatic reactions.
2. Results and discussion
2.1. Effects of noncovalent interactions on the charge density of polar atoms
We focused our study on the enzymatic catalysis contributed by noncovalent interactions because the debate between TS stabilization and GS destabilization is mainly centred on such enzymatic reactions. Moreover, the noncovalent interactions do not change the reaction paths and can accelerate reactions significantly (e.g., >1017 as reported in previous studies).5,28 To explore how enzymes catalyse reactions via enzyme–substrate noncovalent interactions, we first demonstrated how interactions of polar atoms affect the charge densities of these atoms. The relative charge density of atoms of the same element can be determined from the H-bonding capabilities of the atoms. The H-bonding capability of a polar atom is the water to nonpolar solvent phase transfer free energy contributed by the electrostatic interactions of the atom (see Fig. S2†).33,34 An atom with a higher charge density carries stronger electrostatic interactions with water and as such possesses higher water to nonpolar solvent phase transfer free energy. Thus, an atom with higher H-bonding capability has a higher charge density. We validated this relationship by demonstrating a strong linear association between the H-bonding capability of hydrogen atoms in a series of substituted phenols and the Hammett constants of the substituents (Fig. S3†).Because an atom with higher charge density will have higher water to nonpolar solvent phase transfer free energy, we explored how noncovalent interactions impact the charge densities of polar atoms by exploring how the intramolecular H-bonds of organic compounds affect the water to nonpolar solvent partition coefficients (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) of the respective compounds (Fig. S4†). By comparing the logP values of 2-substituted phenols, which may contain intramolecular H-bonds, versus structurally similar 3- or 4-substituted phenols, which do not have intramolecular H-bonds, we found that the electrostatic interaction between two atoms decreases the charge densities of these atoms. Moreover, stronger electrostatic interactions reduced charge densities to a larger extent. For example, the positive charge density of the hydrogen atom in the molecule H–X without any electrostatic interaction (left, Fig. 2A) is larger than that of H–X containing a weak H-bond interaction (middle), which in turn is larger than that of H–X possessing a strong H-bond interaction (right). It was reported that this charge density reduction results from the charge transfer between atoms forming the electrostatic interaction.35–37
P) of the respective compounds (Fig. S4†). By comparing the logP values of 2-substituted phenols, which may contain intramolecular H-bonds, versus structurally similar 3- or 4-substituted phenols, which do not have intramolecular H-bonds, we found that the electrostatic interaction between two atoms decreases the charge densities of these atoms. Moreover, stronger electrostatic interactions reduced charge densities to a larger extent. For example, the positive charge density of the hydrogen atom in the molecule H–X without any electrostatic interaction (left, Fig. 2A) is larger than that of H–X containing a weak H-bond interaction (middle), which in turn is larger than that of H–X possessing a strong H-bond interaction (right). It was reported that this charge density reduction results from the charge transfer between atoms forming the electrostatic interaction.35–37
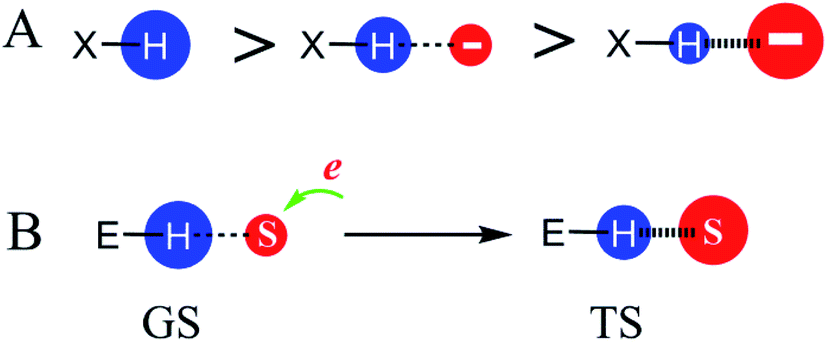 | ||
| Fig. 2 Effects of noncovalent interactions on the charge density of polar atoms. Larger red or blue circles represent higher negative or positive charge densities, respectively. (A) The relative positive charge density of hydrogen atoms that have different electrostatic interactions. (B) The charge density change of an environmental atom (atom H in E–H) from the GS to TS. The substrate atom S has a larger negative charge density in the TS versus GS, enhancing its electrostatic interaction with the positively charged hydrogen atom and reducing the positive charge density of the hydrogen atom. | ||
In reactions involving electron-movement, there are substrate atoms accepting or donating electrons during the reactions. If the substrate atoms have a larger charge density in the TS versus GS, the atoms that form electrostatic interactions with the substrate atoms will have a lower charge density in the TS versus GS. As shown in Fig. 2B, if a negatively charged substrate atom S accepts electrons, S has a larger negative charge density and forms a stronger electrostatic interaction with the positively charged environmental atom H in the TS versus GS. As a result, the positive charge density of H decreases from the GS to the TS.
2.2. Enzyme–substrate noncovalent interactions that modulate ΔG‡s
For an enzyme to catalyze the reaction of a substrate, the substrate enters the enzyme active site to form an enzyme–substrate complex prior to the reaction step during which the catalysis occurs.38Fig. 3A shows the binding and reaction processes (from the GS to the TS) of the isomerization of 5-AND catalyzed by KSI. This is a well-studied example of an enzymatic reaction that supports catalysis via TS stabilization18,20–22 and GS destabilization.24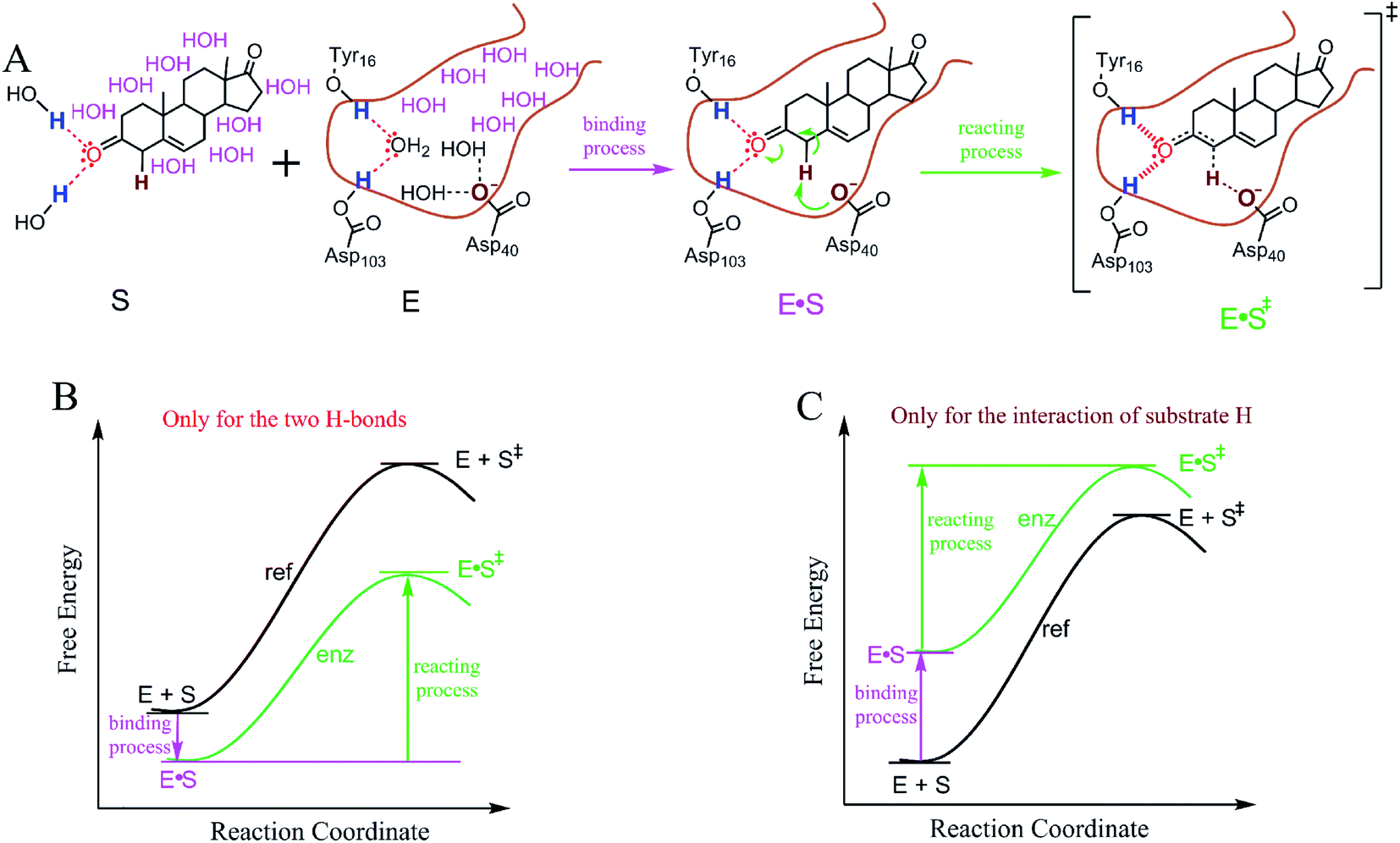 | ||
| Fig. 3 Effects of enzyme–substrate interactions on the free energy difference between enzymatic and reference reactions. (A) The binding and reaction processes of the isomerization of 5-androstene-3,17-dione (5-AND) catalyzed by ketosteroid isomerase (KSI); the processes after the TS are not shown. The water molecules on the nonpolar surfaces of 5-AND and in the KSI active site are colored pink. (B) Theoretical reaction coordinates catalyzed exclusively by electrostatic interactions of the substrate oxygen atom. (C) Theoretical reaction coordinates catalyzed exclusively by the interaction between the substrate α-H and the negatively charged oxygen atom of Asp40. | ||
In the binding process, all enzyme–substrate interactions affect the binding free energy of 5-AND. Among these interactions, the H-bond interactions between the substrate oxygen atom and the polar H-atoms of Tyr16 and Asp103 stabilize the GS to a minor extent (note: free energy contributed by the formation of the H-bonds is partially cancelled by the breaking of the H-bonds of the atoms with water, see Fig. S5†). The interaction between the nonpolar substrate α-H and the negatively charged oxygen atom of Asp40 destabilizes the GS largely. The other enzyme–substrate interactions are mainly hydrophobic, resulting from the exclusion of water from the nonpolar surfaces of 5-AND and in the KSI active site. Because of the large nonpolar surface area of 5-AND and the active site of KSI, the hydrophobic interactions stabilize the GS largely (Fig. S6†). However, the exclusion of water from nonpolar surfaces occurs merely in the binding process, and thus, the hydrophobic interactions have little effect on the ΔG‡ of the reaction. Only the interactions of the substrate atoms experiencing a charge density alteration, which includes the substrate oxygen atom and the α-H, change during the reaction process and affect the ΔG‡ of the reaction, as demonstrated in previous studies.33,39 Thus, we focus our investigation on the interactions of substrate atoms that experience a charge alteration to evaluate their role in ΔG‡ reduction. For the sake of completion, we also consider the roles of the interactions of substrate atoms without charge density alterations.
2.3. Comparison of reaction coordinate diagrams for TS stabilization and GS destabilization
The electrostatic interactions of the substrate oxygen catalyze the reaction via TS stabilization, while desolvation of the COO− group of Asp40, which corresponds to the interaction between the nonpolar substrate α-H and the COO− group, catalyzes the reaction via GS destabilization. The electrostatic interactions enhance enzyme–substrate binding (Fig. S5†) and reduce the ΔG‡ of the reaction. Visual reaction coordinates for demonstrating the free energy difference between the enzymatic and solution reactions caused exclusively by the electrostatic interactions are illustrated in Fig. 3B. The polar–nonpolar interaction reduces enzyme–substrate binding and reduces the ΔG‡ of the reaction as well. Visual reaction coordinates demonstrating the free energy difference between the enzymatic and solution reactions caused exclusively by the interaction of the nonpolar substrate α-H are shown in Fig. 3C. Comparison of these theoretical reaction coordinate diagrams indicates that catalyses by TS stabilization versus GS destabilization are fundamentally different in the binding process, but may yield a similar ΔG‡ reduction. Thus, we next investigated whether TS stabilization and GS destabilization are similar in ΔG‡ reduction. If TS stabilization and GS destabilization demonstrate similarity in reducing ΔG‡, this justifies further investigation into why the binding processes are different.2.4. Similarity between TS stabilization and GS destabilization in ΔG‡ reduction
We used the electrostatic interactions and the desolvation of COO− in the KSI-catalyzed isomerization of 5-AND and the desolvation of Cl− in halogenase-catalyzed halide alkylation,28 to explore whether TS stabilization and GS destabilization possess a common mechanism in reducing ΔG‡. Fig. 4 shows these interactions for GSs and TSs in enzymatic versus reference solution reactions. We focused on factors affecting the free energy of these reactions, which includes the strength of enzyme–substrate interactions and the atomic charge densities of the interactions.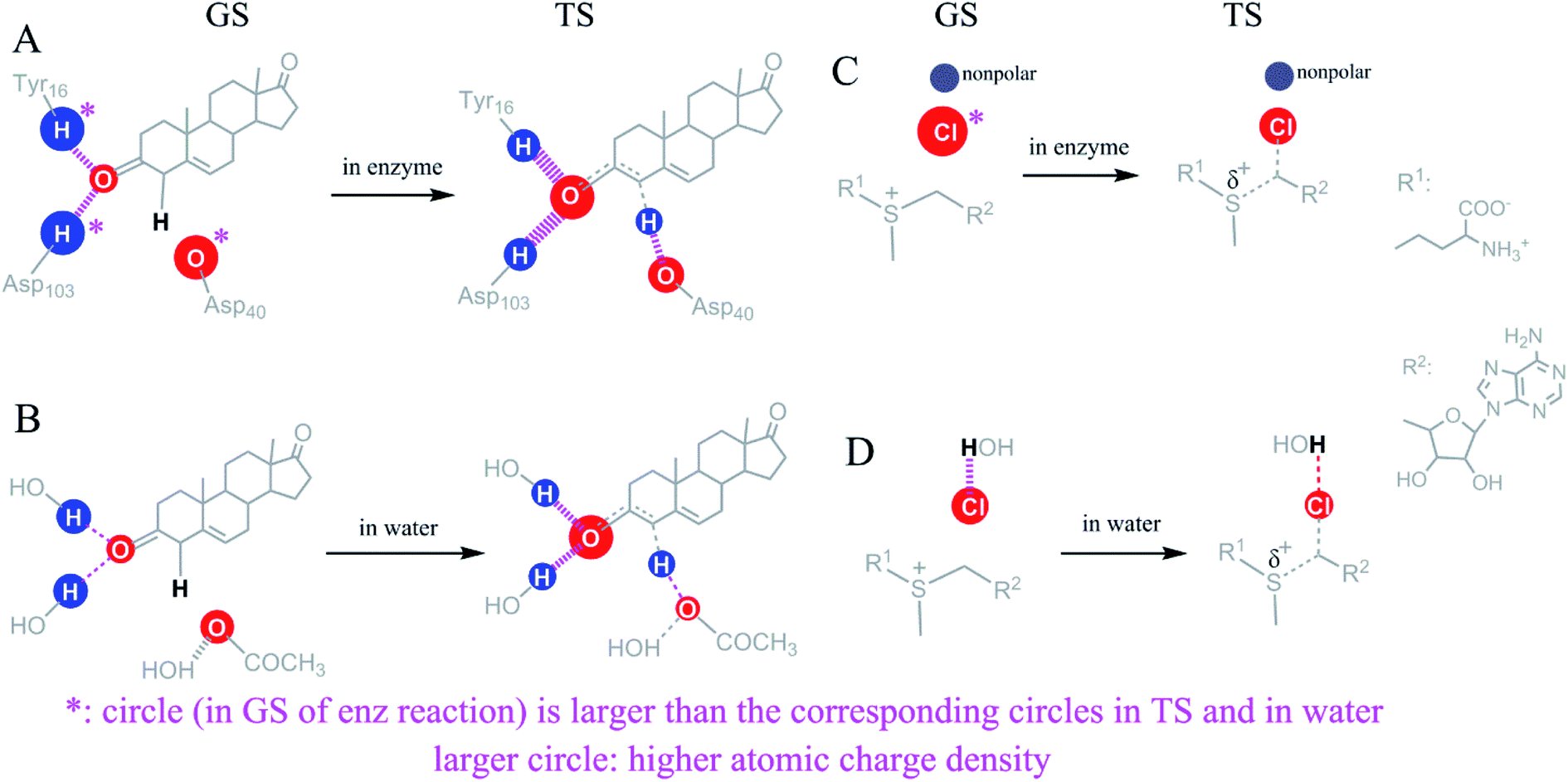 | ||
| Fig. 4 Comparison of the strength and the atomic charge density of enzyme–substrate interactions versus reference reactions for exploring the common ΔG‡ reduction mechanism. Larger red or blue circles represent higher negative or positive charge densities, respectively. Thicker dashed lines represent stronger electrostatic interactions. (A and B) The KSI-catalyzed and reference solution reactions for the isomerization of 5-AND. Interactions explored included the two H-bonds of the substrate oxygen atom and the polar–nonpolar interaction of the substrate α-H; (C and D) the interactions of the Cl− in the halogenase-catalyzed halide alkylation and the reference solution reaction. Common for all the interactions is marked with “*”. The common ΔG‡ reduction mechanism: the charge densities of the atoms undergoing a reduction in charge density between the GSs and TSs are higher than those of the corresponding atoms in the reference reactions in aqueous solution. | ||
2.5. Generality of the common ΔG‡ reduction mechanism
We investigated a large number of diverse enzymatic reactions and found that enhancing the charge densities of atoms experiencing a reduction in charge density between the GSs and TSs reduces the ΔG‡s of the reactions. To further demonstrate its generality, we used theoretical derivation to explore whether the common ΔG‡ reduction mechanism applies to all catalyses mediated by the noncovalent interactions of substrate atoms experiencing a charge density alteration. In this derivation, we focused on a general enzyme–substrate interaction that may reduce ΔG‡via TS stabilization or via GS destabilization. The substrate atom of the interaction is represented by “R” in the GS and “R‡” in the TS. The enzyme-based atom is represented by E. The environmental atom in the reference reaction is represented by W. The interactions change from R⋯E to R‡⋯E in the enzymatic reaction and from R⋯W to R‡⋯W in the reference reaction. The effect of the R‡⋯E interaction on the difference between ΔG‡cat and ΔG‡uncat, which is expressed by ΔΔG‡RE, represents the free energy change of the following H-bond pairing process as has been shown in previous studies.33,34,39| R‡⋯W + R⋯E ⇌ R‡⋯E + R⋯W | (1) |
| ΔΔG‡RE = −(HE − HW)(HR‡ − HR)/7.02 | (2) |
For R⋯E to affect ΔΔG‡RE, R must have a charge density alteration. The charge density of R either becomes higher or lower from the GS to TS. If R has a higher charge density in the TS than in the GS (HR‡ > HR), HE must be larger than HW (E must have a larger charge density than W) for catalysis to occur (ΔΔG‡RE <0). Because R‡ has a higher charge density than R, the charge densities of E and W are lower in the TS than in the GS (Fig. 2B). Therefore, if R has a higher charge density in the TS versus GS, E has a reduction in charge density and must have a larger charge density than W so that the interaction R⋯E contributes to the catalytic power.
If R has a lower charge density in the TS versus GS (HR‡ < HR), HE must be less than HW for catalysis to occur. HE is less than HW in the following cases: (1) E has a lower charge density than W (HW > HE ≥ 0) and (2) E has a similar charge to R, resulting in unfavourable electrostatic repulsion (HE < 0). In both cases, R and R‡ have weaker favourable electrostatic attraction or stronger unfavourable electrostatic repulsion with E than with W. As a result, both R and R‡ have a higher charge density in the enzymatic reaction than in the reference reaction. Thus, if the charge density of R‡ is lower than R, the latter must have a larger charge density than the corresponding substrate atom in the solution reaction so that the enzyme–substrate interaction contributes to the catalytic power.
Therefore, irrespective of whether an enzyme–substrate interaction catalyzes a reaction via a TS stabilization or GS destabilization mechanism, either the substrate or enzyme-based atom of the interaction has a reduction in charge density. This respective atom must have a higher charge density than the corresponding atom in the reference reaction for catalysis to be favourable. For a general reaction in which electrons move from one substrate atom (R1) to another (R2), the negatively charged atom in the electron-donating center has a reduction in charge density and the positively charged atom in the electron-accepting center has a reduction in charge density. The ΔG‡ reduction mechanism is summarized in Fig. 5. In the electron-donating center, either R1 or E1 (the enzyme-based atom interacting with R1) is negative and has a larger negative charge density than the corresponding atom in the reference reaction. In the electron-accepting center, either R2 or E2 (the enzyme-based atom interacting with R2) is positive and has a larger positive charge density than the corresponding atom in the reference reaction.
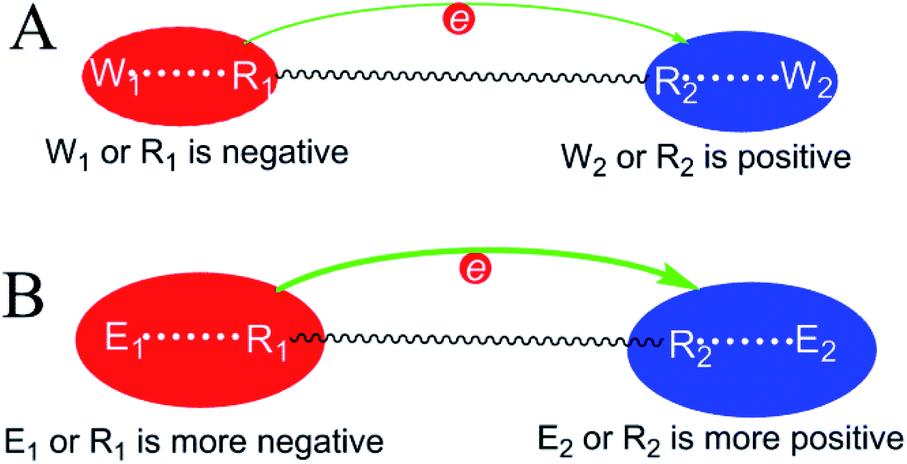 | ||
| Fig. 5 Similarity between TS stabilization and GS destabilization: ΔG‡ reduction mechanism for a general reaction in which electrons move from one substrate atom (R1) to another (R2). E1 and E2 indicate the enzyme-based atoms interacting with R1 and R2. W1 and W2 are the atoms interacting with R1 and R2 in the reference solution reaction. Larger red or blue circles represent higher negative or positive charge densities, respectively. (A) The reference solution reaction; (B) the enzymatic reaction. | ||
2.6. Consistency between the ΔG‡ reduction mechanism versus the charge alteration mechanism
In a previous study, we reported that enzymes reduce the ΔG‡s of reactions by decreasing positive charges and/or increasing negative charges in the electron-donating centers, as well as by decreasing negative charges and/or increasing positive charges in the electron-accepting centers of the reactions.39 We further demonstrate the relationship between the ΔG‡ reduction mechanism identified in this study and the charge alteration mechanism. As shown in Fig. 5B, either E1 or R1 is negatively charged in the electron-donating center. If there are positively charged environmental atoms interacting with the negatively charged atom, the negative charge density of the atom is reduced by the interactions. Decreasing positive charges will decrease the electrostatic interactions and increase the charge density of the negatively charged atom. Similarly, in the electron-accepting center, decreasing negative charges will increase the charge density of the positively charged atom. Therefore, the ΔG‡ reduction mechanism described in the present work is consistent with the charge alteration mechanism reported in our previous work. Because the charge alteration mechanism has been validated using diverse types of enzymatic reactions, e.g., reactions catalysed by electrostatic stress, catalytic triad, and metal complexes, this ΔG‡ reduction mechanism also applies to similarly diverse enzymatic reactions. Thus, this ΔG‡ reduction mechanism commonly applies to the reactions catalysed by TS stabilization and GS destabilization.2.7. Difference between TS stabilization versus GS destabilization in the binding process
Since ΔG‡ reduction mechanisms for TS stabilization and GS destabilization are comparable, it is necessary to demonstrate why some interactions catalyze reactions by TS stabilization and others by GS destabilization. We used the H-bonds in the KSI-catalyzed isomerization of 5-AND, which supports TS stabilization, and the desolvation of Cl− in halogenase-catalyzed halide alkylation, which supports GS destabilization, to explore possible reasons.Fig. 6A shows the interaction change of Cl− during the binding process of the halogenase-catalyzed halide alkylation. Cl− has a favourable interaction with water before binding and this interaction is eliminated after binding. This desolvation event enhances the charge density of Cl−. Without desolvation, Cl− in the GS of the enzymatic reaction would not have a larger charge density than the Cl− in the GS of the reference reaction and catalysis would not be favourable. Thus, GS destabilization appears to enhance the charge density of catalytic atoms that have a reduction in charge density. Fig. 6B shows the interaction change of the hydrogen atom of Tyr16 during the binding process of 5-AND to KSI. The electrostatic interaction of the hydrogen atom becomes stronger and the charge density of the hydrogen atom decreases during the binding process. Even so, the hydrogen atom in the GS still has a higher charge density than the corresponding atom in the reference solution reaction. Thus, for interactions that catalyse reactions by TS stabilization, the atoms involving a reduction in charge density must have a larger charge density preceding enzyme–substrate binding than the corresponding atoms in the reference reactions. Even though the charge density of the catalytic atoms becomes lower following binding, these atoms still have a higher charge density than the corresponding atoms in the reference reaction.
 | ||
| Fig. 6 Difference between TS stabilization versus GS destabilization in the binding process. Larger red or blue circles represent higher negative or positive charge densities, respectively. The values given in red represent the H-bonding capabilities of nearby atoms. (A) GS destabilization: The interaction of Cl− in the halogenase-catalyzed halide alkylation is weakened during the process, which increases the negative charge density of Cl−. The Cl− in the GS of the enzymatic reaction possesses a larger charge density than the Cl− in the GS of the reference reaction. (B) TS stabilization: the charge density of the hydrogen atom in the enzymatic reaction becomes lower during the binding process because its interaction becomes stronger, but is still larger than the charge density of the corresponding atom in the reference reaction in the GS. | ||
An apparent fundamental difference between catalysis by TS stabilization versus that by GS destabilization is the timing of enhancing charge densities of the catalytic atoms experiencing a reduction in charge density. For TS stabilization, the charge density of catalytic atoms is enhanced before enzyme–substrate binding. For example, in the KSI-catalyzed isomerization of 5-AND, the hydrogen atoms from Tyr16 and Asp103 already have higher positive charge densities than the hydrogen atoms of water before the enzyme–substrate binding occurs, which is caused by the electron withdrawing groups bound to the hydrogen atoms (Fig. S7†). For GS destabilization, the charge density of catalytic atoms is totally or partially enhanced during enzyme–substrate binding.
The charge density of the Cl− in the halogenase-catalyzed halide alkylation is totally enhanced during enzyme–substrate binding. While the charge density of the oxygen atom of Asp40 in the KSI-catalysed isomerization of 5-AND is partially enhanced during enzyme–substrate binding because the oxygen atom has fewer electrostatic interactions than the corresponding atom in the reference reaction before the enzyme–substrate binding. In the haloperoxidase-catalyzed halogenation of organic compounds performed in 1-propanol (Fig. S8†),40 the halide has weaker electrostatic interactions with 1-propanol than the halide in water. The charge density of the halide is partially enhanced before the enzyme–substrate binding and partially enhanced during the enzyme–substrate binding process. No matter how the charge density of a catalytic atom is enhanced, the catalytic power contributed by the catalytic atom depends on the relative charge density of the atom in the GS of the enzymatic reaction versus the corresponding atom in the reference reaction, and has little relationship with the charge density of the atom before the enzyme–substrate binding.
2.8. Relationship between TS stabilization of the enzymatic reaction and GS destabilization of enzyme–substrate interactions
For an enzymatic reaction to be catalysed by GS destabilization, the TS of the reaction must always be more stable than the TS of the corresponding reference reaction, as illustrated by the reaction coordinate diagram in Fig. 1. This diagram is quite different from the theoretical reaction coordinate diagram that shows the free energy difference between the enzymatic and reference reactions caused by the interactions via the GS destabilization mechanism (Fig. 3C). This difference derives from enzyme–substrate interactions that have no effect on ΔG‡. In Fig. 1, the free energy difference between the enzymatic and reference solution reactions results from a combination of all the enzyme–substrate interactions, which includes interactions affecting ΔG‡, as well as interactions that do not impact ΔG‡. The interactions that do not affect ΔG‡ can significantly stabilize both the GS and TS. For example, in the KSI-catalysed isomerization of 5-AND, the interactions that do not affect ΔG‡, which are mainly hydrophobic interactions, stabilize both the GS and TS largely (Fig. S6†). Even though the desolvation of negatively charged COO− of Asp40 is unfavourable to enzyme–substrate binding, the interactions that do not affect ΔG‡ contribute to the binding of 5-AND to KSI as being favourable in terms of free energy. Without the interactions that do not affect ΔG‡, 5-AND would not enter the active site of KSI for catalysis. Thus, for the catalysis mediated by the interactions associated with GS destabilization, the interactions of substrate atoms without any charge density alteration are required to provide energy for enhancing the charge densities of the polar atoms experiencing a reduction in charge density in the enzyme–substrate binding process. This phenomenon is defined as the Circe effect in previous studies.41,422.9. TS stabilization versus GS destabilization
Even though the catalysis by TS stabilization or GS destabilization has been studied extensively, it is not easy to resolve the debate between TS stabilization and GS destabilization. The difficulty has much to do with the complexity and diversity of the effects of enzyme–substrate interactions on enzymatic catalysis. First, enzyme–substrate noncovalent interactions in enzymatic reactions include those that increase or decrease ΔG‡s, as well as those that do not impact ΔG‡s. Second, noncovalent interactions have multiple roles in enzymatic catalysis, such as enhancing (or decreasing) enzyme–substrate binding and reducing (or increasing) the ΔG‡s of reactions. Third, enzyme catalysis may require a combination of multiple interactions with diverse roles. For example, the interactions reducing ΔG‡s by GS destabilization require interactions not affecting ΔG‡s for effective catalysis. Experimental and traditional computational data for supporting the TS stabilization or GS destabilization mechanism are usually obtained from specific reactions or specific interactions and do not adequately consider all possible effects of noncovalent interactions on catalytic reactions, making such interpretations difficult to argue for or against distinct enzymatic mechanisms. For example, the TS stabilization mechanism is supported by numerous experimental and computational studies that relate the catalysis to electrostatic interactions, e.g. the electrostatic interactions in the KSI-catalyzed isomerization of 5-AND. However, catalysis by other interactions (e.g. polar–nonpolar interactions) are not considered in this TS stabilization mechanism. Thus, it is difficult to delineate the relationship between TS stabilization and GS destabilization through experimental and traditional computational approaches.In the present study, by comparing similarities in diverse enzymatic catalysis, we contribute a new enzymatic concept that addresses the origin of catalytic power and identifies a common mechanism that is shared by TS stabilization and GS destabilization. Importantly, this common ΔG‡ reduction mechanism is supported by a theoretical approach that applies to diverse examples of catalyses mediated by TS stabilization and GS destabilization. The mechanism of enhancing atomic charge densities can elucidate why some interactions catalyse reactions via TS stabilization and other interactions catalyse reactions via GS destabilization. The full mechanism of enzyme catalysis includes the mechanism of reducing ΔG‡ and the mechanism of enhancing atomic charge densities. This full mechanism represents a significant advance that adds favourably to the TS stabilization versus GS destabilization debate. Although the methods for determining TS stabilization and GS destabilization are different in literature, this full mechanism has no relationship with the methods.
It is worthwhile emphasizing that this study focused exclusively on enzymatic catalysis contributed by the noncovalent interactions of the substrate atoms involving a charge density alteration because the controversy between TS stabilization and GS destabilization is mainly centered on such reactions. The ΔG‡ reduction mechanism developed in this study is targeted for the reaction processes of enzymatic reactions, in which chemical bonds form and break. If the substrate of an enzymatic reaction must undergo a large conformation change prior to the reaction process, the reaction can be catalyzed by facilitating the conformation change in the enzyme–substrate binding process. For example, in the polycyclization of polyisoprene catalyzed by terpene cyclase (Fig. S9†),43 the polyisoprene in its most stable linear conformation cannot undergo polycyclization because the atoms for forming chemical bonds are far from each other. The linear conformation must change to a much less stable productive precyclic conformation so that chemical bonds can form. Terpene cyclase catalyzes the polycyclization of polyisoprene by facilitating the conformation changes. The release of ordered water molecules in the enzyme active site, which is favorable in entropy, provides energy for the conformation change and plays a dominant role in the catalysis.
3. Conclusions
TS stabilization and GS destabilization are the most widely accepted mechanisms for explaining the enormous catalytic power of enzymes contributed by enzyme–substrate noncovalent interactions. In this study, we explored similarities and the differences between TS stabilization and GS destabilization and found that both reduce ΔG‡s through enhancing the charge densities of the catalytic atoms experiencing a reduction in charge density. A notable difference is in their temporal approach to enhance the charge densities of the catalytic atoms. For the catalysis by TS stabilization, the charge density is enhanced prior to enzyme–substrate binding; while for GS destabilization, the charge density is enhanced during enzyme–substrate binding. Increasing atomic charge densities during enzyme–substrate binding requires energy, which is provided by enzyme–substrate interactions not affecting ΔG‡s. Irrespective of how enzymes catalyse reactions, the TSs of enzymatic reactions are always more stable than the TSs of their reference reactions. Our finding could help resolve the current mechanistic debate between TS stabilization and GS destabilization and potentially provide useful guidance in exploring enzymatic catalysis that can aid in the design of potent artificial enzymes.4. Methods
4.1 Methods for calculating H-bonding capabilities
The H-bonding capabilities of the polar hydrogen atoms in the substituted phenols were calculated from the experimental logP16 (logarithm of the hexadecane–water partition coefficient) and logPoct (logarithm of the n-octanol–water partition coefficient) values of the compounds according to the method reported in previous studies.44,45 The H-bonding capabilities of the atoms in other small organic compounds were calculated based on the structures of the compounds according to the approach described in previous studies.33,34 The H-bonding capabilities of the polar hydrogen atoms in the active site of KSI were obtained from a previous study.33
4.2 Experimental water to chloroform partition coefficients
The experimental chloroform partition coefficients of the organic molecules listed in Fig. S4† were obtained from the literature.464.3 Method for calculating the change in surface accessible surface area (ΔSASA) during the binding process of 5-AND to KSI
The total ΔSASA in the binding process of 5-AND to KSI was calculated by summarizing the ΔSASAs of the atoms in the complex of KSI and 5-AND. The ΔSASA of an atom was calculated from the water accessible surface areas (SASAs) of the atom before binding and after binding, based on the crystal structures of the KSI mutant-steroid substrate mimic complex (pdb code: 1E3R (ref. 47)). Before calculation, the nitrogen atom of Asn40 in 1E3R was changed to oxygen. The ligand in 1E3R, androsten 3-betal-ol-17-one, was modified to 5-AND by changing the CHOH in position 3 to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. The 3D structure of 5-AND before binding was created using the SYBYL molecular modeling package (Tripos Inc., St Louis, MO) and minimized with the Tripos force field. The SASAs of atoms were calculated from atomic coordinates and the radii of the solvent was set to 1.4 Å.
O. The 3D structure of 5-AND before binding was created using the SYBYL molecular modeling package (Tripos Inc., St Louis, MO) and minimized with the Tripos force field. The SASAs of atoms were calculated from atomic coordinates and the radii of the solvent was set to 1.4 Å.
4.4 Method for exploring the similarity between TS stabilization and GS destabilization in ΔG‡ reduction
The catalysis contributed by the electrostatic interactions and the desolvation of the COO− of Asp40 in the KSI-catalyzed isomerization of 5-AND and the desolvation of Cl− in halogenase-catalyzed halide alkylation were used to explore the similarity between TS stabilization and GS destabilization in ΔG‡ reduction. We explored the similarity based on the following factors: (1) change in enzyme–substrate interactions; (2) relative strength of enzyme–substrate noncovalent interactions versus the corresponding reference interactions; (3) relative charge density of the atoms that have a larger charge density in the TS versus GS; and (4) relative charge density of the atoms that have a lower charge density in the TS versus GS.The model for quantifying the free energy change of a competitive H-bond pairing process was used to explore whether the common ΔG‡ reduction mechanism applies to all catalyses contributed by the noncovalent interactions of substrate atoms experiencing a charge density alteration. The competitive H-bond pairing process is defined by the general process shown in the following equation, where two H-bond acceptors (A1 and A2) and two H-bond donors (D1 and D2) form mixed pairings.
| A1⋯D2 + A2⋯D1 ⇌ A1⋯D1 + A2⋯D2 | (3) |
The standard free energy change of this process is:
| ΔG = −(HA1 − HA2)(HD1 − HD2)/7.02 | (4) |
4.5 Method for exploring the difference between TS stabilization versus GS destabilization in the binding process
The electrostatic interactions in the KSI-catalyzed isomerization of 5-AND, and the desolvation of Cl− in halogenase-catalyzed halide alkylation were used to explore the difference between TS stabilization and GS destabilization. The difference between TS stabilization and GS destabilization was obtained by analyzing the difference in charge density alteration between the polar hydrogen atoms in the KSI-catalyzed isomerization of 5-AND and the Cl− in the halogenase-catalyzed halide alkylation.Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
D. Chen: conceptualization, methodology, project administration, writing – reviewing and editing, supervision, funding acquisition. Y. Li: investigation, validation, writing – original draft. X. Li: methodology, writing – original draft. X. Hong: validation, writing – review & editing. X. Fang: supervision, writing – review & editing. T. Savidge: methodology, funding acquisition, writing – reviewing & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by grants from the National Natural Science Foundation of China (21763002), the Natural Science Foundation of Jiangxi Province (20202ACBL203008), and the National Institute of Allergy and Infectious Diseases (U01-AI24290 and P01-AI152999).Notes and references
- J. R. Knowles, Nature, 1991, 350, 121–124 CrossRef CAS.
- X. Garrabou, D. S. Macdonald, B. I. M. Wicky and D. Hilvert, Angew. Chem., Int. Ed. Engl., 2018, 57, 5288–5291 CrossRef CAS.
- H. Jiang, X. F. Hu, Y. X. Li, C. Yang and T. Ngai, Chem. Sci., 2021, 12, 12463–12467 RSC.
- L. Benny, A. R. Cherian, A. Varghese, N. Sangwan, P. K. Avti and G. Hegde, Mol. Catal., 2021, 516, 111999 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- V. Vaissier, S. C. Sharma, K. Schaettle, T. R. Zhang and T. Head-Gordon, ACS Catal., 2018, 8, 219–227 CrossRef.
- S. Ekesan and D. M. York, Nucleic Acids Res., 2019, 47, 10282–10295 CrossRef CAS.
- G. Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed.
- W. W. Ren, M. Farren-Dai, N. Sannikova, K. Swiderek, Y. Wang, O. Akintola, R. Britton, V. Moliner and A. J. Bennet, Chem. Sci., 2020, 11, 10488–10495 RSC.
- A. J. Rigual, J. Cantero, M. Risso, P. Rodriguez, S. Rodriguez, M. Paulino, D. Gamenara and N. Veiga, Mol. Catal., 2020, 495, 111131 CrossRef CAS.
- B. D. Weitzner, Y. Kipnis, A. G. Daniel, D. Hilvert and D. Baker, Protein Sci., 2019, 28, 2036–2041 CrossRef CAS.
- W. S. Mak and J. B. Siegel, Curr. Opin. Struct. Biol., 2014, 27, 87–94 CrossRef CAS.
- L. Jiang, E. A. Althoff, F. R. Clemente, L. Doyle, D. Rothlisberger, A. Zanghellini, J. L. Gallaher, J. L. Betker, F. Tanaka, C. F. Barbas, 3rd, D. Hilvert, K. N. Houk, B. L. Stoddard and D. Baker, Science, 2008, 319, 1387–1391 CrossRef CAS PubMed.
- C. Acosta-Silva, J. Bertran, V. Branchadell and A. Oliva, ChemPhysChem, 2020, 21, 295–306 CrossRef CAS PubMed.
- M. H. Olsson and A. Warshel, J. Am. Chem. Soc., 2004, 126, 15167–15179 CrossRef CAS.
- A. M. Richard and J. A. Gascon, Theor. Chem. Acc., 2020, 139, 9 Search PubMed.
- P. Kumar, P. K. Agarwal, M. B. Waddell, T. Mittag, E. H. Serpersu and M. J. Cuneo, Angew. Chem., Int. Ed., 2019, 58, 16260–16266 CrossRef CAS.
- Y. F. Wu, S. D. Fried and S. G. Boxer, J. Am. Chem. Soc., 2020, 142, 9993–9998 CrossRef CAS.
- S. Kraut, D. Bebenroth, A. Nierth, A. Y. Kobitski, G. U. Nienhaus and A. Jaschke, Nucleic Acids Res., 2012, 40, 1318–1330 CrossRef CAS.
- X. W. Wang and X. He, Molecules, 2018, 23, 2410 CrossRef.
- Y. F. Wu and S. G. Boxer, J. Am. Chem. Soc., 2016, 138, 11890–11895 CrossRef CAS.
- S. C. Kamerlin, P. K. Sharma, Z. T. Chu and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4075–4080 CrossRef CAS PubMed.
- M. Biler, R. M. Crean, A. K. Schweiger, R. Kourist and S. C. L. Kamerlin, J. Am. Chem. Soc., 2020, 142, 20216–20231 CrossRef CAS PubMed.
- E. A. Ruben, J. P. Schwans, M. Sonnett, A. Natarajan, A. Gonzalez, Y. S. Tsai and D. Herschlag, Biochemistry, 2013, 52, 1074–1081 CrossRef CAS.
- R. S. Phillips, A. Vita, J. B. Spivey, A. P. Rudloff, M. D. Driscoll and S. Hay, ACS Catal., 2016, 6, 6770–6779 CrossRef CAS.
- I. Kuhn, E. Kellenberger, C. Cakir-Kiefer, H. Muller-Steffner and F. Schuber, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 1317–1331 CrossRef CAS PubMed.
- E. Sugrue, P. D. Carr, C. Scott and C. J. Jackson, Biochemistry, 2016, 55, 6304–6313 CrossRef CAS.
- D. C. Lohman, D. R. Edwards and R. Wolfenden, J. Am. Chem. Soc., 2013, 135, 14473–14475 CrossRef CAS PubMed.
- D. Burschowsky, A. van Eerde, M. Okvist, A. Kienhofer, P. Kast, D. Hilvert and U. Krengel, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17516–17521 CrossRef CAS.
- B. Goryanova, T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 2019, 141, 13468–13478 CrossRef CAS PubMed.
- L. S. Mydy, J. R. Cristobal, R. D. Katigbak, P. Bauer, A. C. Reyes, S. C. L. Kamerlin, J. P. Richard and A. M. Gulick, Biochemistry, 2019, 58, 1061–1073 CrossRef CAS PubMed.
- T. P. Silverstein, Biophys. Chem., 2021, 274, 106606 CrossRef CAS.
- D. Chen, Y. B. Li, X. Li, W. Guo, Y. D. Li, T. Savidge and X. L. Fan, J. Phys. Chem. C, 2019, 123, 13586–13592 CrossRef CAS.
- D. Chen, N. Oezguen, P. Urvil, C. Ferguson, S. M. Dann and T. C. Savidge, Sci. Adv., 2016, 2, e1501240 CrossRef.
- M. Pavlov, P. E. M. Siegbahn and M. Sandstrom, J. Phys. Chem. A, 1998, 102, 219–228 CrossRef CAS.
- A. Rondi, Y. Rodriguez, T. Feurer and A. Cannizzo, Acc. Chem. Res., 2015, 48, 1432–1440 CrossRef CAS PubMed.
- D. Chaturvedi, V. Gupta, P. Tandon, A. Sharma, C. Baraldi and M. C. Gamberini, Spectrochim. Acta, Part A, 2012, 99, 150–159 CrossRef CAS PubMed.
- S. Marti, M. Roca, J. Andres, V. Moliner, E. Silla, I. Tunon and J. Bertran, Chem. Soc. Rev., 2004, 33, 98–107 RSC.
- D. Chen, Y. Li, X. Li, T. Savidge, Y. Qian and X. Fan, Arabian J. Chem., 2022, 15, 103611 CrossRef CAS.
- R. Yamada, T. Higo, C. Yoshikawa, H. China, M. Yasuda and H. Ogino, Biotechnol. Prog., 2015, 31, 917–924 CrossRef CAS PubMed.
- W. P. Jencks, Adv. Enzymol. Relat. Areas Mol. Biol., 1975, 43, 219–410 CAS.
- C. R. Geyer and D. Sen, J. Mol. Biol., 2000, 299, 1387–1398 CrossRef CAS PubMed.
- P. O. Syren, S. C. Hammer, B. Claasen and B. Hauer, Angew. Chem., Int. Ed. Engl., 2014, 53, 4845–4849 CrossRef CAS PubMed.
- D. L. Chen, Q. Y. Wang, Y. B. Li, Y. D. Li, H. Zhou and Y. L. Fan, Chemosphere, 2020, 247, 125869 CrossRef CAS PubMed.
- D. Chen, Y. Li, M. Zhao, W. Tan, X. Li, T. Savidge, W. Guo and X. Fan, Phys. Chem. Chem. Phys., 2018, 20, 24399–24407 RSC.
- M. H. Abraham, J. A. Platts, A. Hersey, A. J. Leo and R. W. Taft, J. Pharm. Sci., 1999, 88, 670–679 CrossRef CAS PubMed.
- N. C. Ha, M. S. Kim, W. Lee, K. Y. Choi and B. H. Oh, J. Biol. Chem., 2000, 275, 41100–41106 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01994a |
| This journal is © The Royal Society of Chemistry 2022 |