
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA tunable full-color lanthanide noncovalent polymer based on cucurbituril-mediated supramolecular dimerization†
Hua-Jiang
Yu
a,
Xiao-Lu
Zhou
a,
Xianyin
Dai
a,
Fang-Fang
Shen
 a,
Qingyang
Zhou
a,
Ying-Ming
Zhang
*a,
Xiufang
Xu
a and
Yu
Liu
*ab
a,
Qingyang
Zhou
a,
Ying-Ming
Zhang
*a,
Xiufang
Xu
a and
Yu
Liu
*ab
aCollege of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn; ymzhang@nankai.edu.cn
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, P. R. China
First published on 17th June 2022
Abstract
The construction of lanthanide multicolor luminescent materials with tunable photoluminescence properties has been developed as one of the increasingly significant topics and shown inventive applications in miscellaneous fields. However, fabricating such materials based on synergistically assembly-induced emission rather than simple blending of different fluorescent dyes together still remains a challenge. Herein, we report a europium-based noncovalent polymer with tunable full-color emission, which is constructed from the 2,6-pyridinedicarboxylic acid-bearing bromophenylpyridinium salt. This rationally designed bifunctional component can concurrently serve as a guest molecule and a chelating ligand to associate with cucurbit[8]uril and europium ions, thus leading to the formation of a trichromatic (red–green–blue, RGB) photoluminescent polypseudorotaxane-type noncovalent polymer in aqueous solution. Meanwhile, the full-color emission enclosed within the RGB color triangle could be readily produced by simply tuning the molar ratio of cucurbit[8]uril and europium ions. The lanthanide supramolecular polymer featuring tricolor emission, long lifetime, high photoluminescence efficiency and low cytotoxicity could be further applied in multicolor imaging in a cellular environment. These results provide a new and feasible strategy for the construction of full-color single lanthanide self-assembled nanoconstructs.
Introduction
Multicolor photoluminescent materials, especially white light-emitting devices with an emission spectrum spanning the whole visible region, have attracted considerable attention1–3 due to their promising applications in optical sensors,4–6 bioimaging agents,7–10 flexible full-color displays,11–14 and light-emitting diodes.15–17 Common methods to obtain multicolor emitting materials rely on simply mixing three primary red–green–blue (RGB) colors or covalent attachment of complementary chromophores in a precise ratio, which often involves tedious synthetic procedures and undesired cross-color interference among the multicomponent emitters.18–20 To alleviate these shortcomings, macrocycle-involved host–guest interactions have been developed as a powerful approach to design multicolor luminescent materials, in which individual luminophores can self-assemble into well-ordered nanoarchitectures with distinct changes in photophysical properties.21–24 Among the most frequently encountered macrocycles, cucurbiturils (CBs), a family of macrocyclic receptors possessing hydrophobic cavities and carbonyl-laced portals, have been explored as versatile building blocks to fabricate supramolecular nanoconstructs with elaborate structures and functions.25–27 In this context, cucurbit[8]uril (CB[8]), an eight-membered CB homologue, has been drawn into the limelight because it can enable two specific guest molecules to stack in close proximity within the rigid cavity and often leads to dramatic photoluminescence enhancement in both inanimate milieus and biological fluids.28–32Lanthanide complexes have been widely utilized in the design of luminescent materials and imaging agents due to their excellent photophysical properties, such as large Stokes shifts, narrow emission bands, and long-lived excited states.33–35 Although multicolor emission by blending discrete lanthanide metal ions and organic ligands has been known in some cases,36–39 full-color emission solely based on a single lanthanide metal ion still remains a challenge in aqueous media, to the best of our knowledge. This is mainly due to the vulnerable excited state of lanthanide complexes that can be severely quenched by the O–H group of water molecules and the established chelating ligands around lanthanide metal ions that lack tunable photoluminescence characteristics.40,41
Recently, we have developed several types of CB-confined supramolecular nanosystems with a wide color range and purely organic room-temperature phosphorescence (RTP).42–44 This motivated us to integrate the host–guest-binding-induced phosphorescence and intrinsic lanthanide photoluminescence into a single self-assembled entity, with the aim of attaining a supramolecular lanthanide assembly possessing tunable full-color emission properties in aqueous solution. In this work, a heteroditopic guest, 2,6-pyridinedicarboxylic acid-modified bromophenylpyridinium salt (PY-DPA) could form a stable biaxial pseudorotaxane with CB[8], accompanied by a conversion from blue fluorescence to green phosphorescence in water. Meanwhile, the coordination of terminal carboxylic groups with europium ions (Eu3+) could further lead to the formation of a lanthanide supramolecular polymer, which would significantly enhance the characteristic luminescence of Eu3+via the complexation-triggered intramolecular energy transfer pathway. As a result, tunable full-color luminescence (including the white-light emission) enclosed within the RGB triangle could be conveniently achieved by varying the proportion of Eu3+ and CB[8]. Furthermore, this nanoassembly proved to be highly biocompatible and capable of multicolor cellular imaging upon excitation at a single wavelength. Therefore, it can be envisioned that the resultant Eu3+@PY-DPA⊂CB[8] assembly featuring full color (red–green–blue), different photophysical origins (photoluminescence from singlet and triplet excited states), and diverse scales in lifetimes (ns-level fluorescence, μs-level phosphorescence, and ms-level lanthanide emission) will offer a new and elegant strategy for the fabrication of multicolor smart light-emitting materials.
Results and discussion
The construction of a full-color emissive lanthanide noncovalent polymer is depicted in Scheme 1. The dual-functional guest PY-DPA was synthesized by the reaction between a 2,6-pyridinedicarboxylic acid derivative and 4-(4-bromophenyl)-pyridine. The structural characterization is shown in the ESI (Chart S1 and Fig. S1–S6).† To investigate the binding behavior between PY-DPA and CB[8], 1H NMR titration experiments were preliminarily conducted. As shown in Fig. 1a, the aromatic protons assigned to the PY moiety (Ha–d) underwent a dramatic upfield shift upon gradual addition of CB[8], while the protons of the DPA unit (He) shifted to downfield. These phenomena suggested that the PY moiety was deeply encapsulated in the cavity of CB[8] and the DPA part was located outside the cavity of CB[8]. Besides, the changes of chemical shifts of all aromatic protons achieved the equilibrium state with 0.5 equivalents of CB[8], implying a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 binding stoichiometry between CB[8] and PY-DPA. In addition, 2D rotating-frame Overhauser effect spectroscopy (ROSEY) was employed to gain more insight into the binding mode. Strong correlated signals between Hb and Hd protons were observed, indicative of the head-to-tail stacking mode of two pyridinium units in the cavity of CB[8] (Fig. 1b). Moreover, the formation of the ternary PY-DPA⊂CB[8] inclusion complex was also evaluated by UV-vis spectroscopy. As shown in Fig. 2a, free PY-DPA exhibited the main absorption peak at 308 nm, which experienced a slight bathochromic shift to 312 nm and a decrease in intensity upon the stepwise addition of CB[8], accompanied by the emergence of two isosbestic points at 247 nm and 330 nm. The binding stoichiometry between CB[8] and PY-DPA was further validated as 1:2 from the Job plot, and the association constant (Ka) in the PY-DPA⊂CB[8] complexation was calculated to be 7.46 × 1011 M−2 by the nonlinear least-squares method (Fig. S7†).
:2 binding stoichiometry between CB[8] and PY-DPA. In addition, 2D rotating-frame Overhauser effect spectroscopy (ROSEY) was employed to gain more insight into the binding mode. Strong correlated signals between Hb and Hd protons were observed, indicative of the head-to-tail stacking mode of two pyridinium units in the cavity of CB[8] (Fig. 1b). Moreover, the formation of the ternary PY-DPA⊂CB[8] inclusion complex was also evaluated by UV-vis spectroscopy. As shown in Fig. 2a, free PY-DPA exhibited the main absorption peak at 308 nm, which experienced a slight bathochromic shift to 312 nm and a decrease in intensity upon the stepwise addition of CB[8], accompanied by the emergence of two isosbestic points at 247 nm and 330 nm. The binding stoichiometry between CB[8] and PY-DPA was further validated as 1:2 from the Job plot, and the association constant (Ka) in the PY-DPA⊂CB[8] complexation was calculated to be 7.46 × 1011 M−2 by the nonlinear least-squares method (Fig. S7†).
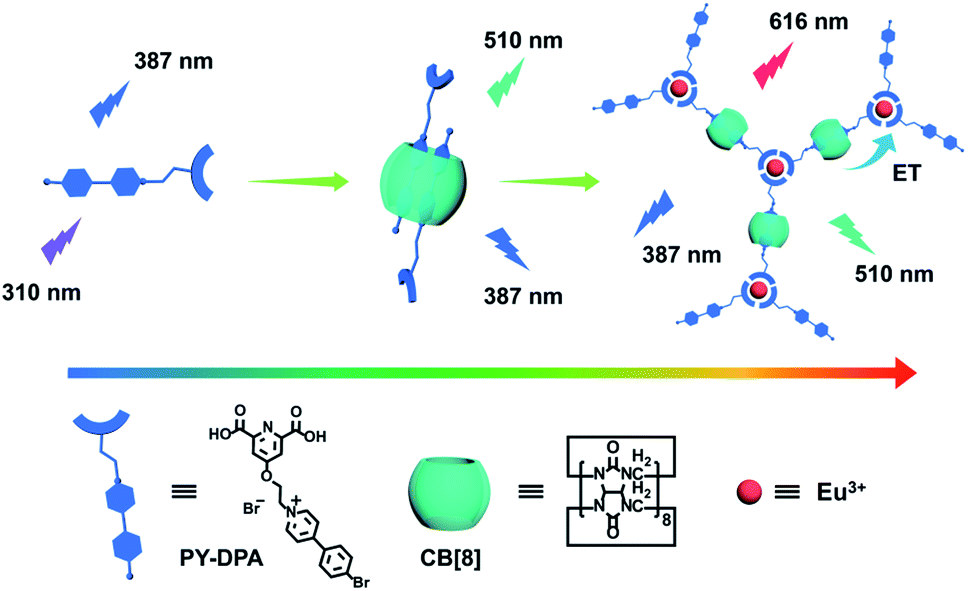 | ||
| Scheme 1 Schematic illustration of a lanthanide noncovalent supramolecular polymer (Eu3+@PY-DPA⊂CB[8] assembly) for tunable full-color emission. | ||
 | ||
| Fig. 1 1H NMR titration spectra (400 MHz, D2O, 298 K) of PY-DPA (5.0 × 10−4 M) upon addition of (a) 0.00, (b) 0.25, (c) 0.50, and (d) 1.0 equivalent of CB[8]; (e) 2D-ROSEY spectrum of the PY-DPA⊂CB[8] complex in D2O ([PY-DPA] = [CB[8]] = 5.0 × 10−4 M). | ||
 | ||
| Fig. 2 (a) UV-vis absorption spectra ([CB[8]] = 0–1.3 × 10−5 M) and (b) prompt photoluminescence spectra of PY-DPA (1.0 × 10−5 M) upon gradual addition of CB[8] ([CB[8]] = 0–1.0 × 10−5 M) in aqueous solution. Inset: photograph showing the changes in luminescent color under light irradiation at 302 nm; (c) prompt and gated photoluminescence spectra (delayed time = 50 μs) of the PY-DPA⊂CB[8] complex; (d) phosphorescence lifetime decay curve of the PY-DPA⊂CB[8] complex at 510 nm ([PY-DPA] = [CB[8]] = 1.0 × 10−5 M, λex = 310 nm, 298 K). | ||
Subsequently, the photoluminescence properties of the PY-DPA⊂CB[8] complex in aqueous solution were further explored. As can be seen from Fig. 2b, free PY-DPA showed blue emission centered at around 387 nm. The appearance of a new emission peak at 510 nm with a decrease in emission at 387 nm was clearly observed upon stepwise addition of CB[8] into the aqueous solution of PY-DPA. The luminescent color changes from blue to green could also be recognized by the naked eye. With a delay time of 50 μs, the emission at 387 nm completely disappeared and only the peak at 510 nm was maintained (Fig. 2c). As revealed by the time-resolved decay curves, the lifetime measured at 387 nm was on a nanosecond scale and the lifetimes were assigned as 0.43 and 0.46 ns for free PY-DPA and the PY-DPA⊂CB[8] complex, respectively (Fig. S8†). In comparison, the lifetime measured at 510 nm for the PY-DPA⊂CB[8] complex was on a microsecond scale (650 μs, Fig. 2d). Moreover, the intensity at 510 nm showed an apparent enhancement after deoxygenation treatment, and the lifetime increased to 2748 μs. Meanwhile, no obvious change was found with respect to the intensity at 387 nm (Fig. S9†). Thus, it could be reasonably inferred that the emission at 510 nm with a relatively longer lifetime came from host–guest-binding-induced RTP.
It is documented that 2,6-pyridinedicarboxylic acid could strongly coordinate with lanthanide metal ions (i.e. Eu3+ and Tb3+) and harvest energy for the sensitization of the characteristic emission.45 Therefore, a UV-vis titration experiment was performed to study the metal-chelating behaviors between PY-DPA and Eu3+. With the addition of Eu3+ to the PY-DPA solution, the absorbance of PY-DPA gradually decreased, as a result of the coordination of Eu3+ with the carboxylic groups of PY-DPA (Fig. S10†). At the same time, the coordination stoichiometry was determined to be 1:3 by analyzing the absorbance at 300 nm versus [Eu3+]/[PY-DPA] molar ratios. In addition, the characteristic emission of Eu3+ with four sharp peaks at 595 nm (5D0 → 7F1), 616 nm (5D0 → 7F2), 650 nm (5D0 → 7F3) and 695 nm (5D0 → 7F4) was observed in the Eu3+@PY-DPA and Eu3+@PY-DPA⊂CB[8] complexes, on account of the efficient energy transfer from the DPA moiety to Eu3+ (Fig. 3c). It was also found that two sets of emission peaks, especially the phosphorescence one, showed an obvious decline by adding Eu3+ to the PY-DPA⊂CB[8] complex (Fig. 3d and e). Thus, we inferred that an energy transfer process might occur between the PY⊂CB[8] part and Eu3+@DPA center. The more direct evidence was provided by the time-resolved curves and excitation spectra. With the gradual addition of Eu3+ to the PY-DPA⊂CB[8] complex, the lifetime at 510 nm remarkably decreased from 544 to 220 μs (Fig. S11†). Meanwhile, the excitation spectra of the Eu3+@PY-DPA⊂CB[8] complex monitored at 616 and 510 nm substantially coincided in the region above 290 nm, and the maximum peak was located at 311 nm, which was identical to the main absorption peak of the PY-DPA⊂CB[8] complex. In clear contrast, the effective excitation of Eu3+@DPA monitored at 616 nm appeared in the region below 290 nm (Fig. S12†). Moreover, the excitation–emission mapping showed that the optimum excitation wavelengths for Eu3+@DPA and the Eu3+@PY-DPA⊂CB[8] complex were 270 and 310 nm, respectively (Fig. 3a and b). The distinct redshift of the excitation wavelength validated the occurrence of energy transfer, which could facilitate the luminescence of the Eu3+ center at the longer excitation wavelength. It was noteworthy that the luminescence intensity of the Eu3+@PY-DPA⊂CB[8] complex at 616 nm was 3.6 times higher than that of Eu3+@PY-DPA under the same experimental conditions (Fig. 3c and d), and the lifetime and quantum yield of the Eu3+@PY-DPA⊂CB[8] complex (τ = 1.64 ms and Φ = 21.1%) were significantly higher than those of Eu3+@PY-DPA (τ = 1.22 ms and Φ = 2.85%, Table S1†). These spectroscopic results jointly demonstrate that the complexation with CB[8] can not only induce the phosphorescence emission but also enhance the photoluminescence efficiency of the lanthanide center via the energy transfer pathway.
 | ||
| Fig. 3 Excitation–emission mapping of (a) Eu3+@DPA ([DPA] = 1.0 × 10−4 M and [Eu3+] = 3.3 × 10−5 M) and (b) Eu3+@PY-DPA ⊂CB[8] complex in H2O ([PY-DPA] = 1.0 × 10−5 M, CB[8] = 5.0 × 10−6 M, and [Eu3+] = 3.3 × 10−6 M); prompt photoluminescence spectra of (c) PY-DPA and (d) PY-DPA with 0.5 equivalents of CB[8] and (e) 1.0 equivalent of CB[8] upon gradual addition 1/3 equivalents of Eu3+ in H2O. Inset: photograph showing the changes in luminescent color under light irradiation at 302 nm. (f) The 1931 CIE chromaticity diagram illustrating the luminescent color changes of DPA-PY with the addition of Eu3+ and CB[8] (λex = 310 nm, 298 K). | ||
To deepen the understanding of CB[8]-induced luminescence enhancement behaviors, we estimated the number of water molecules that coordinate to Eu3+ in the first coordination domain for the Eu3+@PY-DPA complex and Eu3+@PY-DPA⊂CB[8] assembly. The excited-state lifetime of Eu3+@PY-DPA was measured to be 1.22 ms in H2O and 2.59 ms in D2O, while the lifetime of Eu3+@PY-DPA⊂CB[8] was 1.64 ms in H2O and 2.20 ms in D2O (Fig. S13†). Accordingly, the coordination numbers of water molecules (q) were calculated to be −0.11 and 0.22 with and without CB[8], respectively, indicating that the coordination of water molecules was significantly inhibited in the Eu3+@PY-DPA⊂CB[8] assembly (Table S1†).46 The protection of Eu3+ from the quenching of water molecules was attributed to the formation of a close-packed supramolecular nanostructure in the case of the Eu3+@PY-DPA⊂CB[8] assembly. Indeed, compared to the binary Eu3+@PY-DPA or PY-DPA⊂CB[8] complex, the Eu3+@PY-DPA⊂CB[8] assembly showed an obvious Tyndall effect and a clear decline in transmittance, implying the existence of large-sized species (Fig. S14†). In addition, the intuitive morphology of the assembly was revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM and TEM images consistently showed that the Eu3+@PY-DPA⊂CB[8] assembly exhibited a flake-like morphology, which probably contributed to the extensive intermolecular self-assembly of the supramolecular polymer (Fig. S15†).
Given that the Eu3+@PY-DPA⊂CB[8] assembly could emit three-primary-color (RGB) luminescence including blue fluorescence, green phosphorescence and red emission of lanthanide metal ions, multicolor photoluminescence outputs enclosed within the RGB color triangle could be obtained by rationally manipulating the host–guest and metal–ligand interactions. As depicted in the CIE 1931 chromaticity diagram (Fig. 3f), the luminescent color changed linearly from blue to pink with the stepwise addition of Eu3+ to free PY-DPA aqueous solution. Unexpectedly, when Eu3+ was continually added to the solution of the PY-DPA⊂CB[8] complex, a linear change of luminescent color from cyan to red including the white emission was achieved with the chromaticity coordinate (0.34, 0.31). Moreover, benefitting from the CB[8]-induced fluorescence–phosphorescence transition, the luminescent color range could be further expanded by changing the amount of CB[8]. For instance, when the molar ratio of CB[8] was fixed at 1 equivalent, the luminescent color of the PY-DPA⊂CB[8] complex could be tuned linearly from green to red with the addition of Eu3+. On the foundation of these results, a multicolor emission hydrogel was conveniently fabricated by dispersion of agarose into the specifically emissive solution. As shown in Fig. 4a, four luminescence hydrogels with blue, green, red, and white emission could be intuitively recognized by the naked eye under UV light irradiation at 302 nm.
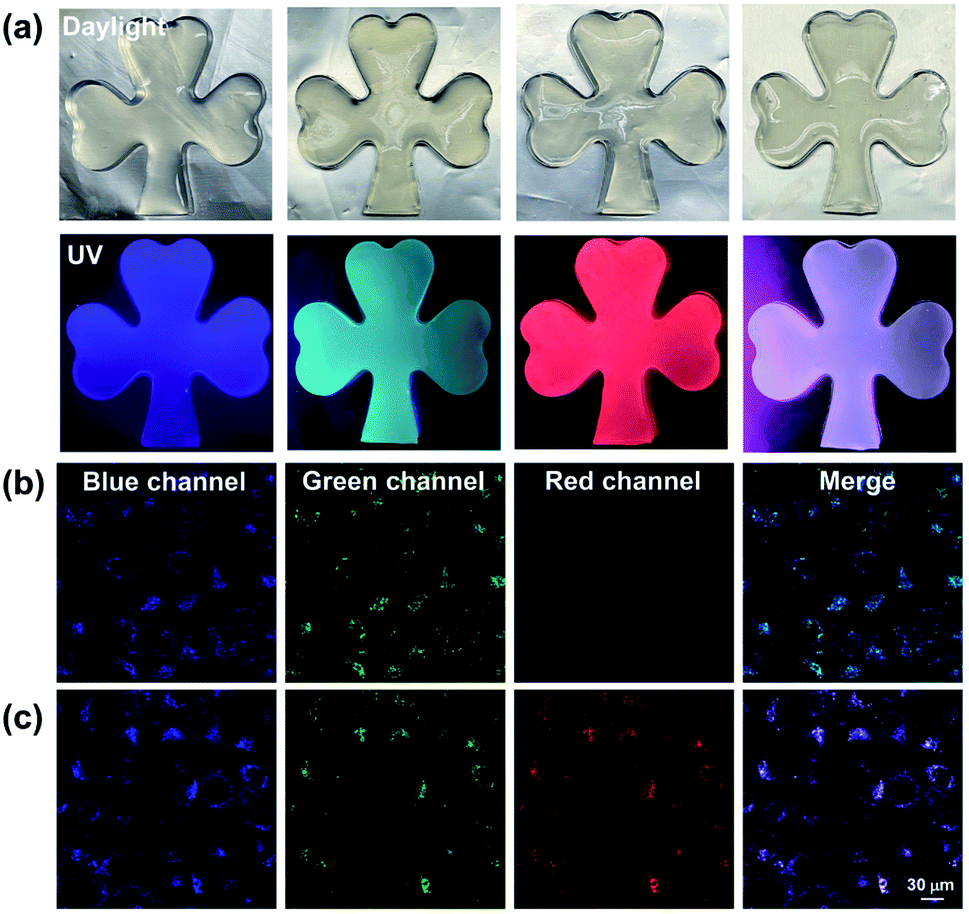 | ||
| Fig. 4 (a) Photographs of multicolor hydrogels under daylight and 302 nm UV light irradiation; confocal microscopy images of HeLa cells treated with the (b) PY-DPA⊂CB[8] complex and (c) Eu3+@PY-DPA⊂CB[8] assembly. The excitation wavelength was set at 405 nm ([PY-DPA] = CB[8] = 1.0 × 10−5 M, [Eu3+] = 6.7 × 10−7 M and scale bar = 30 μm). | ||
Considering that long-lived luminescent materials can significantly distinguish between background fluorescence and autofluorescence in living organisms, we wondered whether the Eu3+@PY-DPA⊂CB[8] assembly with superior photophysical properties could be further applied in cell imaging. Then, HeLa cells were incubated with the PY-DPA⊂CB[8] complex and Eu3+@PY-DPA⊂CB[8] assembly for 12 h, and then observed by confocal laser scanning microscopy. As shown in Fig. 4b, the cells treated with the PY-DPA⊂CB[8] complex showed blue fluorescence and green phosphorescence upon excitation with a 405 nm laser. Besides, benefitting from the efficient energy transfer from PY⊂CB[8] to the Eu3+@DPA center, brighter red emission assigned to the millisecond-level Eu3+ complex was immediately found in the case of the Eu3+@PY-DPA⊂CB[8] assembly at the same excitation wavelength (Fig. 4c). In addition, the cell cytotoxicity of the Eu3+@PY-DPA⊂CB[8] assembly was evaluated by the cell counting kit-8 (CCK8) assay. It was found that more than 90% of the cells survived after being treated with the ternary assembly for 24 h, indicative of the excellent biocompatibility of the assembly (Fig. S16†). As we know, the potential cytotoxicity of metal complexes is considered one of the major obstacles limiting their practical applications. It is necessary to develop novel labelling methods using highly biocompatible metal complexes. Therefore, it is believed that our obtained Eu3+@PY-DPA⊂CB[8] assembly with tricolor emission and multidimensional excited-state lifetimes (ns-level blue, μs-level green, and ms-level red emission) will have promising potential in time-resolved multicolor bio-imaging.
Conclusions
In conclusion, a CB[8]-mediated lanthanide noncovalent polymer was successfully constructed via synergistic host–guest complexation and metal–ligand coordination, which was able to show RGB photoluminescence and cell imaging under a single wavelength excitation. The encapsulation by CB[8] could induce PY-DPA to form a stable homoternary inclusion complex, thus giving rise to dual fluorescence–phosphorescence emission in aqueous solution. Meanwhile, the resultant complex could further assemble with Eu3+ to generate the polypseudorotaxane-type supramolecular aggregate, accompanied by the significant enhancement of red emission via the complexation-assisted energy transfer process. Interestingly, multicolor emission including the unique white light could be produced in aqueous solution and the gel phase through simply adjusting the amounts of CB[8] and Eu3+. More significantly, the obtained supramolecular lanthanide assembly with a long lifetime and full-color emission could also be applied for multicolor cell imaging. Thus, we believe that the present work will bring about new enlightenment for the fabrication of multicolor light-emitting materials and high-resolution reliable bio-imaging probes.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
H.-J. Yu, Y.-M. Zhang and Y. Liu conceived and directed this study. H.-J. Yu conducted the synthesis, characterization, and analysis of all materials described with assistance from X.-L Zhou, F.-F. Shen, and Q. Zhou. The cell imaging experiment was conducted by Y. Dai. H.-J. Yu wrote this manuscript. Y.-M. Zhang, X. Xu and Y. Liu supervised the work and edited the manuscript. All authors discussed and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21871154, 22171148, 22131008, and 21873051), the Natural Science Foundation of Tianjin (no. 21JCZDJC00310), the Fundamental Research Funds for the Central Universities (Nankai University) and the Haihe Laboratory of Sustainable Chemical Transformations.References
- H. Wang, X. Ji, Z. Li and F. Huang, Adv. Mater., 2017, 29, 1606117 CrossRef PubMed.
- H. Wang, X. Ji, Z. Li, C. N. Zhu, X. Yang, T. Li, Z. L. Wu and F. Huang, Mater. Chem. Front., 2017, 1, 167–171 RSC.
- Q. Li, Y. Wu, J. Cao, Y. Liu, Z. Wang, H. Zhu, H. Zhang and F. Huang, Angew. Chem., Int. Ed., 2022, 61, e202202381 CAS.
- H. H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed.
- H. Li and G. C. Bazan, Adv. Mater., 2009, 21, 964 CrossRef CAS.
- H. N. Kim, Z. Guo, W. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79–93 RSC.
- Y. Yang, Q. Zhao, W. Feng and F. Li, Chem. Rev., 2013, 113, 192–270 CrossRef CAS PubMed.
- X. He, Z. Zhao, L.-H. Xiong, P. F. Gao, C. Peng, R. S. Li, Y. Xiong, Z. Li, H. H.-Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, C. Z. Huang, N. Ma and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6904–6911 CrossRef CAS PubMed.
- H.-Q. Peng, C.-L. Sun, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Adv. Funct. Mater., 2016, 26, 5483–5489 CrossRef CAS.
- L. Zhu, X. Li, Q. Zhang, X. Ma, M. Li, H. Zhang, Z. Luo, H. Ågren and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 5175–5182 CrossRef CAS PubMed.
- S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS PubMed.
- S. Huo, P. Duan, T. Jiao, Q. Peng and M. Liu, Angew. Chem., Int. Ed., 2017, 56, 12174–12178 CrossRef CAS PubMed.
- G. Naren, C.-W. Hsu, S. Li, M. Morimoto, S. Tang, J. Hernando, G. Guirado, M. Irie, F. M. Raymo, H. Sundén and J. Andréasson, Nat. Commun., 2019, 10, 3996 CrossRef CAS PubMed.
- D. Li, J. Yang, M. Fang, B. Z. Tang and Z. Li, Sci. Adv., 2022, 8, eabl8392 CrossRef CAS PubMed.
- G. M. Farinola and R. Ragnia, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC.
- Y. I. Park, O. Postupna, A. Zhugayevych, H. Shin, Y.-S. Park, B. Kim, H.-J. Yen, P. Cheruku, J. S. Martinez, J. W. Park, S. Tretiak and H.-L. Wang, Chem. Sci., 2015, 6, 789–797 RSC.
- B. Liu, B. Chu, Y.-L. Wang, L.-F. Hu, S. Hu and X.-H. Zhang, Green Chem., 2021, 23, 422–429 RSC.
- S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed.
- T. Vandana, A. Karuppusamy and P. Kannan, Polymer, 2017, 124, 88–94 CrossRef CAS.
- J. E. Kwon, S. Park and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 11239–11246 CrossRef CAS PubMed.
- G. Sun, J. Pan, Y. Wu, Y. Liu, W. Chen, Z. Zhang and J. Su, ACS Appl. Mater. Interfaces, 2020, 12, 10875–10882 CrossRef CAS PubMed.
- X.-Y. Lou, N. Song and Y.-W. Yang, Chem.–Eur. J., 2019, 25, 11975–11982 CrossRef CAS PubMed.
- J. Wang, X. Yao, Y. Liu, H. Zhou, W. Chen, G. Sun, J. Su, X. Ma and H. Tian, Adv. Opt. Mater., 2018, 6, 1870049 CrossRef.
- Q.-W. Zhang, D. Li, X. Li, P. B. White, J. Mecinovic, X. Ma, H. Ågren, R. J. M. Nolte and H. Tian, J. Am. Chem. Soc., 2016, 138, 13541–13550 CrossRef CAS PubMed.
- Y.-H. Liu, Y.-M. Zhang, H.-J. Yu and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 3870–3880 CrossRef CAS PubMed.
- X. Yang, R. Wang, A. Kermagoret and D. Bardelang, Angew. Chem., Int. Ed., 2020, 59, 21280–21292 CrossRef CAS PubMed.
- H. Nie, Z. Wei, X.-L. Ni and Y. Liu, Chem. Rev., 2022, 122, 9032–9077 CrossRef CAS PubMed.
- G. Wu, Y. J. Bae, M. Olesińska, D. Antón-García, I. Szabó, E. Rosta, M. R. Wasielewski and O. A. Scherman, Chem. Sci., 2020, 11, 812–825 RSC.
- T. Jiang, X. Wang, J. Wang, G. Hu and X. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 14399–14407 CrossRef CAS PubMed.
- H. Wu, Y. Wang, L. O. Jones, W. Liu, B. Song, Y. Cui, K. Cai, L. Zhang, D. Shen, X.-Y. Chen, Y. Jiao, C. L. Stern, X. Li, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 16849–16860 CrossRef CAS PubMed.
- X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef PubMed.
- W.-L. Zhou, Y. Chen and Y. Liu, Acta Chim. Sin., 2020, 78, 1164–1176 CrossRef CAS.
- W.-L. Zhou, Y. Chen, W. Lin and Y. Liu, Chem. Commun., 2021, 57, 11443–11456 RSC.
- O. Kotova, S. Comby, C. Lincheneau and T. Gunnlaugsson, Chem. Sci., 2017, 8, 3419–3426 RSC.
- Y. Ou, W. Zhou, Z. Zhu, F. Ma, R. Zhou, F. Su, L. Zheng, L. Ma and H. Liang, Angew. Chem., Int. Ed., 2020, 59, 23810–23816 CrossRef CAS PubMed.
- H. Sun, X. Li, Y. Zhu, X. Wang, Q. Zhang and X. Hao, J. Mater. Chem. C, 2019, 7, 5782–5791 RSC.
- H. Liu, T. Chu, Z. Rao, S. Wang, Y. Yang and W.-T. Wong, Adv. Opt. Mater., 2015, 3, 1545–1550 CrossRef CAS.
- T. Kai, M. Kishimoto, M. Akita and M. Yoshizawa, Chem. Commun., 2018, 54, 956–959 RSC.
- G. Weng, S. Thanneeru and J. He, Adv. Mater., 2018, 30, 1706526 CrossRef PubMed.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed.
- F.-F. Shen, Y. Chen, X. Dai, H.-Y. Zhang, B. Zhang, Y. Liu and Y. Liu, Chem. Sci., 2021, 12, 1851–1857 RSC.
- X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
- M. R. George, C. A. Golden, M. C. Grossel and R. J. Curry, Inorg. Chem., 2006, 45, 1739–1744 CrossRef CAS PubMed.
- M.-a. Morikawa, S. Tsunofuri and N. Kimizuka, Langmuir, 2013, 29, 12930–12935 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization. See https://doi.org/10.1039/d2sc02384a |
| This journal is © The Royal Society of Chemistry 2022 |