DOI:
10.1039/D2SC02176E
(Edge Article)
Chem. Sci., 2022,
13, 7755-7764
Total synthesis of Lentinus giganteus glycans with antitumor activities via stereoselective α-glycosylation and orthogonal one-pot glycosylation strategies†
Received
18th April 2022
, Accepted 26th May 2022
First published on 27th May 2022
Abstract
The accessibility to long, branched and complex glycans containing many 1,2-cis glycosidic linkages with precise structures remains a challenging task in chemical synthesis. Reported here is an efficient, stereoselective and orthogonal one-pot synthesis of a tetradecasaccharide and shorter sequences from Lentinus giganteus polysaccharides with antitumor activities. The synthetic strategy consists of: (1) newly developed merging reagent modulation and remote anchimeric assistance (RMRAA) α-(1→6)-galactosylation in a highly stereoselective manner, (2) DMF-modulated stereoselective α-(1→3)-glucosylation, (3) RMRAA stereoselective α-(1→6)-glucosylation, (4) several orthogonal one-pot glycosylations on the basis of N-phenyltrifluoroacetimidate (PTFAI) glycosylation, Yu glycosylation and ortho-(1-phenylvinyl)benzoate (PVB) glycosylation to streamline oligosaccharide synthesis, and (5) convergent [7 + 7] glycosylation for the final assembly of the target tetradecasaccharide. In particular, this new RMRAA α-galactosylation method has mild reaction conditions, broad substrate scopes and significantly shortened step counts for the heptasaccharide synthesis in comparison with 4,6-di-tert-butylsilyene (DTBS) directed α-galactosylation. Furthermore, DFT calculations shed light on the origins of remote anchimeric assistance effects (3,4-OBz > 3,4-OAc > 4-OBz > 3-OBz) of acyl groups.
Introduction
Carbohydrates are essential and the most abundant biomolecules with crucial roles in various biological processes, including cell growth and proliferation, viral and bacterial infection, and immune responses.1,2 In comparison with the synthesis of proteins and DNA via the gene-regulated process, glycan biosynthesis is a stepwise and non-template-driven process, which often involves post-translational modifications in the Golgi apparatus and endoplasmic reticulum, and often results in heterogeneous and complex carbohydrate sequences. Isolation of homogeneous and pure glycans from natural sources is a formidable task. Chemical synthesis is a scalable and reliable means to prepare pure and well-defined glycans for developing new therapeutic agents and deciphering their functions.3–16 However, compared with the routine and efficient automated solid-phase synthesis of oligonucleotides and peptides, the synthesis of glycans is much more challenging and lags behind. The major issues in glycan synthesis are regio- and stereochemical problems, and it is often time-consuming and requires tedious carbohydrate synthesis.17,18 Indeed, cases of the total synthesis of long, branched and complex glycans with many 1,2-cis glycosidic linkages and over 10 units are still limited.19–27 Therefore, the development of efficient strategies for the synthesis of long, branched and complicated carbohydrates containing diverse 1,2-cis glycosidic linkages is direly needed and remains challenging in chemical synthesis.
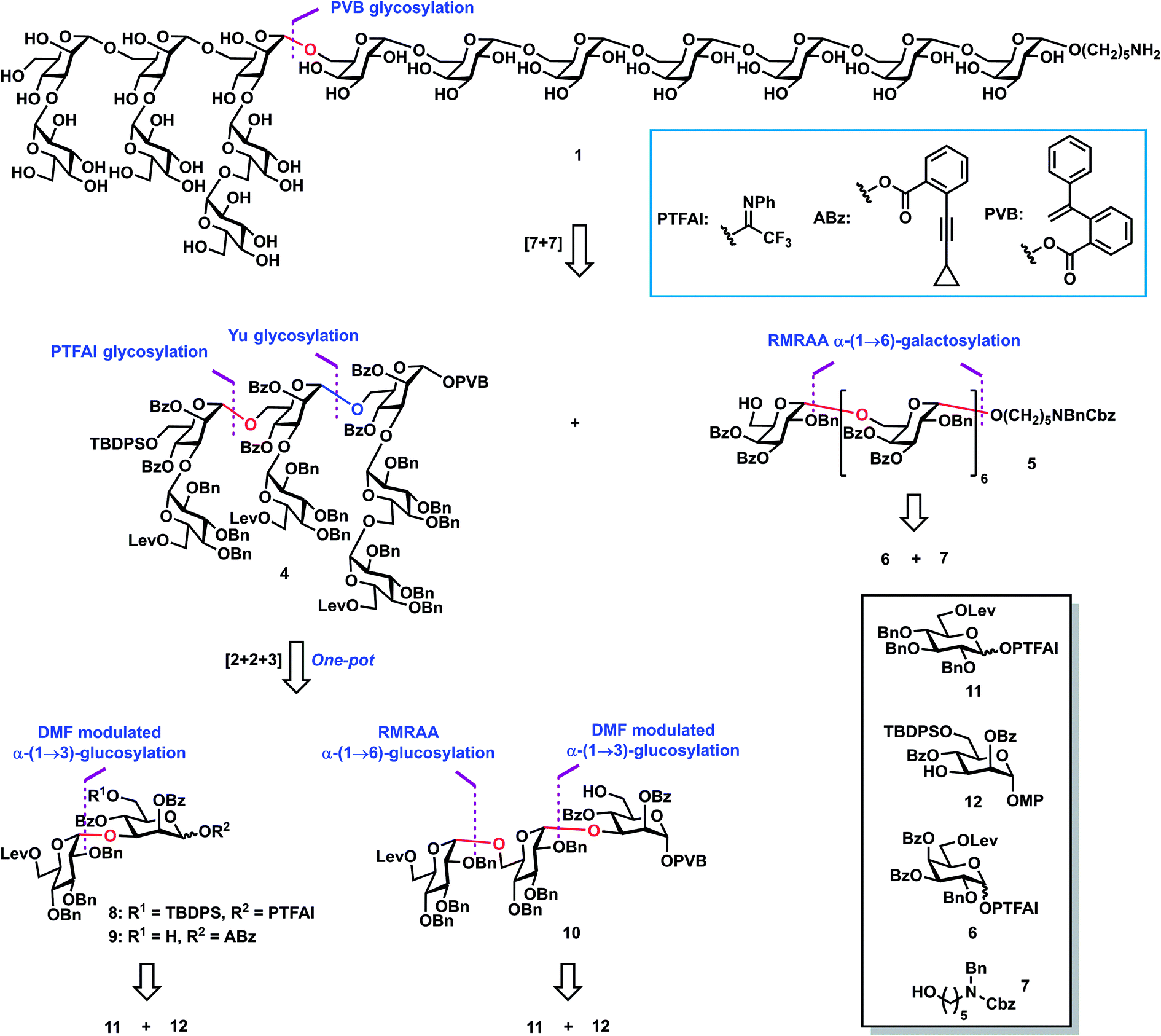
In 2016, a novel neutral polysaccharide LGPS-1 (Fig. 1) with a tetradecasaccharide repeating unit was isolated from Lentinus giganteus, which is an edible mushroom and widely distributed in South China.28 LGPS-1 glycan has the following unique structural characteristics: (1) a long glycan chain with seven 1,2-cis-galactosidic linkages at the reducing end, which are challenging to construct in an efficient and highly stereoselective manner; (2) four challenging 1,2-cis glucosidic bonds in the side chain; (3) two 3,6-branched mannose motifs at the nonreducing end. LGPS-1 could induce HepG2 cell apoptosis via intrinsic mitochondrial apoptosis and PI3K/Akt signaling pathways, which involved caspase-3 activation, RARP-1 cleavage, the loss of mitochondrial membrane potential, the release of cytochrome c into the cytoplasm and the inhibition of the phosphorylation of Akt. Intrigued by their interesting biological activities as well as novel and challenging structures, we selected the tetradecasaccharide repeating unit 1 and shorter sequences 2 and 3 of LGPS-1 as synthetic targets for in-depth biological studies on the structure–activity relationships and exploitation of their unknown biological functions.
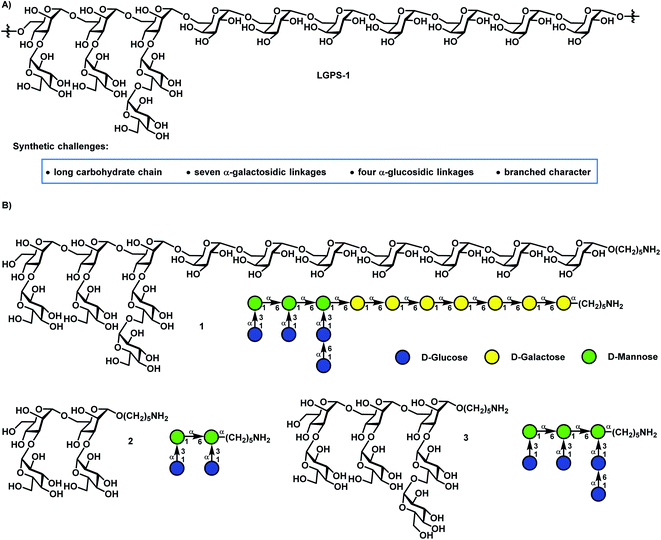 |
| | Fig. 1 (A) The structure of polysaccharide LGPS-1 from Lentinus giganteus, and (B) the structures of the tetradecasaccharide repeating unit 1 and shorter sequences (2 and 3). | |
Different from the traditional synthesis of glycans, one-pot glycan assembly not only omits the workups of glycosylation interval and purification of the intermediates, but also accelerates the synthesis and reduces chemical waste.17a,25,29 Herein, through the orthogonal one pot glycosylation strategy with glycosyl N-phenyltrifluoroacetimidate30 (PTFAI) as the donor, and glycosyl ortho-alkynylbenzoates31 (ABz) (Yu glycosylation) and glycosyl ortho-(1-phenylvinylbenzoates)32 (PVB) as the bifunctional acceptors, we report the stereoselective synthesis of Lentinus giganteus glycans 1–3. The following steps are highlighted in our synthetic strategies: (1) a new stereoselective α-galactosylation method via a merging reagent modulation and remote anchimeric assistance (RMRAA) glycosylation strategy, (2) DMF-modulated α-(1→3)-glucosylation in a highly stereoselective manner, (3) the RMRAA glycosylation strategy for highly stereoselective α-(1→6)-glucosylation, (4) several orthogonal one-pot glycosylations to streamline oligosaccharide synthesis, and (5) convergent [7 + 7] glycosylation for the final assembly of the target tetradecasaccharide 1.
Results and discussion
Retrosynthesis
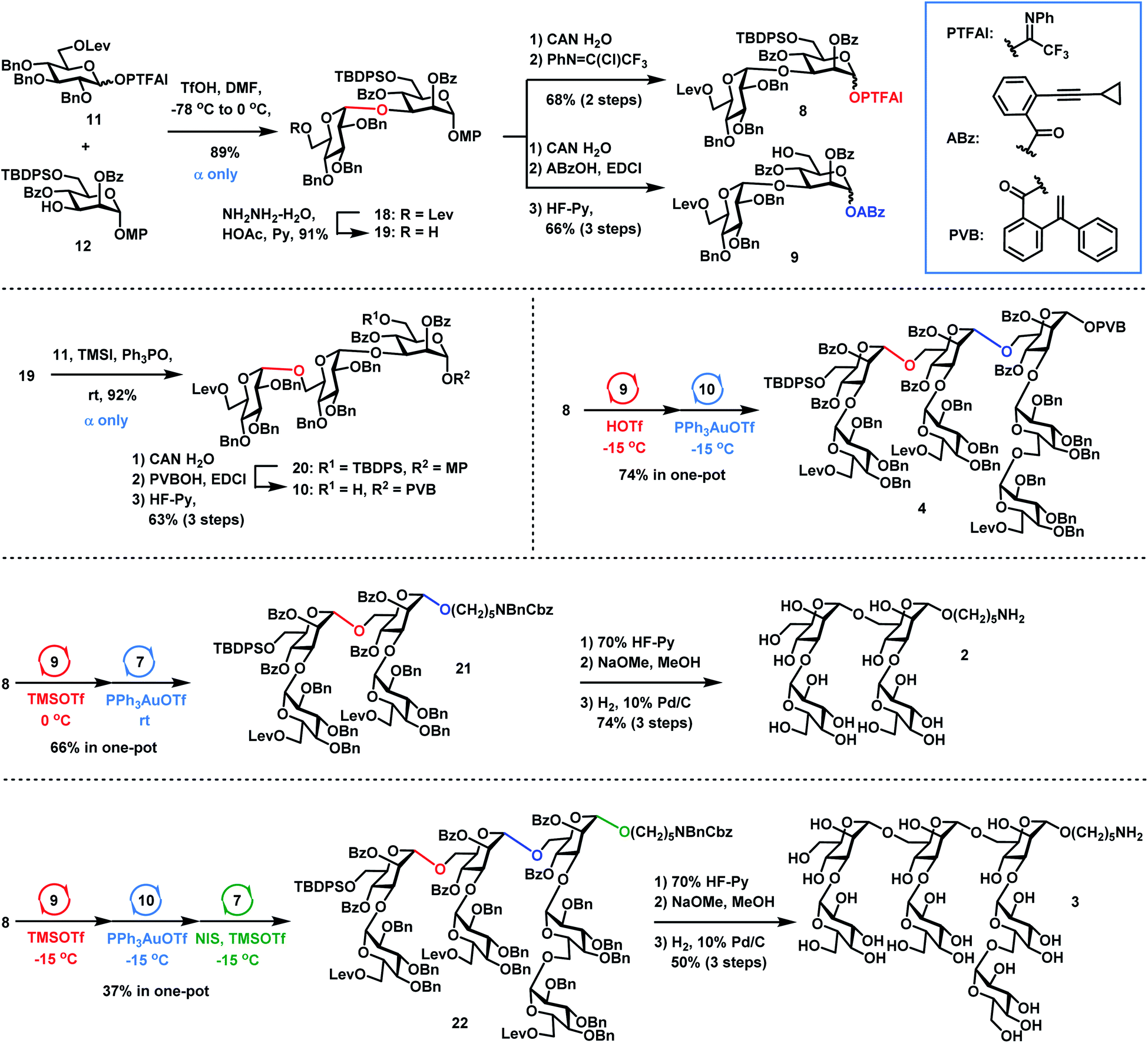
Retrosynthetic analysis (Scheme 1) of tetradecasaccharide 1 showed that the assembly can be achieved via the convergent [7 + 7] 1,2-trans stereoselective mannosylation of glycosyl PVB 4 and the heptasaccharide acceptor 5. As for the preparation of heptasaccharide acceptor 5, galactosyl PTFAI 6 and the linker HO(CH2)5NBnCbz 7 were selected to undergo the iterative and stereoselective α-galactosylation and deprotection of Lev for chain elongation. The design of monosaccharide building block 6 was based on stereoselective α-galactosylation studies, as described below. As for the synthesis of another heptasaccharide donor 4, three-component orthogonal one-pot 1,2-trans stereoselective mannosylation of disaccharide PTFAI 8, disaccharide ABz 9, and trisaccharide PVB 10 was designed to assemble 4. As for the synthesis of α-Glc-(1→3)-Man disaccharides 8–9 and α-Glc-(1→6)-α-Glc-(1→3)-Man trisaccharide 10, an α-(1→3) glucosyl linkage can be built by DMF-modulated stereoselective 1,2-cis glucosylation of glucosyl PTFAI donor 11 and the mannoside acceptor 12,33 while an α-(1→6) glucosyl bond can be stereoselectively constructed via the recently developed RMRAA glycosylation of 6-O-levulinoyl (Lev) glucosyl PTFAI donor 11.25b
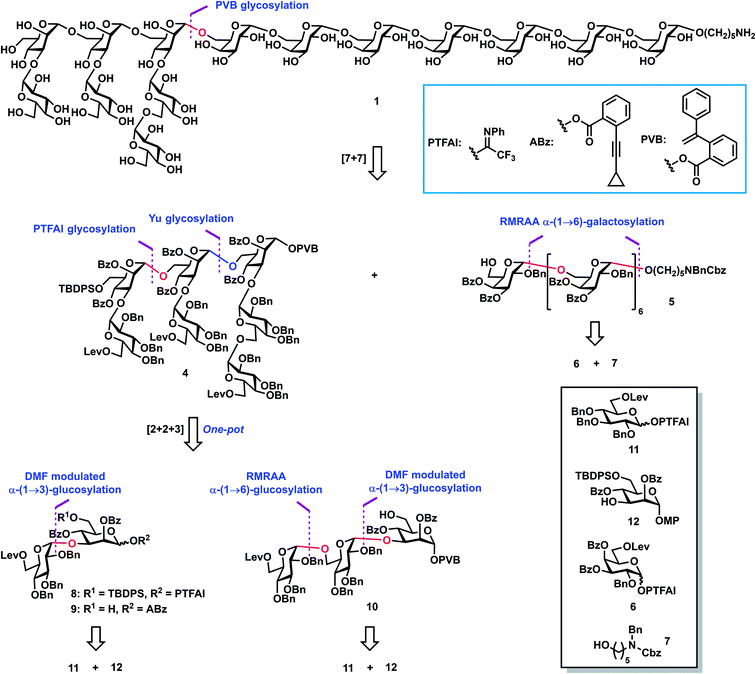 |
| | Scheme 1 Retrosynthetic analysis of the tetradecasaccharide 1. | |
Reaction optimization of α-galactosylation
Recently, we introduced the RMRAA glycosylation strategy for the highly stereoselective assembly of 1,2-cis glucosyl linkages.25b We wondered whether this RMRAA glycosylation strategy could be extended to α-galactosylation. The highly reactive primary alcohol linker 7 as a strong nucleophile acceptor was selected to investigate 1,2-cis galactosylation with galactosyl donors 13 (Scheme 2A). When perbenzyl group-protected galactosyl PTFAI 13a was coupled with 7 using a TMSI and Ph3PO reagent combination, the monosaccharide 14a was generated in 94% yield with a low selectivity (α/β = 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). To our surprise, when the C-6-Bn group in 13a was replaced with the Lev group with the remote anchimeric assistance effect or the TBDPS group with the steric shielding effect in 13b or 13c, low stereoselectivities (α/β = 2:1 for Lev; α/β = 1.5:1 for TBDPS) were also obtained, which was totally different from that of 1,2-cis glucosylation.25b Interestingly, when the C-4-Bn group was exchanged with the Bz group in 13d, the stereoselectivity was dramatically improved (α/β = 13:1).34 The additional replacement of the C-6-Bn group with the Lev group or the TBDPS group in 13e or 13f did not have beneficial effects on the stereoselectivities (α/β = 12:1 for 13e and 13f). When the C-3-Bn group was replaced with the Bz group in 13g, an improved stereoselectivity (α/β = 7:1) was also obtained. To our delight, when both C-3 and C-4 Bn groups were exchanged with Bz groups in 13h, the monosaccharide 14h was prepared in an excellent yield (95%) with an excellent stereoselectivity (α/β > 50:1). In comparison, without reagent modulation (TMSI and Ph3PO), when 13h was coupled with 7 in the activation of TMSOTf, a moderate stereoselectivity (α/β = 5:1) of 14h was obtained. Furthermore, when the corresponding galactosyl bromide 13i was glycosylated with 7 under Demchenko's protocol (Ag2SO4, HOTf, CH2Cl2, 0 °C to rt),35 a moderate stereoselectivity (α/β = 7:1 for 13i) was obtained. A low stereoselectivity (α/β = 1.5:1 for 13j) was also obtained when the thioglycoside 13j was coupled with 7 under Seeberger's protocol (NIS, TMSOTf, CH2Cl2, −20 °C).36 These results clearly demonstrate that highly stereoselective 1,2-cis galactosylation can be achieved via the RMRAA glycosylation strategy. Moreover, the scalability of this RMRAA α-galactosylation protocol was demonstrated by the efficient synthesis of 14h on a 1.4 g scale in 92% isolated yield.
:1). To our surprise, when the C-6-Bn group in 13a was replaced with the Lev group with the remote anchimeric assistance effect or the TBDPS group with the steric shielding effect in 13b or 13c, low stereoselectivities (α/β = 2:1 for Lev; α/β = 1.5:1 for TBDPS) were also obtained, which was totally different from that of 1,2-cis glucosylation.25b Interestingly, when the C-4-Bn group was exchanged with the Bz group in 13d, the stereoselectivity was dramatically improved (α/β = 13:1).34 The additional replacement of the C-6-Bn group with the Lev group or the TBDPS group in 13e or 13f did not have beneficial effects on the stereoselectivities (α/β = 12:1 for 13e and 13f). When the C-3-Bn group was replaced with the Bz group in 13g, an improved stereoselectivity (α/β = 7:1) was also obtained. To our delight, when both C-3 and C-4 Bn groups were exchanged with Bz groups in 13h, the monosaccharide 14h was prepared in an excellent yield (95%) with an excellent stereoselectivity (α/β > 50:1). In comparison, without reagent modulation (TMSI and Ph3PO), when 13h was coupled with 7 in the activation of TMSOTf, a moderate stereoselectivity (α/β = 5:1) of 14h was obtained. Furthermore, when the corresponding galactosyl bromide 13i was glycosylated with 7 under Demchenko's protocol (Ag2SO4, HOTf, CH2Cl2, 0 °C to rt),35 a moderate stereoselectivity (α/β = 7:1 for 13i) was obtained. A low stereoselectivity (α/β = 1.5:1 for 13j) was also obtained when the thioglycoside 13j was coupled with 7 under Seeberger's protocol (NIS, TMSOTf, CH2Cl2, −20 °C).36 These results clearly demonstrate that highly stereoselective 1,2-cis galactosylation can be achieved via the RMRAA glycosylation strategy. Moreover, the scalability of this RMRAA α-galactosylation protocol was demonstrated by the efficient synthesis of 14h on a 1.4 g scale in 92% isolated yield.
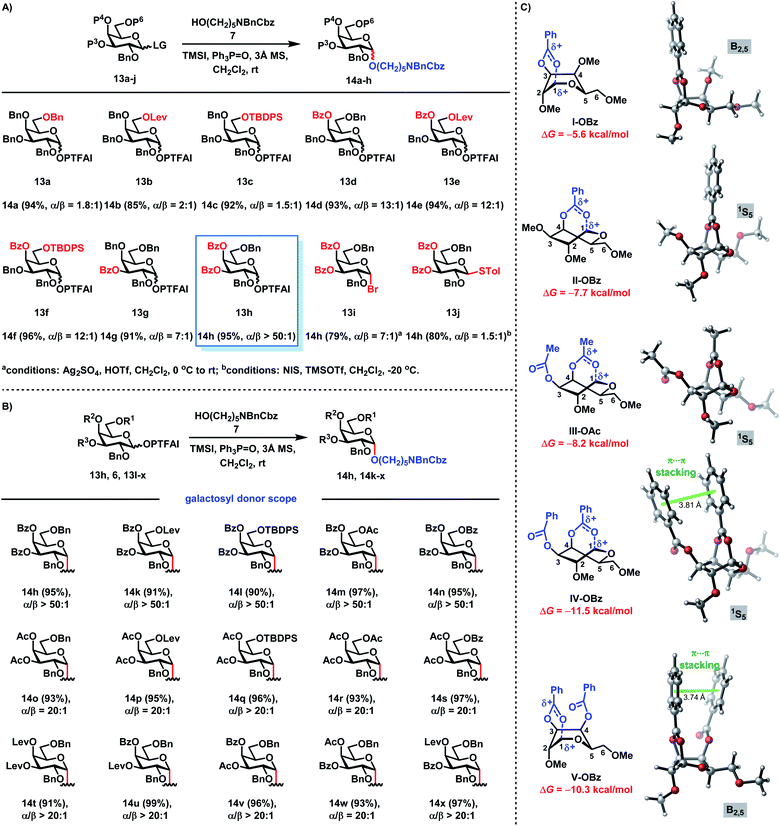 |
| | Scheme 2 (A) Reaction optimization of α-galactosylation; (B) the scope of galactosyl PTFAI donors; (C) computed geometries of β-oxocarbenium stabilized by remote acyl groups. The relative free energies (ΔG) are with respect to the oxocarbenium without the stabilization of remote acyl groups. | |
The scope of galactosyl PTFAI donors
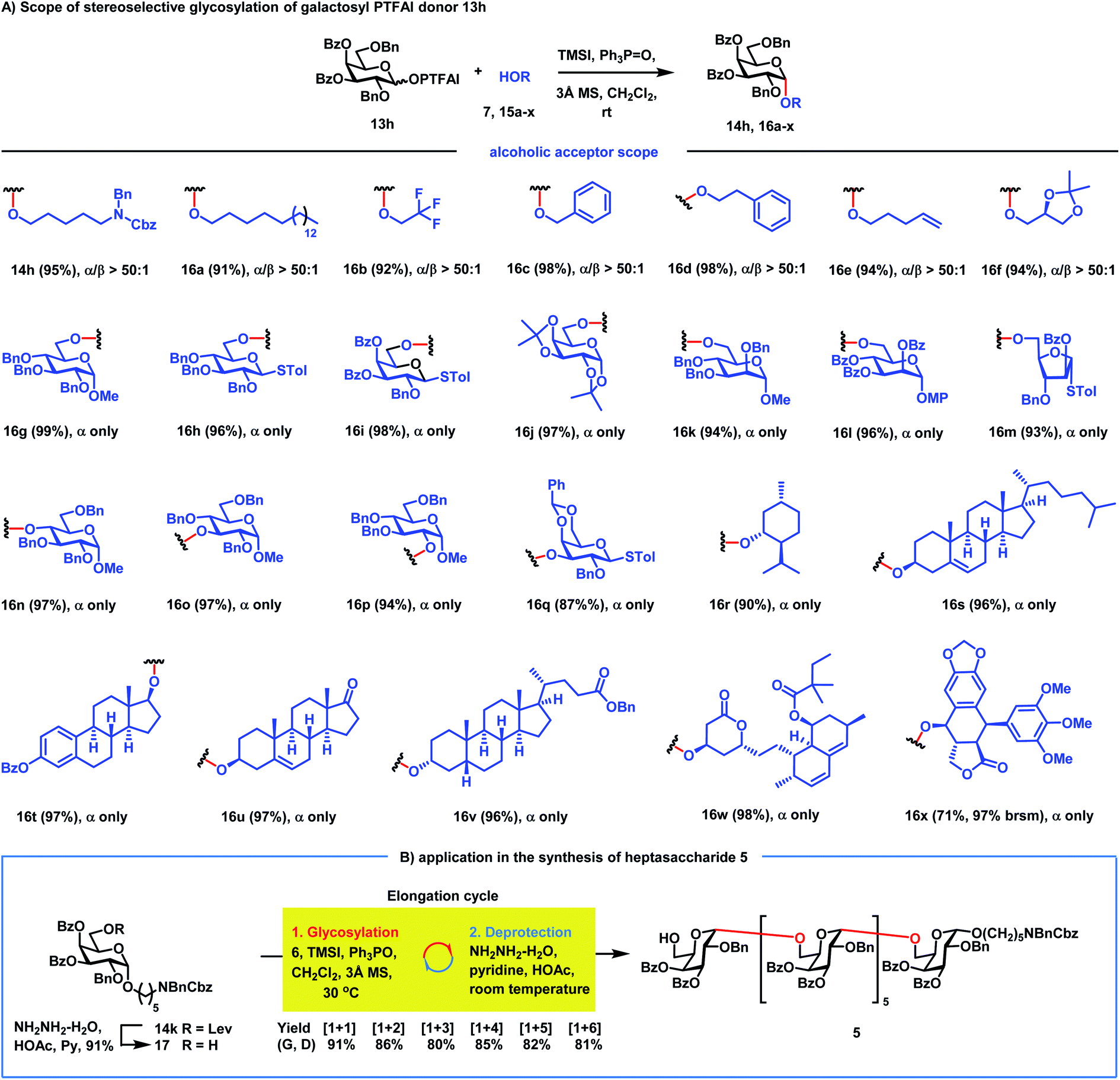
We next investigated the scope of galactosyl PTFAI donors using the linker 7 as a strong nucleophile and TMSI and Ph3PO as the reagent combination (Scheme 2B). When both C3 and C4 positions were fixed for Bz groups, the C6 position was suitable for various groups including Lev, TBDPS, Ac and Bz in galactosyl donors (6, 13l–n), providing the corresponding α-galactosides 14k–n in excellent yields (90–97%) with outstanding stereoselectivities (α/β > 50:1). Besides the Bz group, Ac or Lev groups were also suitable acyl groups for both C3 and C4 positions in donors (13o–s), and the corresponding α-galactosides 14o–s were obtained in 91–97% yields with excellent stereoselectivities (α/β ≥ 20:1). Interestingly, when C3 and C4 positions were installed with different acyl groups in donors (13t–x), α-galactosides 14t–x were also prepared in 93–99% yields with great stereocontrol (α/β ≥ 20:1).
DFT calculations
In order to better understand the remote anchimeric assistance with acyl groups, we performed DFT calculations at the M06-2X/6-311+G(d,p)-SMD//M06-2X/6-31G(d)-SMD level of theory to rationalize the origins of highly stereoselective α-galactosylation (Scheme 2C). The results indicate that, compared to the oxocarbenium without remote anchimeric assistance, the acyl groups (OAc or OBz) at C3 and/or C4 positions can thermodynamically favor the formation of β-oxocarbenium (Scheme 2C). This kind of structure shields the β-face and favors the α-face SN2 nucleophilic attack to afford 1,2-cis glycosylation. Moreover, the participation of remote acyl groups exhibits different capacities for stabilizing β-oxocarbenium. The general trend is that the two acyl groups at C3 and C4 positions (IV-OBz, V-OBz and III-OAc) are superior to one acyl group (I-OBz and II-OBz). In particular, although having two acyl groups, the 3,4-OBz groups (in IV-OBz and V-OBz) are more effective than the 3,4-OAc groups (in III-OAc) to stabilize the formed β-oxocarbenium. This is mostly due to the attractive π⋯π stacking interactions between phenyl groups in IV-OBz with the 1S5 conformation and V-OBz with the B2,5 conformation. The computed stability of the β-oxocarbenium reveals that the choice of remote acyl groups is critical for the high-level stereocontrol of galactosylation. The relative stability order of the β-oxocarbenium (3,4-OBz > 3,4-OAc > 4-OBz > 3-OBz) is fully consistent with the experimental observations (Scheme 2A and B). Together with the β-phosphonium iodide modulated by Ph3PO and TMSI, the thermodynamically favored β-oxocarbenium with a rational choice of remote anchimeric acyl groups can significantly suppress the undesired oxocarbenium species, thus favoring the nucleophilic SN2 displacement for excellent 1,2-cis galactosylation.
The scope of alcoholic acceptors in α-galactosylation
Next, the scope of α-galactosylation with 3,4-di-O-Bz galactosyl PTFAI donor 13h was investigated (Scheme 3A). We were pleased to discover that a wide range of primary alcohols 15a–f including such strong nucleophiles as 1-octadecanol, benzyl alcohol, 2-phenylethanol and 4-penten-1-ol were coupled smoothly with 13h, providing α-galactosides 16a–f in 91–98% yields with excellent stereoselectivities (α/β > 50:1). It is worth noting that other glycosylation methods often failed to achieve highly stereoselective 1,2-cis glycosylation when highly reactive primary alcohols were used as strong nucleophile acceptors.37–43 Glycosylation of carbohydrate acceptors 15g–m including D-glucopyranosyl, D-galactopyranosyl, D-mannopyranosyl and D-arabinofuranosyl primary alcohols with 13h also proceeded smoothly, generating the corresponding disaccharides 16g–m in excellent yields (93–99%) as single α isomers. Carbohydrate secondary alcohols 15n–q including the hindered O4-position of glucose were also glycosylated efficiently with 13h, producing α-(1→4), (1→3), and (1→2)-disaccharides 16n–q in 87–97% yields with complete stereocontrol. Gal-α-(1→3)-Gal disaccharide 16q is a part of the structural motif of the α-Gal epitope, which could produce acute rejections during xenotransplantations.44 Besides carbohydrate acceptors, bioactive molecules 15r–v including menthol 15r, cholesterol 15s, estradiol benzoate 15t, epiandrosterone 15u and lithocholic benzoate 15v were also amenable to this galactosylation method, furnishing the corresponding α-galactosides 16r–v in excellent yields with complete stereoselectivities. Impressively, even complex bioactive molecules like simvastatin 15w with antihyperlipidemic and anti-inflammatory activities and podophyllotoxin 15x with antitumor and antiviral activities are also suitable acceptors, generating galactosylated simvastatin 16w and galactosylated podophyllotoxin 16x with only an α isomer in 98% and 71% yields, respectively. It is worth noting that diverse functional groups including acetal (15f, 15j, 15q and 15x), alkene (15e, 15s and 15u), thioacetal (15h, 15i, 15m and 15q), ketone (15u), 1,3-diene (15w), lactone (15w and 15x) and electron-rich phenyl (15x) moieties were well tolerated in the reactions, which clearly proved the mild reaction conditions of this new method.
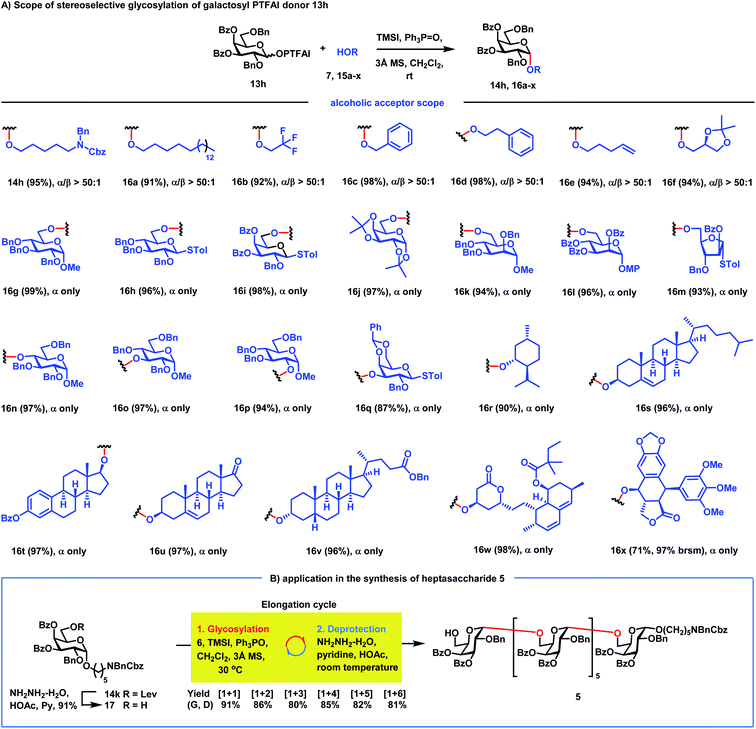 |
| | Scheme 3 (A) The scope of RMRAA 1,2-cis galactosylation; (B) application to the synthesis of heptasaccharide 5. | |
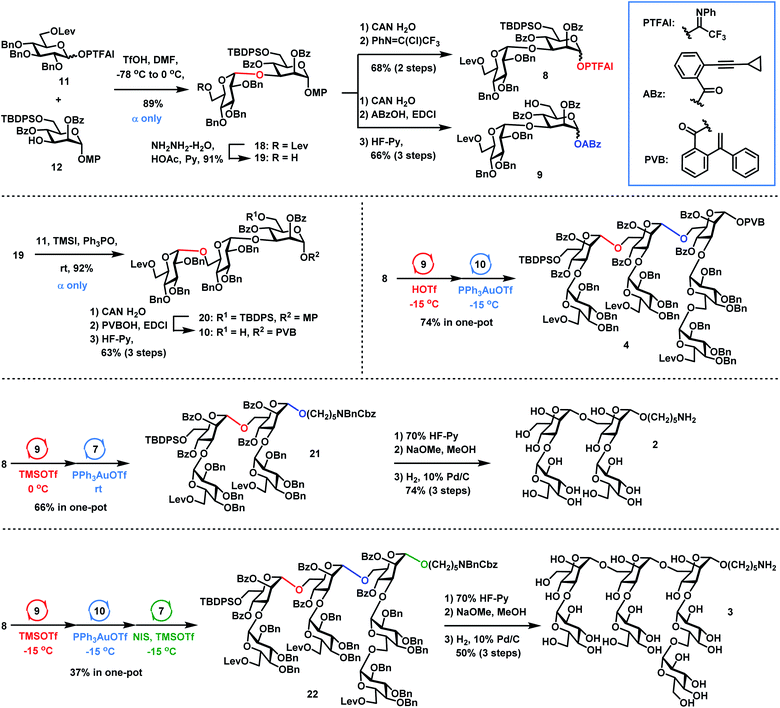 |
| | Scheme 4 Orthogonal one-pot synthesis of shorter sequences 2 and 3 and heptasaccharide donor 4. | |
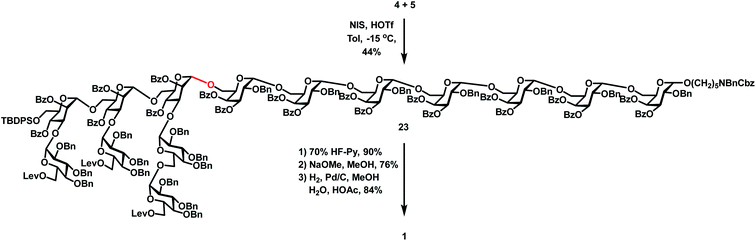 |
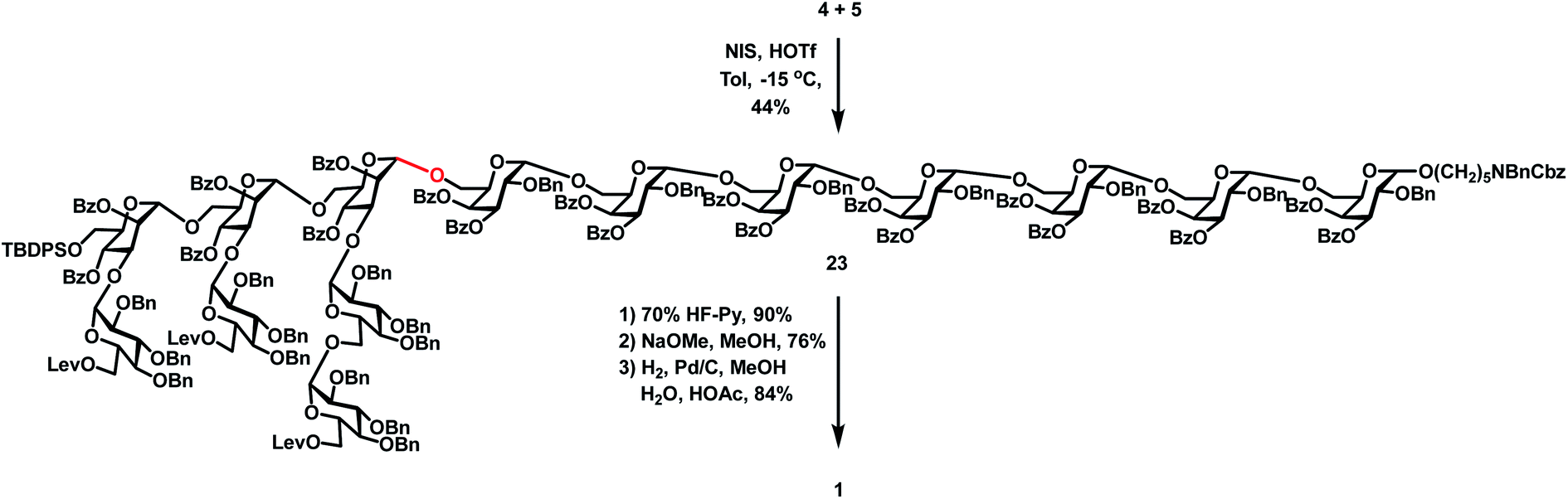
| | Scheme 5 Synthesis of the target tetradecasaccharide 1. | |
Synthesis of heptasaccharide 5
We next applied this stereoselective α-galactosylation method to the synthesis of the heptasaccharide acceptor 5. As can be seen from Scheme 3B, the iterative application of this RMRAA α-(1→6)-galactosylation of 6 and deprotection of the Lev group proceeded successfully, providing the elongated heptasaccharide 5 with excellent yields and complete stereoselectivities, which is ready for convergent [7 + 7] glycosylation. All α-Gal configurations of 5 were confirmed by the chemical shifts of anomeric carbon centers between 97.1 ppm and 97.8 ppm. Although 4,6-di-tert-butylsilyene (DTBS) directed galactosylation could also achieve highly selective α-galactosylation,45 it usually needs five steps to achieve one sugar unit elongation for the synthesis of the heptasaccharide acceptor 5, including: (1) glycosylation; (2) deprotection of the 4,6-di-tert-butylsilyene protecting group; (3) selective protection of the C6–OH group; (4) protection of the resulting C4–OH group; and (5) deprotection of the C6–OH group for the next sugar elongation cycle. In contrast, the current RMRAA α-galactosylation only needs two steps (glycosylation and deprotection of the 6-O-Lev group) for elongation of one sugar unit, which significantly reduces the step counts.46
Synthesis of 2–4
The synthesis of Lentinus giganteus glycans 2 and 3 and heptasaccharide donor 4 commenced with the α-Glc-(1→3)-Man disaccharide 18, which was obtained in 89% yield by DMF-modulated α-(1→3)-glucosylation between 11 (1.5 equiv.) and 12 under Codee's conditions (TfOH, DMF, −78 °C to 0 °C) (Scheme 4). The α-Glc configuration was confirmed by the chemical shift of the newly formed anomeric carbon center of 18 (99.2 ppm). Oxidative deprotection of the anomeric 4-methoxyphenyl group in 18 with ceric ammonium nitrate (CAN) afforded the hemiacetal intermediate, which was converted to disaccharide PTFAI 8via the imidate installation, and disaccharide ABz 9 by esterification with ABzOH and deprotection of the TBDPS group with 70% HF-Py. The further coupling of 6-O-Lev equipped donor 11 (1.5 equiv.) with disaccharide 19 derived from 18 under TMSI–Ph3PO reaction conditions successfully furnished the α-Glc-(1→6)-α-Glc-(1→3)-Man trisaccharide 20 as a single α isomer in 92% yield, which was converted to trisaccharide PVB 10 in 63% overall yield via the following functional groups transformation: (1) removal of the anomeric para-methoxyphenyl group with CAN in H2O; (2) condensation of the resulting hemiacetal with PVBOH; (3) deprotection of the TBDPS group with 70% HF-Py. As for the synthesis of 2, coupling of disaccharide PTFAI donor 8 (1.2 equiv.) with disaccharide ABz 9 (1.0 equiv.) in the catalysis of TMSOTf at 0 °C furnished the tetrasaccharide intermediate, which underwent further Yu glycosylation with HO(CH2)5NBnCbz 7, providing the desired tetrasaccharide 21 in 66% yield in one-pot. Removal of the TBDPS group in 21 with 70% HF-Py, deprotection of all Bz groups and Lev groups with NaOMe in MeOH, and global removal of the Cbz group and all Bn groups (H2, 10% Pd/C) successfully generated the desired shorter sequence 2 in 74% overall yield. As for the synthesis of 4 and 3, sequential coupling of PTFAI donor 8 (1.2 equiv.), ABz 9 (1.0 equiv.) and PVB 10 (0.9 equiv.) afforded the desired heptasaccharide 4 in 74% yield in a one-pot manner. Alternatively, orthogonal one-pot glycosylation of PTFAI donor 8 (1.2 equiv.), ABz 9 (1.0 equiv.), PVB 10 (0.9 equiv.) and HO(CH2)5NBnCbz 7 (1.5 equiv.) successfully produced the desired heptasaccharide 22 in 37% overall yield. The desired shorter sequence 3 was prepared in 50% overall yield via removal of the TBDPS group with 70% HF-Py, deprotection of all Lev and Bz groups with NaOMe in MeOH, and hydrogenolysis of 13 Bn groups and one Cbz group with 10% Pd/C. It was noted that orthogonal one-pot glycosylation based on PTFAI glycosylation, Yu glycosylation and PVB glycosylation precluded such issues as aglycon transfer inherent to thioglycoside-based orthogonal one-pot glycosylation.
Synthesis of the target tetradecasaccharide 1
With the desired heptasaccharides 4 and 5 in hand, we embarked on the final assembly of tetradecasaccharide 1. The identification of conditions for efficient [7 + 7] glycosylation between donor 4 and acceptor 5 was not trivial. Various conditions failed to achieve effective coupling due to the formation of significant amounts of donor hydrolyzed and eliminated products or an acceptor TMS-capped side product. Ultimately, glycosylation of heptasaccharide donor 4 and heptasaccharide acceptor 5 in the presence of NIS and HOTf in toluene at −15 °C successfully generated the desired tetra-decasaccharide 23 in an acceptable 44% yield (Scheme 5). Finally, removal of the TBDPS group with 70% HF-Py, deprotection of three Lev groups and twenty Bz groups with NaOMe in MeOH, and global hydrogenolysis of 20 Bn groups and one Cbz group with 10% Pd/C in 23 successfully produced tetra-decasaccharide 1 in 57% overall yield over three steps.
The 1H NMR spectra of tetra-decasaccharide 1 and the shorter sequences 2 and 3 with the highlighted chemical shifts of the anomeric protons are shown in Fig. 2. The structures of 1–3 were confirmed by their NMR signal assignment and MAIDI-TOF and ESI mass spectra. In-depth biological studies could be carried out for the synthetic Lentinus giganteus glycans 1–3 installed with a pentyl amine linker at the reducing end.
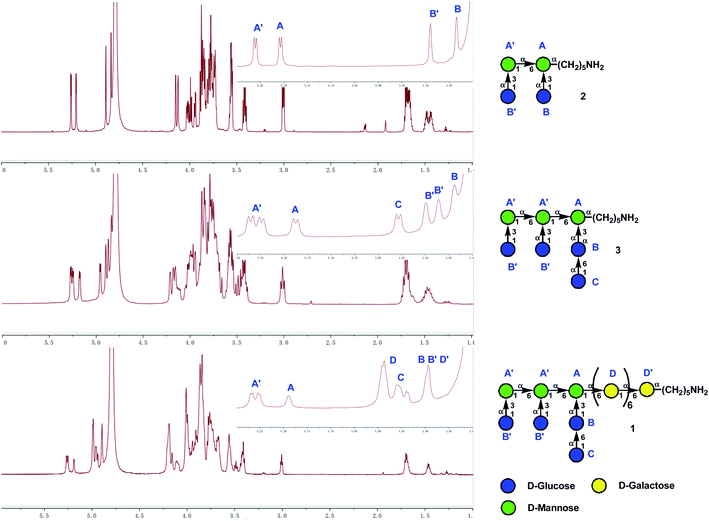 |
| | Fig. 2 Evidence for structures of 1 and the shorter sequences 2 and 3 by NMR spectroscopy. | |
Conclusions
Herein we describe highly stereoselective α-glycosylation and orthogonal one-pot glycosylation strategies for the first chemical synthesis of the tetra-decasaccharide repeating unit 1 and shorter sequences 2 and 3 from Lentinus giganteus glycans, which have antitumor activities through inducing apoptosis. Several steps are highlighted in our synthetic approach: (1) newly developed highly stereoselective RMRAA α-(1→6)-galactosylation, (2) stereoselective α-(1→3)-glucosylation via DMF-modulation, (3) highly stereoselective α-(1→6)-glucosylation via the RMRAA glycosylation strategy, (4) several one-pot glycosylations to facilitate glycan synthesis, and (5) convergent [7 + 7] glycosylation for the synthesis of the final target tetradecasaccharide. In particular, the new RMRAA α-galactosylation method enjoys mild reaction conditions, broad substrate scopes and highly shortened step counts in the synthesis of heptasaccharide 5 in comparison with DTBS directed α-galactosylation. Furthermore, DFT calculations uncovered the origins of remote anchimeric assistance effects (3,4-OBz > 3,4-OAc > 4-OBz > 3-OBz) of acyl groups. We believe that the present work will inspire the advances of other synthetic strategies towards streamlining the chemical synthesis of long, branched and complex glycans with many 1,2-cis-glycosidic linkages.47
Data availability
Experimental data including experimental procedures, characterization data, and NMR spectra for new compounds; mechanistic experiments; detailed computational studies, calculated structures, and energies is accessible in the ESI.†
Author contributions
G. X. conceived the idea, designed the research, supervised the work and wrote the manuscript. Y. Z. made the initial discovery and performed all experiments. S. L., H. H. and R. S. performed some experiments. Y. H. and G. L. performed DFT calculations. Y. H. and G. L. performed DFT calculations and wrote part of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
G. X. acknowledges the financial support from the Key Project of the Natural Science Foundation of Yunnan Province (No. 202101AS070030), the National Natural Science Foundation of China (21907097), the Young Talents Project of High-level Talent Introduction Program of Yunnan Province, the CAS Pioneer Hundred Talents Program (No. 2017-128), the State Key Laboratory of Phytochemistry and Plant Resources in West China (P2021-ZZ02) and the Kunming Institute of Botany. G. L. acknowledges the financial support from the National Natural Science Foundation of China (No. 21973055), the Natural Science Foundation of Shandong Province (ZR2019MB049), the Taishan Scholar of Shandong Province (No. tsqn201812013) and the Qilu Young Scholar of Shandong University. DFT calculations were performed at the HPC Cloud Platform of Shandong University. Y. Z. acknowledges the financial support from the China Postdoctoral Science Foundation, No. 70 General Fund, 2021 (No. E11W4212), and the CAS Special Research Assistant Project (2021).
Notes and references
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- P. H. Seeberger, Chem. Rev., 2021, 121, 3598–3626 CrossRef CAS PubMed.
- D. Crich, J. Am. Chem. Soc., 2021, 143, 17–34 CrossRef CAS PubMed.
-
(a) L. Krasnova and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 3735–3754 CrossRef CAS PubMed;
(b) D. A. Williams, K. Pradhan, A. Paul, I. R. Olin, O. T. Tuck, K. D. Moulton, S. S. Kulkarni and D. H. Dube, Chem. Sci., 2020, 11, 1761–1774 RSC.
- G. Xiao, X. Shao, D. Zhu and B. Yu, Nat. Prod. Rep., 2019, 36, 769–787 RSC.
- C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed.
- M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed.
- W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed.
- P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS PubMed.
- S. J. Danishefsky, Y.-K. Shue, M. N. Chang and C.-H. Wong, Acc. Chem. Res., 2015, 48, 643–652 CrossRef CAS.
- T. J. Boltje, T. Buskas and G. J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- B. Ernst and J. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
-
C.-H. Wong, Carbohydrate-Based Drug Discovery, Wiley-VCH, Weinheim, 2003 Search PubMed.
-
(a) S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed;
(b) H.-Y. Wang, S. A. Blaszczyk, G. Xiao and W. Tang, Chem. Soc. Rev., 2018, 47, 681–701 RSC.
-
(a) A. V. Demchenko, Curr. Org. Chem., 2003, 7, 35–79 CrossRef CAS;
(b) A. V. Demchenko, Synlett, 2003, 1225–1240 CAS;
(c)
A. E. Christina, G. A. van der Marel and J. D. C. Codee, Modern Synthetic Methods in Carbohydrate Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014 Search PubMed;
(d) S. Crotti and R. Adamo, Curr. Org. Synth., 2013, 10, 501–524 CrossRef CAS;
(e) S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC.
- Y. Matsuzaki, Y. Ito, Y. Nakahara and T. Ogawa, Tetrahedron Lett., 1993, 34, 1061–1064 CrossRef CAS.
- V. Pozsgay, Tetrahedron: Asymmetry, 2000, 11, 151–172 CrossRef CAS.
-
(a) M. Joe, Y. Bai, R. C. Nacario and T. L. Lowary, J. Am. Chem. Soc., 2007, 129, 9885–9901 CrossRef CAS PubMed;
(b) K. Shen, B. Bai, Y. H. Liu and T. L. Lowary, Angew. Chem., Int. Ed., 2021, 60, 24859–24863 CrossRef CAS;
(c) L. Wang and T. L. Lowary, Chem. Sci., 2021, 12, 12192–12200 RSC.
- A. Ishiwata and Y. Ito, J. Am. Chem. Soc., 2011, 133, 2275–2291 CrossRef CAS PubMed.
- M. Islam, G. P. Shinde and S. Hotha, Chem. Sci., 2017, 8, 2033–2038 RSC.
- Y. Wu, D. Xiong, S. Chen, Y. Wang and X. Ye, Nat. Commun., 2017, 8, 14851 CrossRef CAS PubMed.
-
(a) Y. Zhang, Z. Chen, Y. Huang, S. He, X. Yang, Z. Wu, X. Wang and G. Xiao, Angew. Chem., Int. Ed., 2020, 59, 7576–7584 CrossRef CAS PubMed;
(b) Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS.
- Y. Zhu, M. Delbianco and P. H. Seeberger, J. Am. Chem. Soc., 2021, 143, 9758–9768 CrossRef CAS PubMed.
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 4142 CrossRef CAS PubMed.
- Y. Tian, Y. Zhao, H. Zeng, Y. Zhang and B. Zheng, Carbohydr. Polym., 2016, 154, 231–240 CrossRef CAS.
-
(a) Y. Zhang, G. Xiang, S. He, Y. Hu, Y. Liu, L. Xu and G. Xiao, Org. Lett., 2019, 21, 2335–2339 CrossRef CAS PubMed;
(b) H. He, L. Xu, R. Sun, Y. Zhang, Y. Huang, Z. Chen, P. Li, R. Yang and G. Xiao, Chem. Sci., 2021, 12, 5143–5151 RSC;
(c) R. Yang, H. He, Z. Chen, Y. Huang and G. Xiao, Org. Lett., 2021, 23, 8257–8261 CrossRef CAS PubMed;
(d) G. Xiao and H. He, Eur. J. Org. Chem., 2021, 2021, 3681–3689 CrossRef CAS;
(e) Y. Zhang and G. Xiao, J. Carbohydr. Chem., 2021, 40, 327–338 CrossRef CAS.
-
(a) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC;
(b) B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
-
(a) B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed;
(b) W. Li and B. Yu, Chem. Soc. Rev., 2018, 47, 7954–7984 RSC;
(c) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS.
- P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405 CrossRef CAS PubMed.
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
-
(a) M. Marianski, E. Mucha, K. Greis, S. Moon, A. Pardo, C. Kirschbaum, D. A. Thomas, G. Meijer, G. von Helden, K. Gilmore, P. H. Seeberger and K. Pagel, Angew. Chem., Int. Ed., 2020, 59, 6166–6171 CrossRef CAS;
(b) K. Upadhyaya, Y. P. Subedi and D. Crich, Angew. Chem., Int. Ed., 2021, 60, 25397–25403 CrossRef CAS.
- M. Shadrick, Y. Singh and A. V. Demchenko, J. Org. Chem., 2020, 85, 15936–15944 CrossRef CAS PubMed.
- H. S. Hahm, M. Hurevich and P. H. Seeberger, Nat. Commun., 2016, 7, 12482 CrossRef CAS PubMed.
- D. K. Njeri, E. A. Valenzuela and J. R. Ragains, Org. Lett., 2021, 23, 8214–8218 CrossRef CAS PubMed.
-
(a) B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira and P. H. Seeberger, Angew. Chem., Int. Ed., 2016, 55, 14431–14434 CrossRef CAS;
(b) B. Schumann, R. Pragani, C. Anish, C. L. Pereira and P. H. Seeberger, Chem. Sci., 2014, 5, 1992–2002 RSC;
(c) C. E. Martin, F. Broecker, S. Eller, M. A. Oberli, C. Anish, C. L. Pereira and P. H. Seeberger, Chem. Commun., 2013, 49, 7159–7161 RSC.
-
(a) S. R. Shenoy, D. M. Smith and K. A. Woerpel, J. Am. Chem. Soc., 2006, 128, 8671–8677 CrossRef CAS PubMed;
(b) M. G. Beaver and K. A. Woerpel, J. Org. Chem., 2010, 75, 1107–1118 CrossRef CAS PubMed.
- D. Safari, A. T. Dekker Huberta, A. F. Joosten John, D. Michalik, C. de Souza Adriana, R. Adamo, M. Lahmann, A. Sundgren, S. Oscarson, P. Kamerling Johannis and H. Snippe, Infect. Immun., 2008, 76, 4615–4623 CrossRef CAS PubMed.
- T. J. Boltje, W. Zhong, J. Park, M. A. Wolfert, W. Chen and G.-J. Boons, J. Am. Chem. Soc., 2012, 134, 14255–14262 CrossRef CAS PubMed.
- M. J. L. Thijssen, M. N. van Rijswijk, J. P. Kamerling and J. F. G. Vliegenthart, Carbohydr. Res., 1998, 306, 93–109 CrossRef CAS PubMed.
- Y. Jiao, Z. Ma, D. Hodgins, B. Pequegnat, L. Bertolo, L. Arroyo and M. A. Monteiro, Carbohydr. Res., 2013, 378, 15–25 CrossRef CAS PubMed.
- U. Galili, Immunol. Today, 1993, 14, 480–482 CrossRef CAS.
-
(a) A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef CAS;
(b) A. Imamura, H. Ando, H. Ishida and M. Kiso, Heterocycles, 2008, 76, 883–908 CrossRef CAS;
(c) A. Imamura, H. Ando, H. Ishida and M. Kiso, Org. Lett., 2005, 7, 4415–4418 CrossRef CAS;
(d) A. Imamura, A. Kimura, H. Ando, H. Ishida and M. Kiso, Chem.–Eur. J., 2006, 12, 8862–8870 CrossRef CAS PubMed;
(e) A. Imamura, N. Matsuzawa, S. Sakai, T. Udagawa, S. Nakashima, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2016, 81, 9086–9104 CrossRef CAS PubMed;
(f) N. Yagami and A. Imamura, Rev. Agric. Sci., 2018, 6, 1–20 CrossRef.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
-
(a) X. Ma, Z. Zheng, Y. Fu, X. Zhu, P. Liu and L. Zhang, J. Am. Chem. Soc., 2021, 143, 11908–11913 CrossRef CAS PubMed;
(b) Q. Li, S. M. Levi and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 11865–11872 CrossRef CAS PubMed;
(c) A. B. Mayfield, J. B. Metternich, A. H. Trotta and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 4061–4069 CrossRef CAS PubMed;
(d) N. Komura, K. Kato, T. Udagawa, S. Asano, H.-N. Tanaka, A. Imamura, H. Ishida, M. Kiso and H. Ando, Science, 2019, 364, 677–680 CrossRef CAS PubMed;
(e) Y. Park, C. Harper Kaid, N. Kuhl, E. Kwan Eugene, Y. Liu Richard and N. Jacobsen Eric, Science, 2017, 355, 162–166 CrossRef CAS PubMed;
(f) K. Meng Liu, P. Y. Wang, Z. Y. Guo, D. C. Xiong, X. J. Qin, M. Liu, M. Liu, W. Y. Xue and X. S. Ye, Angew. Chem., Int. Ed., 2022, 61, e202114726 Search PubMed;
(g) X. Liu, Y. Song, A. Liu, Y. Zhou, Q. Zhu, Y. Lin, H. Sun, K. Zhu, W. Liu, N. Ding, W. Xie, H. Sun, B. Yu, P. Xu and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202201510 CAS;
(h) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q. D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022 DOI:10.1038/s41557-022-00918-z.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Roujing
Sun
a,
Gang
Lu
a,
Roujing
Sun
a,
Gang
Lu