
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMutual functionalization of dinitrogen and methane mediated by heteronuclear metal cluster anions CoTaC2−†
Li-Hui
Mou‡
ac,
Yao
Li‡
bc,
Gong-Ping
Wei‡
ac,
Zi-Yu
Li
*ac,
Qing-Yu
Liu
ac,
Hui
Chen
 *bc and
Sheng-Gui
He‡
*ac
*bc and
Sheng-Gui
He‡
*ac
aState Key Laboratory for Structural Chemistry of Unstable and Stable Species, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: shengguihe@iccas.ac.cn
bCAS Key Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
cBeijing National Laboratory for Molecular Sciences and CAS Research/Education Center of Excellence in Molecular Sciences, Beijing 100190, P. R. China
First published on 14th July 2022
Abstract
The direct coupling of dinitrogen (N2) and methane (CH4) to construct the N–C bond is a fascinating but challenging approach for the energy-saving synthesis of N-containing organic compounds. Herein we identified a likely reaction pathway for N–C coupling from N2 and CH4 mediated by heteronuclear metal cluster anions CoTaC2−, which starts with the dissociative adsorption of N2 on CoTaC2− to generate a Taδ+–Ntδ− (terminal-nitrogen) Lewis acid–base pair (LABP), followed by the further activation of CH4 by CoTaC2N2− to construct the N–C bond. The N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N cleavage by CoTaC2− affording two N atoms with strong charge buffering ability plays a key part, which facilitates the H3C–H cleavage via the LABP mechanism and the N–C formation via a CH3 migration mechanism. A novel Nt triggering strategy to couple N2 and CH4 molecules using metal clusters was accordingly proposed, which provides a new idea for the direct synthesis of N-containing compounds.
N cleavage by CoTaC2− affording two N atoms with strong charge buffering ability plays a key part, which facilitates the H3C–H cleavage via the LABP mechanism and the N–C formation via a CH3 migration mechanism. A novel Nt triggering strategy to couple N2 and CH4 molecules using metal clusters was accordingly proposed, which provides a new idea for the direct synthesis of N-containing compounds.
Introduction
The construction of N–C bonds directly from N2 offers the potential to reduce the enormous fossil-fuel consumption for the current synthesis of N-containing organic compounds through NH3, which is mainly produced via the energy-intensive Haber–Bosch process (N2 + H2 → NH3).1 Considerable research efforts have been devoted to this subject since the initial observation of N–C bond formation in the reaction of N2 complexes with organic halides.2 A long-sought goal for this chemistry has been the direct coupling of N2 with abundant carbon-based molecules that are viable in industry. However, activation of these stable molecules remains a challenge, and only CO,3–5 CO2,6–8 and unsaturated hydrocarbons9,10 have been reported to form N–C bonds with N2 induced by appropriate reactive species. CH4, the major component of widely available natural gas, is closely linked to industrial NH3 synthesis in which H2 is produced through the energy-intensive CH4-reforming process. Therefore, the direct coupling of N2 and CH4 is of great importance for economic and environmental reasons. However, the appropriate species that can mediate the coupling of inert N2 and CH4 molecules to form an N–C bond has not been revealed.Isolated gas-phase atomic clusters are important model systems for the precise design of desired active sites and the fundamental understanding of bond activation and formation processes.11 The activation and individual functionalization of N2 or CH4,12–18 and the formation of N–C bonds19–25 are also extensively studied issues in the gas-phase field. Typical examples of N–C bond formation from N2 or CH4 in gas-phase studies can be classified as follows: (i) reactions of metal carbide clusters with N222–25 or metal nitride clusters with CH4,26,27 and (ii) coupling reactions of CH4 with NH328,29 or N2 with CO2.6 In addition to reactions involving N2 or CH4, the N–C bond formation was also observed in a few other reaction systems.30–32 Compared with these N–C formation processes, the direct coupling of N2 and CH4 is rather challenging and of particular interest.
Herein, we report the first example of N–C bond formation from the mutual functionalization of N2 and CH4 mediated by heteronuclear metal cluster anions CoTaC2− under thermal collision conditions. A terminal-nitrogen (Nt) triggering strategy for the coupling of N2 and CH4 was accordingly proposed (Scheme 1), which starts with the dissociative adsorption of N2 on metal-based substrates to generate a Mδ+–Ntδ− Lewis acid–base pair (LABP), followed by the H3C–H cleavage of CH4via the LABP mechanism and the N–C formation via a CH3 migration mechanism. Considering that the activation of CH4 by some metal nitrides follows the LABP mechanism,26,27 we infer that the proposed strategy can be quite general, which is confirmed by the study on the FeTaC2−/N2/CH4 reaction system. The important roles of producing substrate-Nt complexes in the coupling of N2 and CH4, as well as the strength of M–Nt bonds in the activity of the N2 complexes were discussed.
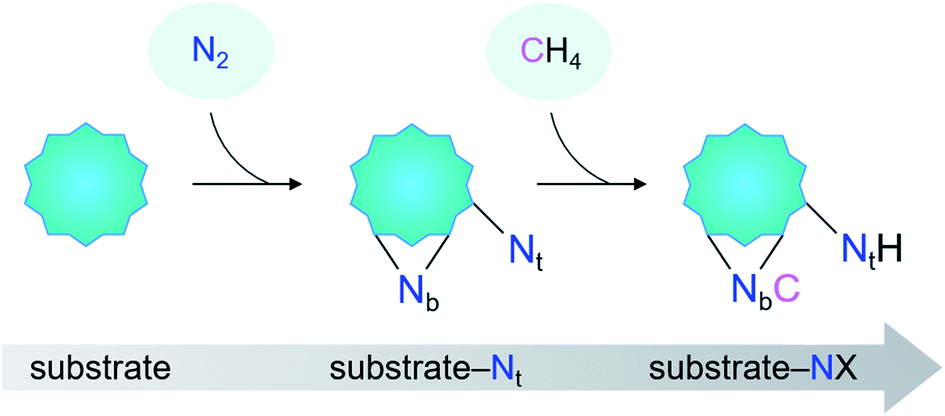 | ||
| Scheme 1 The proposed terminal-nitrogen (Nt) triggering strategy for the coupling of N2 and CH4. The key to this strategy is the predissociation of N2 on an appropriate substrate to generate a Nt and a bridging-N (Nb). | ||
Results
Cluster reactivity
The spectra in Fig. 1 have been obtained by using an online time-of-flight (TOF) mass spectrometer and show the results for the reactions of CoTaC2− (m/z = 264) with N2 and CoTaC2N2− (m/z = 292) with CH4. Reference spectra with inert He as the reactant gas were also recorded (Fig. 1a and d). The CoTaC2− ions were generated by laser ablation of a mixed Co–Ta disk target (molar ratio Co/Ta = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in the presence of 0.05% CD4 diluted with He carrier gas, and then mass-selected and thermalized to room temperature to react with N2 in a linear ion trap (LIT) reactor. As shown in Fig. 1b, on pulsing 0.56 Pa N2 into the LIT reactor, a strong product peak assigned as CoTaC2N2− appeared, suggesting the following reaction channel:
:1) in the presence of 0.05% CD4 diluted with He carrier gas, and then mass-selected and thermalized to room temperature to react with N2 in a linear ion trap (LIT) reactor. As shown in Fig. 1b, on pulsing 0.56 Pa N2 into the LIT reactor, a strong product peak assigned as CoTaC2N2− appeared, suggesting the following reaction channel:| CoTaC2− + N2 → CoTaC2N2− | (1) |
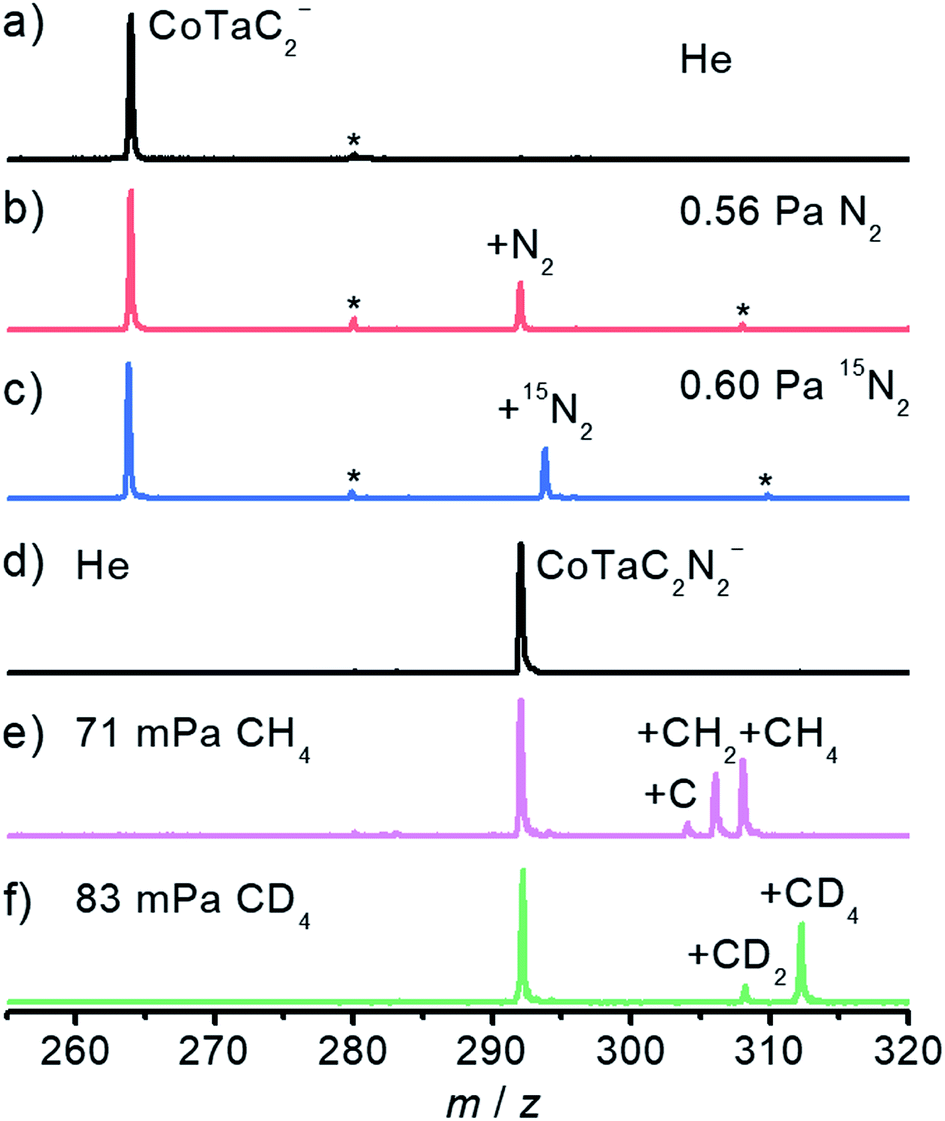 | ||
| Fig. 1 Time-of-flight mass spectra of the reactions of CoTaC2− with (a) He, (b) N2, and (c) 15N2, and CoTaC2N2− with (d) He, (e) CH4, and (f) CD4. The reaction times are 4.8 ms for (b and c), 1.8 ms for (e), and 1.9 ms for (f). Peaks marked with asterisks are due to water impurities in the gas handling system. | ||
The isotopic labeling experiment using 15N2 as the reactant gas (Fig. 1c) confirmed the above reaction channel. We should mention in passing that, a very tiny metal atom ejection channel (less than 1%) producing TaC2N2− was also observed (Fig. S1a–c†). Based on a least-squares fitting procedure (Fig. S2a†), the rate constant k1(CoTaC2−/N2) of the pseudo first-order reaction between CoTaC2− and N2 was estimated to be (8.0 ± 1.6) × 10−13 cm3 molecule−1 s−1, corresponding to a reaction efficiency (Φ = k1/kc) of about 0.1% relative to the theoretical collision rate (kc).33
To study the reaction of the N2 association product CoTaC2N2− with CH4, a newly-developed double ion trap apparatus (Scheme S1 in the ESI†) was used to first generate the CoTaC2N2− from the reaction of CoTaC2− with N2 in the first LIT reactor and then mass select CoTaC2N2− to interact with CH4 in the second LIT reactor. Upon the interaction of CoTaC2N2− with CH4 (Fig. 1e), three product peaks assigned as CoTaC2N2CH4− (m/z = 308), CoTaC2N2CH2− (m/z = 306), and CoTaC2N2C− (m/z = 304) were observed, suggesting the following reaction channels:
| CoTaC2N2− + CH4 → CoTaC2N2CH4− 49% | (2) |
| CoTaC2N2− + CH4 → CoTaC2N2CH2− + H2 41% | (3) |
| CoTaC2N2− + CH4 → CoTaC2N2C− + 2H2 10% | (4) |
The rate constant k1(CoTaC2N2−/CH4) for the reaction of CoTaC2N2− with CH4 was estimated to be (1.4 ± 0.3) × 10−11 cm3 molecule−1 s−1, corresponding to a Φ of about 1.4%. Noticeably, the reaction channel of ejecting two D2 molecules was negligible when using isotope-labeled CD4 in place of CH4 (Fig. 1f). The branching ratio of reaction channel (2) to reaction channel (3) changed to 80:20 in the CD4 experiment, and the intermolecular kinetic isotopic effect (KIE) calculated using k1(CoTaC2N2−/CH4)/k1(CoTaC2N2−/CD4) was estimated to be 1.3 (Fig. S2b and c†). The loss of H2 (D2) in the reaction of CoTaC2N2− with CH4 (CD4) suggests that activation of C–H bonds must have occurred. To determine the mechanisms of CoTaC2− + N2 and CoTaC2N2− + CH4 reactions, structural characterization of the reactant cluster ions should be performed.
Structural characterization
Photoelectron imaging spectroscopy (PEIS)34 combined with quantum chemistry calculations was employed to characterize the structures of CoTaC2− and CoTaC2N2−. The structures were optimized at the density functional theory (DFT) level,35 and their relative energies were then refined by high-level RCCSD(T) (partially spin-adapted open-shell coupled cluster method with single, double, and perturbative triple excitations) and DMRG-SC-NEVPT2 (density matrix renormalization group strongly-contract n-electron valence perturbation theory) methods.36,37 The experimental spectrum of CoTaC2− recorded with 670 nm photons at 10 K reveals a sharp peak centered at 1.63 eV, followed by three discernible peaks with electron binding energies of 1.70, 1.73, and 1.76 eV (Fig. 2a, top). The calculated lowest-lying isomer (2IS1) of CoTaC2− features a Co–Ta double bond with a Wiberg bond index (WBI) of 2.04 and a C2 ligand. The presence of two d–d bonding orbitals between Co and Ta atoms provides further evidence for the Co–Ta double bond (Fig. S12†). The Franck–Condon (FC)-simulated spectrum of the 2A → 3A vibrational transition for 2IS1 can reasonably reproduce the experimental spectrum from 1.70 to 1.76 eV (Fig. 2a, middle),38 and the calculated adiabatic electron detachment energy (ADE) of 2IS1 is close to the experimental value (1.83 eV vs. 1.70 eV), suggesting that 2IS1 is the most probable species of CoTaC2− generated in the experiment. The first spectral peak centered at 1.63 eV might come from the minor population of the isomer 4IS2 with a relative energy of 0.59 eV higher than 2IS1, considering that its calculated ADE (1.59 eV) and simulated profile of the 4A → 3A transition match well with the first spectral peak (Fig. 2a, bottom). A more detailed discussion of the structural assignment of CoTaC2− is provided in the ESI (Fig. S3†).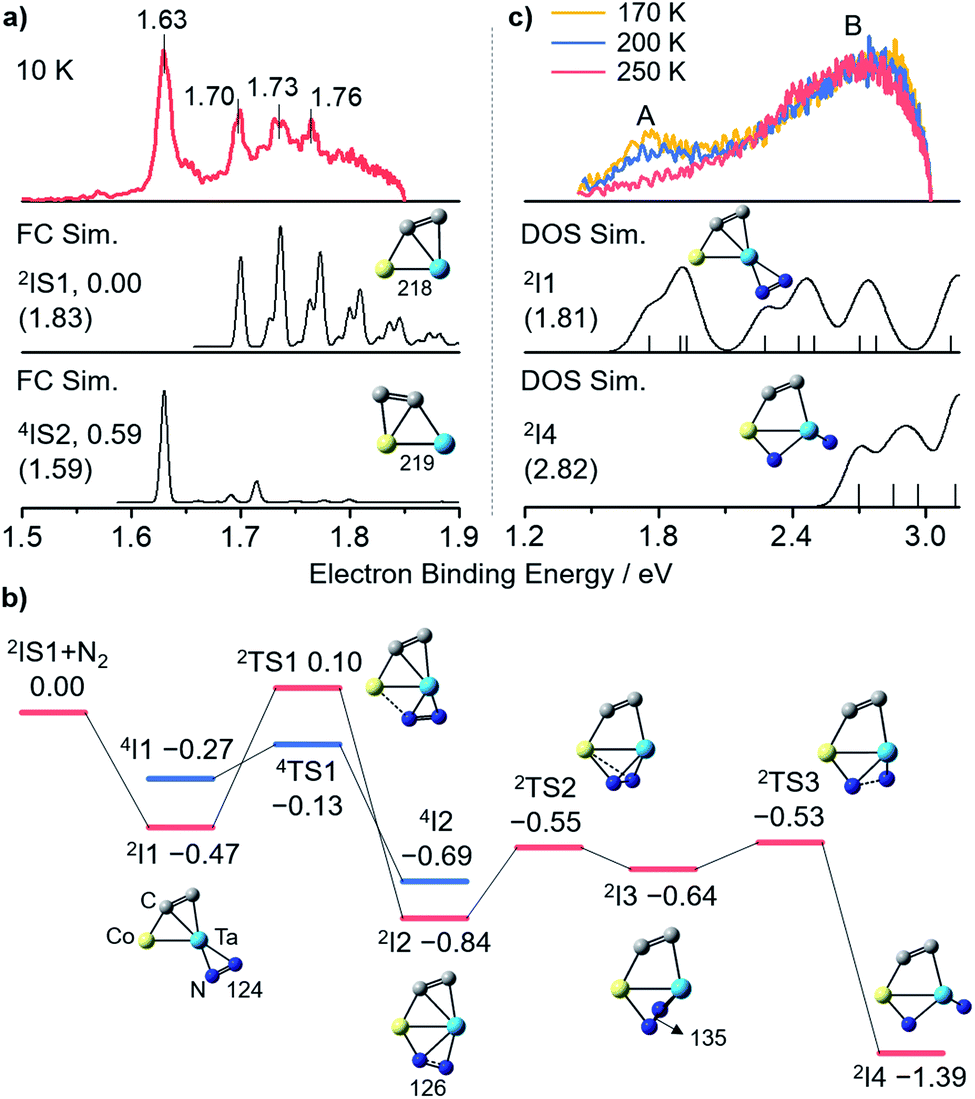 | ||
| Fig. 2 (a) Experimental and Franck–Condon (FC)-simulated photoelectron spectra of CoTaC2−. (b) RCCSD(T)-calculated potential energy profile for the reaction of CoTaC2− (2IS1) with N2. (c) Experimental and density of states (DOS)-simulated photoelectron spectra of CoTaC2N2−. The 670 nm (1.85 eV) and 410 nm (3.02 eV) photons were used for CoTaC2− and CoTaC2N2−, respectively. The relative energies, ADEs and VDEs (in brackets) are given in eV. The bond lengths are in pm. The superscripts indicate spin multiplicities. The simulated spectra of 2IS1, 2I1, 2I4 and 4IS2 are red shifted by 0.13, 0.05, 0.12 eV and blue shifted by 0.04 eV, respectively. | ||
Reaction mechanism for the CoTaC2−/N2 couple
The reaction pathway of CoTaC2− (2IS1) with N2 calculated at the RCCSD(T) level is shown in Fig. 2b. The N2 molecule first approaches the Ta atom through a side-on (η2) mode to form the encounter complex 2I1 (−0.47 eV). Further binding of N2 to Co is impeded by a high energy barrier with the doublet spin state (2TS1/0.10 eV); however, this process can be accomplished through transiting to the quartet spin state (4TS1/−0.13 eV) and then a more stable intermediate 2I2 (−0.84 eV) with a side-on-end-on (η2:η1) bounded N2 unit is formed. By surmounting 2TS2, N2 is coordinated to the Co–Ta center in a distorted-η2:η1 mode (2I3/−0.64 eV) with an N–N bond length of 135 pm. The N–N bond is disrupted entirely after overcoming a slight energy barrier of 0.11 eV (2TS3/−0.53 eV), yielding one terminal-N (Nt) and one bridging-N (Nb) in 2I4 (−1.39 eV). The subsequent N–Ccluster coupling would encounter a highly positive energy barrier (2TS17/0.10 eV, Fig. S4†) that has little chance of being surmounted under thermal collision conditions, and thus the 2I4 would be stabilized as the adsorption product CoTaC2N2− through collisions with bath gas He. Note that the very minor reaction channel of generating TaC2N2− in the reactivity experiment might come from the reaction of the high-lying isomer 4IS2 with N2, in which two steps of N–Ccluster coupling take place before ejecting a neutral Co atom (Fig. S5†).The PEIS characterization of CoTaC2N2− at different reaction temperatures and photon energies supported the assignment of 2I4 as the adsorption product and also proved the reliability of our RCCSD(T) calculations. As shown in Fig. 2c, only one spectral peak (B) was observed in the experimental spectrum recorded with 410 nm photons at 250 K (note that the residual water in the room-temperature ion trap will deplete the CoTaC2− signal under the condition of a long trapping time of 80 ms, so the room-temperature reaction was not performed in the PEIS experiment); however, two spectral peaks (A and B) could be observed when the reaction temperature decreased to 200 K and 170 K. This implies that an additional intermediate was stabilized as the adsorption product CoTaC2N2− at lower temperatures and accounted for the appearance of peak A. Considering that the process of 2I1 → 4TS1 → 2I2 with the highest energy barrier and a spin crossing39 is the rate-limiting step (Fig. 2b), 2I1 is the most probable species that could be stabilized at lower temperatures. When the temperature is decreased to a certain value, all of the I1 might be stabilized and cannot transform into I4. However, this situation is not the focus of this work because we aim to study the reactions of CoTaC2−/N2 and CoTaC2N2−/CH4 couples at room temperature. The simulated spectra of CoTaC2N2− isomers based on density of states (DOS) simulations40 confirmed the contribution of 2I1 to peak A and indicated that peak B in the 250 K spectrum was contributed by 2I4 (Fig. 2c). Moreover, the employment of 365 nm photons for the PEIS characterization of CoTaC2N2− provides further evidence for the agreement of the DOS-simulated spectrum of 2I4 with the experimental one (Fig. S6b and d†). Other CoTaC2N2− isomers with the N–N or N–C bonds should be excluded due to their mismatched VDE values and spectral patterns with the experimental spectrum (Fig. S6e–k†) and their theoretically predicted inertness toward CH4 (Fig. S7†). Therefore, a Nt-containing complex was successfully prepared through the dissociative adsorption of N2 on CoTaC2− at room temperature.
Reaction mechanism for the CoTaC2N2−/CH4 couple
As shown in Fig. 3, the CoTaC2N2− (2I4) interacts with CH4 by first anchoring it on the Ta atom to form 2I5 with a binding energy of 0.44 eV. The first H3C–H bond cleavage preferably proceeds via the cooperative mechanism of a LABP composed of Ta (natural charge: 1.31e) and Nt (natural charge: −0.79e) atoms, generating a Nt–H bond and a Ta–CH3 moiety in 2I6 (−2.52 eV). After that, the CH3 group tends to migrate from Ta to Nb to liberate the Ta site (2I6 → 2TS5 → 2I7). On the basis of Rice–Ramsperger–Kassel–Marcus (RRKM) theory,41 the conversion rate of 2I6 → 2TS5 is estimated to be 6.9 × 104 s−1, which is one order of magnitude smaller than the collision rate (6.7 × 105 s−1) that a cluster experiences with the bath gas He in the LIT reactor. This suggests that only a small part of 2I6 could overcome 2TS5 to form 2I7, while most of the 2I6 would be stabilized as the adsorption product CoTaC2N2CH4− through collisions with bath gas. If the barrier height of 2TS5 is decreased by 0.1 eV, which could be the uncertainty of RCCSD(T) calculations,42 the rate of 2I6 → 2TS5 is increased to 3.3 × 105 s−1, which is of the same order of magnitude as the collision rate and can lead to the stabilization of about half of the 2I6 (P1) as CoTaC2N2CH4−. This agrees well with the ratio of CoTaC2N2CH4− to CoTaC2N2CH2,0− (49:51) in the reactivity experiment (Fig. 1).
 | ||
| Fig. 3 Potential energy profile for the reaction of CoTaC2N2− (2I4) with CH4. The structures are optimized at the DFT level. The zero-point vibration corrected energies in eV relative to the separated reactants are calculated at the RCCSD(T) level. The C atoms from CoTaC2− and CH4 are shown in different colours. | ||
After the formation of 2I7, the reaction proceeds with a H atom of the CH3 group transferred to the Ta atom to form 2I8. The resultant CH2 unit then forms a chemical bond with the Co atom, enabling the consecutive activation of the remaining two C–H bonds via the transfer of H atoms to Co (2I9 → 2I10 → 2I11). Along with two steps of structural rearrangements (2I11 → 2I12 → 2I13), two H atoms on Co make a H2 unit to generate the lowest-lying isomer (2IS3) of CoTaC2N2CH2− concomitant with loss of H2. The RRKM-theory calculated rates of traversing 2TS6–2TS11 and H2 desorption from 2I13 are at least two orders of magnitude larger than the collision rate (ESI Table 3†), indicating the impossible stabilization of intermediates 2I7–2I13 and the facile formation of 2IS3. The generation of P2 (2IS3 + H2) is highly exothermic (−2.71 eV), so 2IS3 has enough internal energy to undergo further transformation to evaporate the second H2 molecule and yield the lowest-lying isomer (2IS4) of CoTaC2N2C− (Fig. S8†). The most favorable pathway to generate P3 (2IS4 + H2) from 2IS3 involves a Co–NH bond-forming process (2I14 → 2I15) and a Co–CN bond-breaking process (2I15 → 2I16), followed by the transfer of a H atom from N to Ta to generate 2I17, from which a H2 molecule can be evaporated. The above high-level RCCSD(T) calculations indicate that the N–CCH4 bond is formed in the dehydrogenation product CoTaC2N2CH2,0−. Note that it is very difficult to perform the PEIS characterization of CoTaC2N2CH4,2,0− with the current apparatus due to the very weak signals of these species and the mass overlap with other species such as CoTaC4H2O− and CoTaC2ON2− (Fig. S9†). Quantum chemistry calculations confirmed that the formation of the N–CCH4 bond is necessary for the experimentally observed reactions and the migration of the CH3 group from Ta to Nb is the most favorable pathway.
Discussion
As shown in Fig. 4, the coupling reaction of N2 with CH4 mediated by CoTaC2− starts with cleavage of the NN bond by CoTaC2− to generate CoTaC2N2− with a Taδ+–Ntδ− LABP, followed by further activation of CH4 by CoTaC2N2−, in which the first C–H bond is cleaved via a Taδ+–Ntδ− LABP mechanism and the N–C coupling is subsequently achieved via a CH3 migration mechanism. The order of activation of the two molecules is crucial to this coupling reaction, as reflected by the low reactivity of CoTaC2− toward CH4 to produce CoTaC3H2− (Fig. S1d and e,†k1 = 1.2 × 10−14 cm3 molecule−1 s−1) and the theoretically predicted impossibility of functionalizing N2 by CoTaC3H2− (Fig. S10c†). Comparative studies on CoTaC2−/CH4 and CoTaC2N2−/CH4 reaction couples indicate that the approach of CH4 to CoTaC2N2− is overall barrierless, while the approach of CH4 to CoTaC2− encounters a positive energy barrier of 0.05 eV (Fig. S10a†). Moreover, cleavage of the H3C–H bond by CoTaC2− follows the oxidative addition mechanism, which is kinetically less favorable than the LABP mechanism in the CoTaC2N2−/CH4 couple (Fig. S10b†). Therefore, the design of first activating N2 not only constructs a Taδ+–Ntδ− LABP to facilitate the initial activation of CH4, but also generates a sufficiently reactive Nb atom that can accept the migrating CH3 group to form the N–C bond.
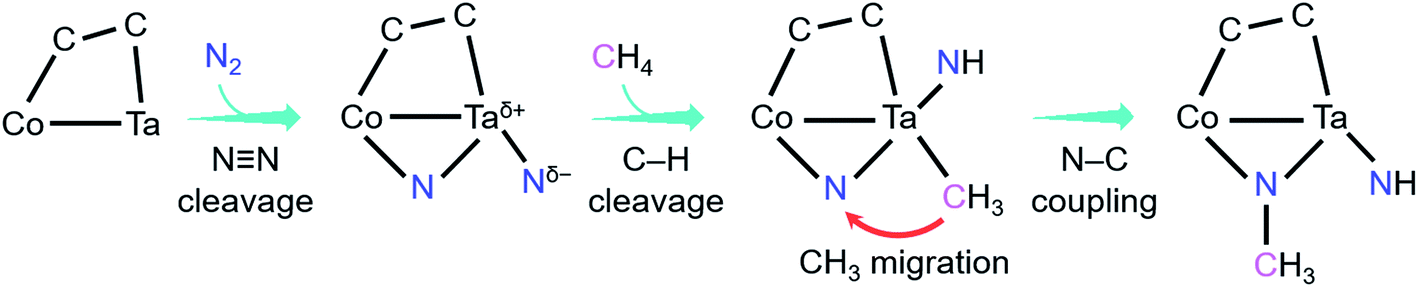 | ||
| Fig. 4 Key events and mechanisms for the coupling reaction of N2 with CH4 mediated by CoTaC2− cluster anions. | ||
Natural charge analysis of the CoTaC2N2−/CH4 reaction system reveals that the two N atoms in CoTaC2N2− exhibit strong charge buffering ability throughout the activation of CH4: they store a large number of negative charges (ΔQ = −0.47e) during the processes of H3C–H cleavage (Nt–H formation) and Nb–C formation, while releasing all of the stored negative charges (ΔQ = 0.52e) during the activation of the remaining three C–H bonds (Fig. S11†). Such charge buffering behavior largely reduces the kinetic barriers of rate-limiting steps and thus drives the N–C bond formation. Noticeably, the N–C bond formation from N2 outlined here differs fundamentally from that in previous gas-phase studies. Generally, the carbon ligands in transition metal carbide clusters, such as FeTaC2−, FeV2C2−, V3C4−, and Ta2C4−, were employed to construct N–Ccluster bonds after the NN bond was completely cleaved by the metal center.22–25 This study provides a likely pathway for N–C bond formation from the product of N2 cleavage through migrating a CH4-derived CH3 group to a N2-derived N atom, which opens a new window for the reactions of N2-derived metal nitrides.
Finally, we infer that N2-derived metal nitrides with a M–Nt LABP might be promising species for further activation of CH4. As expected, the previously reported FeTaC2N2− with a similar Taδ+–Ntδ− LABP (natural charge: 1.33e/−0.89e) is demonstrated to react with CH4 to produce FeTaC2N2CH4− and FeTaC2N2CH2− (ratio = 70:30) by using a newly-designed ship-lock-type reactor (Fig. S13†).43 More interestingly, the reaction rate of the FeTaC2N2−/CH4 couple (k1 = 2.8 × 10−15 cm3 molecule−1 s−1) is about four orders of magnitude smaller than that of CoTaC2N2−/CH4 couple, indicating a large difference in the initial activation of CH4 by the Taδ+–Ntδ− LABP. A key difference between the two systems is that the degree of N2 reduction by CoTaC2− is properly reduced compared to FeTaC2−, as reflected by fewer negative charges on N atoms and smaller electron occupancy on N2p orbitals in CoTaC2N2− than those in FeTaC2N2− (Fig. S14†). This leads to a slightly weaker Ta–Nt bond in CoTaC2N2− (WBI: 2.4) than that in FeTaC2N2− (WBI: 2.6), which has more possibility to buffer the charge variation during the initial H3C–H cleavage process. This agrees with the fact that it is difficult for N2-derived nitrides with strong M–N bonds to activate other molecules. Further research on the optimal design of highly reactive Mδ+–Ntδ− LABP during the activation of N2 to trigger efficient N–C coupling in the further activation of CH4 is in progress.
Conclusions
A possible N–C bond formation from the mutual functionalization of N2 and CH4 mediated by heteronuclear metal cluster anions CoTaC2− was studied by using mass spectrometry, photoelectron spectroscopy and a modelled reaction pathway. As verified by employing mass spectrometry, photoelectron imaging spectroscopy, and quantum chemistry calculations, the coupling reaction of N2 with CH4 starts with the dissociative adsorption of N2 on CoTaC2− to generate a Taδ+–Ntδ− Lewis acid–base pair, which then triggers the H3C–H cleavage (Nt–H formation) and N–CCH4 formation in the reaction of CoTaC2N2− with CH4. Cleaving the NN bond to generate two N atoms with large charge buffer capacity underlies the ability of CoTaC2− to mediate the coupling of N2 and CH4. Based on this finding, a universal Nt triggering strategy was proposed to couple inert N2 and CH4 molecules, which may inspire the rational design of catalysts to produce N-containing organic compounds.
Author contributions
L.-H. M. and S.-G. H. conceived the ideas. L.-H. M. carried out the experiments and calculations and wrote the original manuscript. Y. L. and H. C. assisted with high-level quantum calculations. G. P. W. and Q.-Y. L. assisted with the experiments. S.-G. H., Z.-Y. L. and H. C. supervised the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21833011, 22173111 and 92161205), the Youth Innovation Promotion Association CAS (No. 2020034), and the K. C. Wong Education Foundation.References
- S. Kim, F. Loose and P. J. Chirik, Chem. Rev., 2020, 120, 5637–5681 CrossRef CAS PubMed.
- J. Chatt, A. A. Diamantis, G. A. Heath, N. E. Hooper and G. J. Leigh, J. Chem. Soc., Dalton Trans., 1977, 7, 688–697 RSC.
- D. J. Knobloch, E. Lobkovsky and P. J. Chirik, Nat. Chem., 2010, 2, 30–35 CrossRef CAS.
- S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 11373–11383 CrossRef CAS PubMed.
- Z. J. Lv, Z. Huang, W. X. Zhang and Z. Xi, J. Am. Chem. Soc., 2019, 141, 8773–8777 CrossRef CAS.
- M. Wang, L.-Y. Chu, Z.-Y. Li, A. M. Messinis, Y.-Q. Ding, L. Hu and J.-B. Ma, J. Phys. Chem. Lett., 2021, 12, 3490–3496 CrossRef CAS PubMed.
- Y. Nakanishi, Y. Ishida and H. Kawaguchi, Angew. Chem., Int. Ed., 2017, 56, 9193–9197 CrossRef CAS PubMed.
- D. J. Knobloch, H. E. Toomey and P. J. Chirik, J. Am. Chem. Soc., 2008, 130, 4248–4249 CrossRef CAS.
- S. F. McWilliams, D. L. J. Broere, C. J. V. Halliday, S. M. Bhutto, B. Q. Mercado and P. L. Holland, Nature, 2020, 584, 221–226 CrossRef CAS PubMed.
- L. Morello, J. B. Love, B. O. Patrick and M. D. Fryzuk, J. Am. Chem. Soc., 2004, 126, 9480–9481 CrossRef CAS PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- L.-H. Mou, G.-D. Jiang, Z.-Y. Li and S.-G. He, Chin. J. Chem. Phys., 2020, 33, 507–520 CrossRef CAS.
- Y.-X. Zhao, Z.-Y. Li, Y. Yang and S.-G. He, Acc. Chem. Res., 2018, 51, 2603–2610 CrossRef CAS PubMed.
- H. Schwarz, S. Shaik and J. Li, J. Am. Chem. Soc., 2017, 139, 17201–17212 CrossRef CAS.
- D. V. Fries, M. P. Klein, A. Steiner, M. H. Prosenc and G. Niedner-Schatteburg, Phys. Chem. Chem. Phys., 2021, 23, 11345–11354 RSC.
- F. Mafune, Y. Tawaraya and S. Kudoh, J. Phys. Chem. A, 2016, 120, 4089–4095 CrossRef CAS PubMed.
- G. Liu, I. R. Ariyarathna, S. M. Ciborowski, Z. Zhu, E. Miliordos and K. H. Bowen, J. Am. Chem. Soc., 2020, 142, 21556–21561 CrossRef CAS PubMed.
- N. Levin, J. Lengyel, J. F. Eckhard, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2020, 142, 5862–5869 CrossRef CAS.
- R. Kretschmer, M. Schlangen and H. Schwarz, Understanding Organometallic Reaction Mechanisms and Catalysis, ed. V. P. Ananikov, Wiley-VCH, Weinheim, 2014, pp. 1–16 Search PubMed.
- S. Zhou, J. Li, M. Schlangen and H. Schwarz, Acc. Chem. Res., 2016, 49, 494–502 CrossRef CAS.
- R. Kretschmer, M. Schlangen, M. Kaupp and H. Schwarz, Organometallics, 2012, 31, 3816–3824 CrossRef CAS.
- L.-H. Mou, Y. Li, Z.-Y. Li, Q.-Y. Liu, H. Chen and S.-G. He, J. Am. Chem. Soc., 2021, 143, 19224–19231 CrossRef CAS PubMed.
- L.-H. Mou, Y. Li, Z.-Y. Li, Q.-Y. Liu, H. Chen and S.-G. He, J. Phys. Chem. Lett., 2020, 11, 9990–9994 CrossRef CAS.
- Z.-Y. Li, Y. Li, L.-H. Mou, J.-J. Chen, Q.-Y. Liu, S.-G. He and H. Chen, J. Am. Chem. Soc., 2020, 142, 10747–10754 CrossRef CAS.
- Z.-Y. Li, L.-H. Mou, G.-P. Wei, Y. Ren, M.-Q. Zhang, Q.-Y. Liu and S.-G. He, Inorg. Chem., 2019, 58, 4701–4705 CrossRef CAS.
- S. Zhou, J. Li, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2016, 55, 14863–14866 CrossRef CAS.
- S. Zhou, J. Li, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2016, 55, 11678–11681 CrossRef CAS PubMed.
- K. Koszinowski, D. Schroder and H. Schwarz, J. Am. Chem. Soc., 2003, 125, 3676–3677 CrossRef CAS PubMed.
- M. Diefenbach, M. Brönstrup, M. Aschi, D. Schröder and H. Schwarz, J. Am. Chem. Soc., 1999, 121, 10614–10625 CrossRef CAS.
- R. Kretschmer, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 5387–5391 CrossRef CAS PubMed.
- R. Kretschmer, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 3483–3488 CrossRef CAS PubMed.
- M. Schlangen, J. Neugebauer, M. Reiher, D. Schröder, J. P. López, M. Haryono, F. W. Heinemann, A. Grohmann and H. Schwarz, J. Am. Chem. Soc., 2008, 130, 4285–4294 CrossRef CAS.
- G. Gioumousis and D. P. Stevenson, J. Chem. Phys., 1958, 29, 294–299 CrossRef CAS.
- Q.-Y. Liu, L. Hu, Z.-Y. Li, C.-G. Ning, J.-B. Ma, H. Chen and S.-G. He, J. Chem. Phys., 2015, 142, 164301 CrossRef PubMed.
- J. Kohanoff and N. I. Gidopoulos, Handbook of Molecular Physics and Quantum Chemistry, ed. S. Wilson, John Wiley & Sons, Ltd, Chichester, 2003, pp. 532–568 Search PubMed.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- S. Guo, M. A. Waston, W. F. Hu, Q. M. Sun and G. K. L. Chan, J. Chem. Theory Comput., 2016, 12, 1583–1591 CrossRef CAS PubMed.
- V. Mozhayskiy and A. I. Krylov, ezSpectrum v3.0, see https://iopenshell.usc.edu/downloads Search PubMed.
- D. Schroder, S. Shaik and H. Schwarz, Acc. Chem. Res., 2000, 33, 139–145 CrossRef CAS.
- D. J. Tozer and N. C. Handy, J. Chem. Phys., 1998, 109, 10180–10189 CrossRef CAS.
- J. I. Steinfeld, J. S. Francisco and W. L. Hase, Chemical Kinetics and Dynamics. Prentice-Hall, Upper Saddle River, NJ, 1999, pp. 231–313 Search PubMed.
- W. Jiang, N. J. DeYonker and A. K. Wilson, J. Chem. Theory Comput., 2012, 8, 460–468 CrossRef CAS PubMed.
- G.-P. Wei, Q.-Y. Liu, Y. Ren and S.-G. He, Rev. Sci. Instrum., 2021, 92, 104104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Method details and additional experimental and theoretical results (spectra, data analysis, and calculated structures and reaction mechanisms). See https://doi.org/10.1039/d2sc02416k |
| ‡ Present address. University of Chinese Academy of Sciences, Beijing 100049, P. R. China. |
| This journal is © The Royal Society of Chemistry 2022 |