
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photomediated reductive coupling of nitroarenes with aldehydes for amide synthesis†
Qingyao
Li
,
Peng
Dai
 ,
Haidi
Tang
,
Muliang
Zhang
* and
Jie
Wu
*
,
Haidi
Tang
,
Muliang
Zhang
* and
Jie
Wu
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543, Singapore. E-mail: muliang0206@foxmail.com; chmjie@nus.edu.sg
First published on 3rd August 2022
Abstract
In view of the widespread significance of amide functional groups in organic synthesis and pharmaceutical studies, an efficient and practical synthetic protocol that avoids the use of stoichiometric activating reagents or metallic reductants is highly desirable. A straight-forward pathway to access amides from abundant chemical feedstock would offer a strategic advantage in the synthesis of complex amides. We herein disclose a direct reductive amidation reaction using readily available aldehydes and nitroarenes enabled by photo-mediated hydrogen atom transfer catalysis. It avoids the use of metallic reductants and production of toxic chemical waste. While aldehydes represent a classic class of electrophilic synthons, the corresponding nucleophilic acyl radicals could be directly accessed by photo hydrogen atom transfer catalysis, enabling polarity inversion. Our method provides an orthogonal strategy to conventional amide couplings, tolerating nucleophilic substituents such as free alcohols and sensitive functional groups to amines such as carbonyl or formyl groups. The synthetic utilization of this reductive amidation is demonstrated by the late-stage modification of complex biologically active molecules and direct access of drug molecules leflunomide and lidocaine.
Introduction
The amide functional group is ubiquitous in nature and the significance of amides in chemical biology has been well recognized and widely investigated in the past century.1 The physiological properties of amides and their unique ability to obstruct natural neurotransmission pathways have made them prevalent pharmaceutical agents.2 About 25% of natural and synthetic medicines contain amide linkages, which can be found in widely used drugs such as leflunomide, bupivacaine, roflumilast, fentanyl, and fominoben (Fig. 1A).3 Conventionally, the synthesis of amides is generally based on activated carboxylic acid derivatives such as acid chlorides and anhydrides or rearrangement reactions which require tedious extra prefunctionalization steps, or from carboxylic acids assisted by stoichiometric amounts of activating agents that usually lead to toxic chemical waste byproducts.4 These processes are also typically water or air sensitive, and require conditions which are incompatible with sensitive functional groups, limiting their use in organic synthesis and chemical biology studies.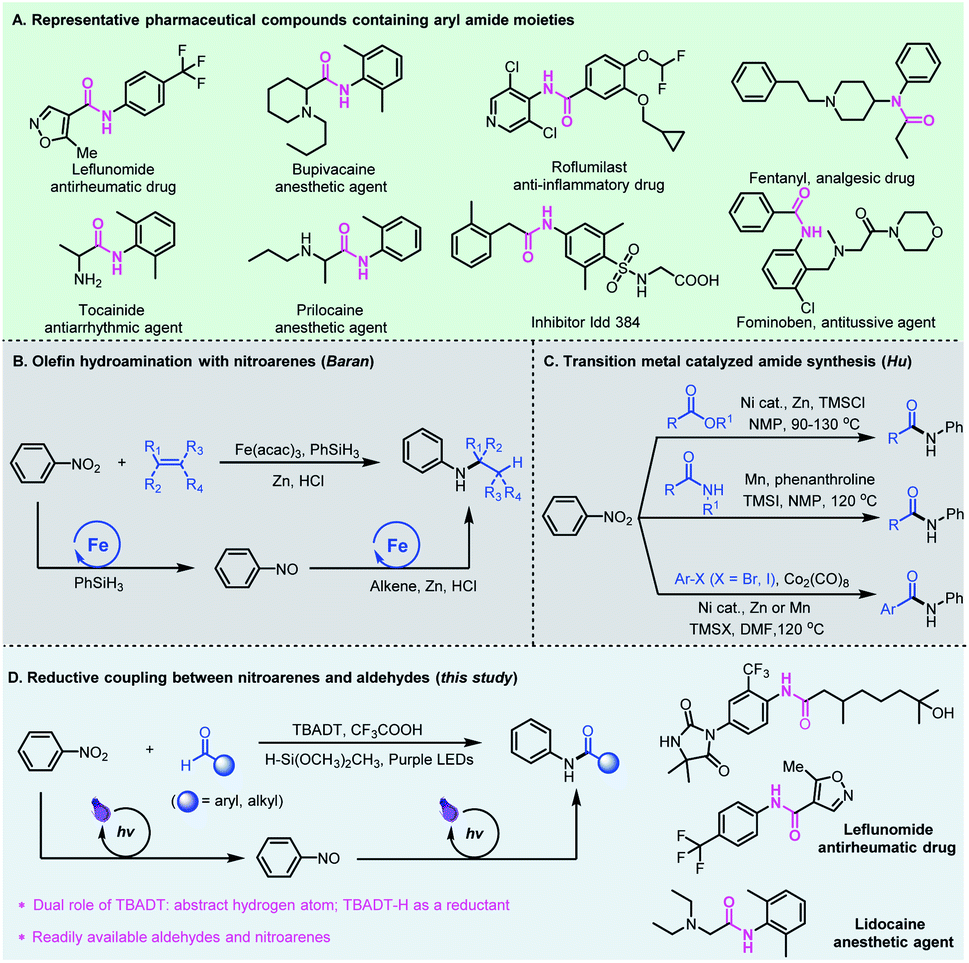 | ||
| Fig. 1 Representative aryl amides and their proposed synthesis from aldehydes and nitroarenes. | ||
The need for amide-containing molecules has led to the development of straightforward pathways to access complex amides from readily available feedstock chemicals. Recent attractive protocols include PIII/PV-catalyzed three-component condensation of amines, carboxylic acids and pyridine N-oxides,5 photocatalyzed oxime isomerization and subsequent Beckmann rearrangement,6 and re-routing of unactivated N,N-dialkylamide bonds through reactive acyl iodide intermediates.7 Chang and co-workers developed a photoinduced amidation of arylboronic acids using 1,4,2-dioxazol-5-ones as an amide coupling partner.8 Later, preparation of functionalized amides using dicarbamoylzincs was also reported by Knochel.9 Alternatively, the cross coupling for amide synthesis has been realized from alkynes,10 alcohols,11 or α-keto acids12 and aldehydes13 using amines as the starting materials. Compared to amines, nitroarenes as the nitrogen source are often more accessible, less expensive and more stable.14 In 2015, Baran et al. reported an efficient hydroamination protocol which synthesizes aryl amines from nitroarenes and alkenes with an iron salt as a catalyst, PhSiH3 as a hydrogen source, and zinc metal as the reductant (Fig. 1B).15 Later, Zhu and co-workers developed nickel-catalyzed remote C(sp3)–H amination of alkenes with nitroarenes for the synthesis of amines.16 In these catalytic cycles, the transition metal-hydrides formed in situ play an important role, and promote the initial coordination of the Lewis acidic metal center with unsaturated bonds, followed by reductive coupling with nitrosoarenes. Recently, Hu et al. developed a series of novel amidation reactions of esters,17 halides18 or amides19 with nitroarenes as the nitrogen source (Fig. 1C). However, these reductive amidation reactions usually require stoichiometric amounts of metallic reductants such as Zn or Mn, and proceed only at high temperatures.
Photomediated hydrogen atom transfer (HAT) catalysis is a powerful strategy for aldehydic C–H functionalization.20 We have developed direct HAT photocatalysis with aldehydes to access the corresponding acyl radicals, which could participate in a series of useful transformations such as asymmetric 1,4-addition with Michael acceptors,21 radical Smiles rearrangements,22 and nucleophilic substitution reactions with sulfone reagents.23 Among the reported photo-HAT catalysts, tetrabutylammonium decatungstate (TBADT) catalytically cleaves the aldehydic C–H bond under near-UV light irradiation, giving rise to acyl radicals and TBADT–H species.24 We envisioned that the TBADT–H species generated in situ could be regarded as an analogue of transition metal-hydrides,25 which could trigger the reduction of nitroarenes, realizing reductive coupling of nitroarenes with aldehydes (Fig. 1D). Specifically, tetrabutylammonium decatungstate abstracts the aldehydic C–H bond to deliver nucleophilic acyl radicals by direct photo hydrogen atom transfer catalysis, enabling polarity inversion. The nucleophilic acyl radicals generated could further undergo a radical addition–elimination process for nitroarene reduction. Such a reductive process is independent of the reductive potential of nitroarenes, thus broadening the substrate scope of nitroarenes without redox limitation. The TBADT plays critical and multifaceted roles in this reductive catalytic process, not only activating the aldehydic C–H bond to deliver acyl radicals, but also using the transferred hydrogen to facilitate the overall reductive coupling. Aldehydes and nitroarenes are both widely used, inexpensive and readily available feedstock chemicals. The reductive coupling of aldehydes with nitroarenes represents an attractive and sustainable strategy for the direct generation of bioactive aryl amide compounds. Even though direct access to amides from aromatic aldehydes and nitroarenes has been reported,26 the pathways relied heavily on ionic reactions using much excess metal reductants with very limited substrate scope and no practical applications.
Results and discussion
We began our investigation with nitrobenzene (1a) and cyclopropanecarbaldehyde (2a) as the model reactants. As shown in Table 1, with 2 mol% TBADT, 2.5 equiv. of dimethoxy-(methyl)silane, 30 mol% trifluoroacetic acid and CH3CN as the solvent, the desired amide product (3a) was obtained in 78% yield under 390 nm light irradiation (entry 1). The use of other silanes such as TMS3Si–H, PhSiH3, and Et3SiH led to inferior reaction yields (entry 2, also see ESI Table S2†). When eosin Y was used as the HAT photocatalyst,27 the reductive amidation did not occur, highlighting the unique role of the TBADT–H generated in situ (Table 1, entry 3). With TFA absent or replaced by another acid such as acetic acid a substantial decrease of the yield was noted (entries 4 and 5). We believe that the addition of TFA could accelerate the photocatalytic process and increase the reactivity.28 A higher reaction temperature resulted in more over-reduced amine byproducts, and a negative influence on the reaction outcome (entry 6). Control experiments demonstrated that both the photocatalyst and light irradiation are essential for this reductive amidation (entries 7 and 8).|
|
||
|---|---|---|
| Entry | Variation from standard conditionsa | Yieldb (%) |
| a Standard conditions: 1a (0.1 mmol), 2a (3 equiv.), TBADT (2 mol%), CF3COOH (30 mol%), H–Si(OCH3)2CH3 (2.5 equiv.), CH3CN (2 mL), 2 × 40 W LEDs (390 nm), 30 °C, 24 h. b Yields are based on the analysis of the 1H NMR spectra of the crude product mixture using CH2Br2 as an internal standard. c Isolated yields. | ||
| 1 | None | 81 (78)c |
| 2 | TMS3Si–H instead of CH3(OCH3)2Si–H | 60 |
| 3 | Eosin Y instead of TBADT | <2 |
| 4 | Without CF3COOH | 40 |
| 5 | CH3COOH instead of CF3COOH | 41 |
| 6 | 60 °C instead of 30 °C | 58 |
| 7 | Without TBADT | 6 |
| 8 | Without light irradiation | 0 |

With the optimized conditions in hand, we sought to evaluate the scope of this reductive amidation. A variety of amides were effectively synthesized from the corresponding nitroarenes and aldehydes as shown in Fig. 2. Secondary and primary alkyl aldehydes proved to be competent starting materials, offering the desired amides (3a–3j) in moderate to good yields. However, tertiary alkyl aldehydes failed to produce the desired product, and this was complicated by the facile decarbonylation of the tertiary acyl radicals generated in situ. A series of aromatic aldehydes and nitroarene partners participated smoothly in the reductive amidation reaction, tolerating a wide range of functional groups, such as halo (3l–3o, 3z–3ff), cyano (3p–3q, 3jj), methoxy (3t, 3mm), trifluoromethyl (3r) and ester (3nn) derivatives. Heteroaromatic aldehydes such as furfural (3x), pyridine (3y) and thiophene (3w) delivered the amide products in moderate to good yields, and reduction-sensitive functional groups such as aldehyde (3hh) and ketone (3gg) and nucleophilic groups such as the hydroxyl group (3j, 3u, 3kk, 3ll) were well tolerated under the reaction conditions. These functionalities may cause compatibility issues when preparing the required aryl amines or activated acid derivatives in conventional amide coupling methods.
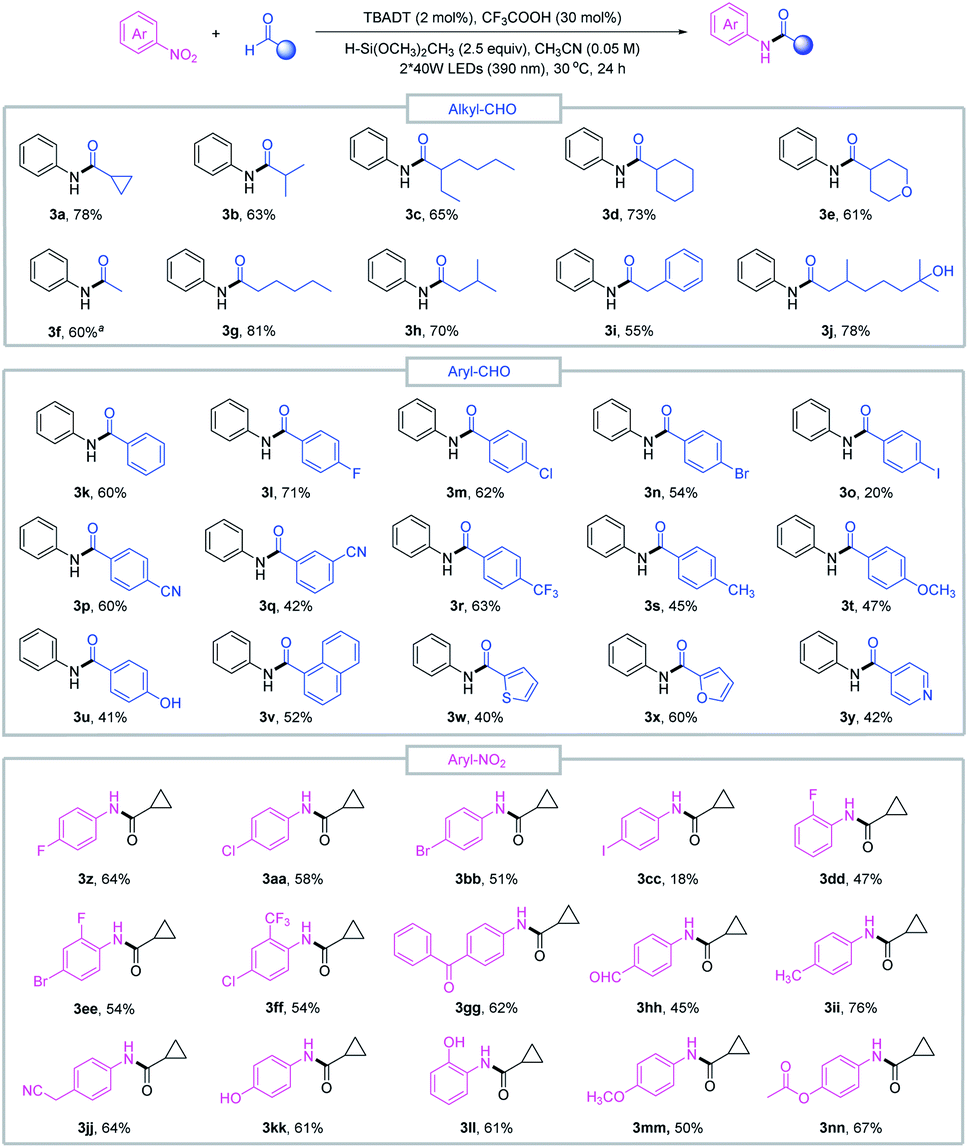 | ||
| Fig. 2 Substrate scope of reductive amidation. Standard conditions: nitroarene 1 (0.1 mmol), aldehyde 2 (3 equiv.), TBADT (2 mol%), CF3COOH (30 mol%), H–Si(OCH3)2CH3 (2.5 equiv.), CH3CN (2 mL), purple LEDs (390 nm), 30 °C, 24 h. aThe amount of aldehyde was 20 equiv. | ||
The good functional group tolerance enables the synthesis of complex amides through late-stage functionalization of biologically active molecules (Fig. 3). This amide bond formation method can be used in the synthesis of pharmaceutical nitroarene compounds, such as flutamide (3oo), nilutamide (3pp), nitroxinil (3qq), nimesulide (3rr), and chloramphenicol (3ss), which are formed in moderate to good yields. Aldehydes derived from natural products such as (+)-fenchol (3tt and 3uu) and menthol (3vv) participated in the reductive coupling smoothly. Notably, the direct coupling of the nitroarene drug molecule nilutamide and the commercially available odiferous hydroxycitronellal delivered the amide product (3ww) in 52% yield. Next, by taking advantage of aldehyde and nitroarene feedstock, leflunomide (3xx), an antirheumatic drug, and lidocaine (3yy), an anesthetic agent, were directly accessed using our protocol. These examples suggest the potential wide utility of this method in medicinal chemistry. Finally, the reductive amidation reaction was transferred to an operationally simple continuous-flow reactor to achieve the amide product (3j) with 6.3 g per 8 h from readily available nitrobenzene and hydroxycitronellal, indicating the potential for large-scale synthesis.
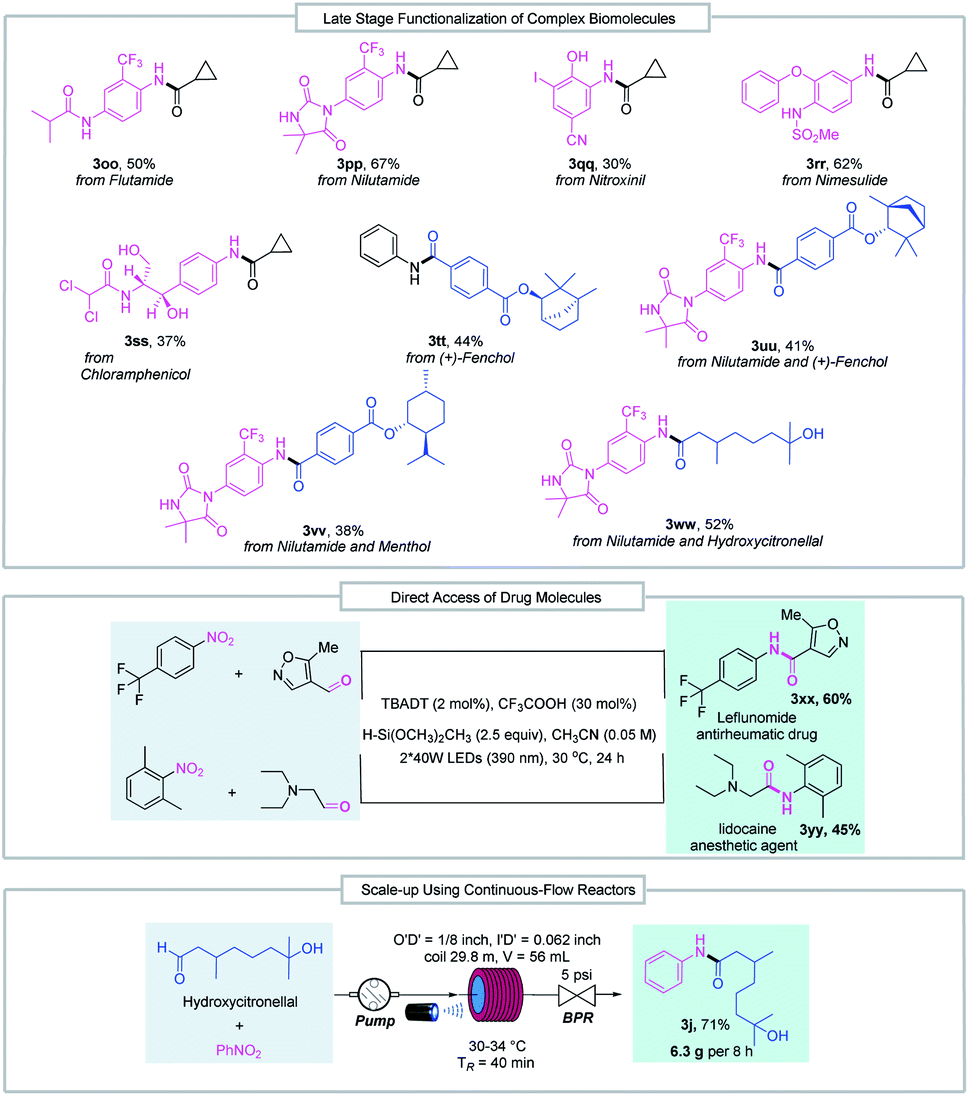 | ||
| Fig. 3 Late-stage functionalization of complex biomolecules, direct access to drug molecules and reaction scale-up in continuous-flow reactors. BPR = back pressure regulator. | ||
Various control experiments were performed to investigate the mechanism of this transformation. To probe the possible nitrogen-containing intermediates involved in this reductive coupling, several nitrogen-based compounds were tested as the starting materials under the standard reaction conditions. Among the reagents tested, nitrosobenzene afforded the desired amide product in 82% yield (Fig. 4A). However, anilines, azobenzene and phenylhydroxyamine gave a significantly lower yield (10%) or no product. This indicates that nitrosobenzene is one of the active intermediates during the reaction process. The addition of TEMPO (2,2,6,6-tetramethyl-1-piperidi-nyloxy) inhibited the transformation, and the TEMPO-acyl radical adduct was detected by ESI-MS, supporting the presence of acyl radicals during the reaction process (Fig. 4B). Moreover, N-hydroxy amide (6) could be obtained if the transformation was quenched after 1 h reaction time (Fig. 4C). Subjecting the N-hydroxy amide (6) to the standard reaction conditions produced the amide quantitatively (Fig. 4D), and this suggested that an N-hydroxy amide was also involved in the reaction process. After reaction, siloxane or silanol derived from the silane was detected (Fig. 4E). The quantum yield of a model reaction was determined to be 0.46 (Fig. 4F).
 | ||
| Fig. 4 Control experiments to elucidate the mechanisms. | ||
Mechanistic rationale
Based on the experimental data, a plausible mechanistic pathway of the reductive coupling is proposed in Fig. 5. Under 390 nm light irradiation, the excited photocatalyst TBADT abstracts a hydrogen atom from the aldehydes or the silanes to generate the corresponding acyl radicals, silyl radicals and reductive intermediate H+[W10O32]5− (Ered = −1.27 V vs. Ag/Ag+ in MeCN).29 The acyl radicals could directly undergo a radical addition on nitroarenes to give the intermediates (I), followed by decomposition to offer nitrosoarenes and carboxyl radicals (II).30 This carboxyl radical species (II) is capable of abstracting another hydrogen atom from aldehydes to offer acyl radicals.20 Next, the nitrosoarenes will react with the acyl radicals to form radical species (III), followed by a SET process with H+[W10O32]5− to give rise to the N-hydroxy amide.31 The highly reducing H+[W10O32]5− can reduce the N-hydroxy amide (Ered = −0.65 V vs. Ag/Ag+ in MeCN, ESI Fig. S12†) to the corresponding radical anion intermediate (VI), which will release OH− to generate an amidyl radical (VII), followed by another SET with H+[W10O32]5− and protonation to accomplish the final amide product. The in situ generated silyl radical may undergo a single electron oxidation by the excited *[W10O32]4− (E ∼ +2.5 V vs. SCE in MeCN)32 to furnish a silyl cation intermediate,33 which is captured by nucleophiles such as H2O and silanols to give silanols or siloxane after deprotonation (for a more detailed description of the plausible mechanism, see ESI Fig. S14†).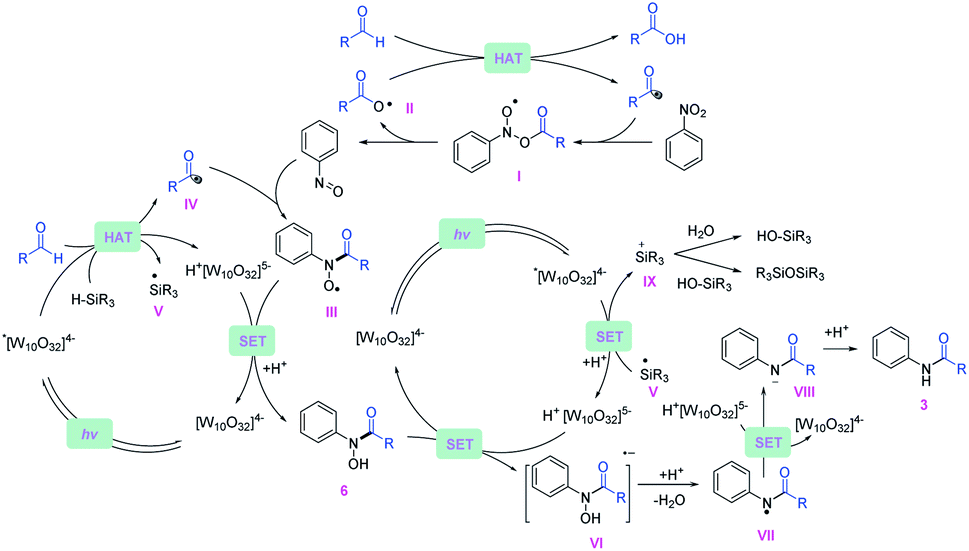 | ||
| Fig. 5 Proposed plausible mechanisms of the reductive amidation reaction. | ||
Conclusions
We have developed an efficient TBADT-based photocatalytic system for a reductive amidation of aldehydes and nitroarenes. A broad range of readily available aldehydes and nitroarenes are competent coupling partners with high functional group tolerance, and furnish a broad scope of functionally diverse aryl amides. This reductive amidation method is an improvement on the conventional amide bond-forming pathways, which require acid activation and pre-reduction of nitroarenes to amines. Moreover, the nitro group exhibits orthogonal reactivity to the amine group, tolerating nucleophilic substituents such as free alcohols. Functional groups sensitive to amines such as carbonyl or formyl groups are also well-tolerated when using nitroarenes as the starting materials, which are difficult to directly access by the conventional amide coupling strategy. This method is amenable to late-stage functionalization of complex molecules and provides direct access to pharmaceuticals, making it promising to support applications in organic materials science and medicinal chemistry.Data availability
The ESI† contains method description, product characterization data, NMR spectra, and mechanism study details.Author contributions
QL, MZ and JW conceived and designed the project. QL, PD, and HT performed all experimental and mechanism studies. QL, MZ, and JW wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the Ministry of Education (MOE) of Singapore (MOET2EP10120-0014), Pharma Innovation Programme Singapore (A*STAR, SERCA19B3a0014), the National University of Singapore (R-143-000-B60-114), and the National Natural Science Foundation of China (22071170).References
- (a) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396–1399 CrossRef CAS PubMed; (b) C. L. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (c) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS; (d) X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef.
- (a) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 2020, 4641–4651 CrossRef CAS; (b) A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: structural significance in chemistry, biochemistry, and materials science, Wiley-Interscience, New York, 2000 Search PubMed.
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed.
- (a) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (b) M. B. Smith, Compendium of Organic Synthetic Methods, Wiley, New York, vol. 9, 2001, pp. 100–116 Search PubMed; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) Y. Zhang, M. L. Blackman, A. B. Leduc and T. F. Jamison, Angew. Chem., Int. Ed., 2013, 52, 4251–4255 CrossRef CAS PubMed; (e) L. Hu, S. Xu, Z. Zhao, Y. Yang, Z. Peng, M. Yang, C. Wang and J. Zhao, J. Am. Chem. Soc., 2016, 138, 13135–13138 CrossRef CAS PubMed; (f) P. V. Ramachandran and H. J. Hamann, Org. Lett., 2021, 23, 2938–2942 CrossRef CAS PubMed; (g) F. S. Movahed, D. N. Sawant, D. B. Bagal and S. Saito, Synthesis, 2020, 52, 3253–3262 CrossRef CAS; (h) Y. Du, T. Barber, S. E. Lim, H. S. Rzepa, I. R. Baxendale and A. Whiting, Chem. Commun., 2019, 55, 2916–2919 RSC; (i) D. N. Sawant, D. B. Bagal, S. Ogawa, K. Selvam and S. Saito, Org. Lett., 2018, 20, 4397–4400 CrossRef CAS PubMed.
- J. M. Lipshultz and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 14487–14494 CrossRef CAS PubMed.
- X. Zhang and T. Rovis, J. Am. Chem. Soc., 2021, 143, 21211–21217 CrossRef CAS.
- D. Zuo, Q. Wang, L. Liu, T. Huang, M. Szostak and T. Chen, Angew. Chem., Int. Ed., 2022, 61, e202202794 CAS.
- Z. Zhou, J. Kweon, H. Jung, D. Kim, S. Seo and S. Chang, J. Am. Chem. Soc., 2022, 144, 9161–9171 CrossRef CAS.
- D. Djukanovic, M. A. Ganiek, k. Nishi, K. Karaghiosoff, K. Mashima and P. Knochel, Angew. Chem., Int. Ed., 2022, 61, e202205440 CrossRef CAS PubMed.
- (a) W. K. Chan, C. M. Ho, M. K. Wong and C. M. Che, J. Am. Chem. Soc., 2006, 128, 14796–14797 CrossRef CAS PubMed; (b) L. Zeng, H. Li, J. Hu, D. Zhang, J. Hu, P. Peng, S. Wang, R. Shi, J. Peng, C. W. Pao, J. L. Chen, J. F. Lee, H. Zhang, Y. H. Chen and A. Lei, Nat. Catal., 2020, 3, 438–445 CrossRef CAS.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- J. Liu, Q. Liu, H. Yi, C. Qin, R. Bai, X. Qi, Y. Lan and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 502–506 CrossRef CAS PubMed.
- (a) J. W. Bode and S. S. Sohn, J. Am. Chem. Soc., 2007, 129, 13798–13799 CrossRef CAS; (b) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796–13797 CrossRef CAS PubMed; (c) N. F. Nikitas, M. K. Apostolopoulou, E. Skolia, A. Tsoukaki and C. G. Kokotos, Chem.–Eur. J., 2021, 27, 7915–7922 CrossRef CAS PubMed.
- (a) G. Li, T. V. Nykaza, J. C. Cooper, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 6786–6799 CrossRef CAS PubMed; (b) A. Bhunia, K. Bergander, C. G. Daniliuc and A. Studer, Angew. Chem. Int. Ed., 2021, 60, 8313–8320 CrossRef CAS; (c) G. Li, L. Yang, J. J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS; (d) C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef PubMed; (e) M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570–11574 CrossRef CAS PubMed; (f) Y. Ning, S. Wang, M. Li, J. Han, C. Zhu and J. Xie, Nat. Commun., 2021, 12, 4637 CrossRef CAS PubMed.
- J. Gui, C. M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS.
- J. Xiao, Y. He, F. Ye and S. Zhu, Chem, 2018, 4, 1645–1657 CAS.
- C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef.
- C. W. Cheung, M. Leendert Ploeger and X. Hu, Chem. Sci., 2018, 9, 655–659 RSC.
- C. W. Cheung, J. A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789–6792 CrossRef CAS.
- (a) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem. Catal., 2021, 1, 523–598 CrossRef; (b) S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 14723–14726 CrossRef CAS PubMed; (c) X. Zhang and D. W. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed; (d) E. Voutyritsa and G. Kokotos, Angew. Chem., Int. Ed., 2020, 59, 1735–1741 CrossRef CAS PubMed.
- Y. Kuang, K. Wang, X. Shi, X. Huang, E. Meggers and J. Wu, Angew. Chem., Int. Ed., 2019, 58, 16859–16863 CrossRef CAS PubMed.
- J. Yan, H. W. Cheo, W. K. Teo, X. Shi, H. Wu, S. B. Idres, L. W. Deng and J. Wu, J. Am. Chem. Soc., 2020, 142, 11357–11362 CrossRef CAS.
- J. Yan, H. Tang, E. J. R. Kuek, X. Shi, C. Liu, M. Zhang, J. L. Piper, S. Duan and J. Wu, Nat. Commun., 2021, 12, 7214 CrossRef CAS PubMed.
- H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS.
- S. W. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- (a) S. K. Jain, K. A. A. Kumar, S. B. Bharate and R. A. Vishwakarma, Org. Biomol. Chem., 2014, 12, 6465 RSC; (b) G. Sheng, X. Wu, X. Cai and W. Zhang, Synthesis, 2015, 47, 949–954 CrossRef CAS.
- (a) D. M. Yan, J. R. Chen and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378–380 CrossRef CAS PubMed; (b) X. Z. Fan, J. W. Rong, H. L. Wu, Q. Zhou, H. P. Deng, J. D. Tan, C. W. Xue, L. Z. Wu, H. R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS.
- (a) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS PubMed; (b) D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS.
- R. F. Renneke, M. Pasquali and C. L. Hill, J. Am. Chem. Soc., 1990, 112, 6585–6594 CrossRef CAS.
- E. G. Janzen and U. M. Oehler, Tetrahedron Lett., 1983, 24, 669–672 CrossRef CAS.
- F. Xu, H. W. Deussen, B. Lopez, L. Lam and K. Li, Eur. J. Biochem., 2001, 268, 4169 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- H. Liang, L. J. Wang, Y. X. Ji, H. Wang and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1839–1844 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and NMR spectra of all new compounds. See https://doi.org/10.1039/d2sc03047k |
| This journal is © The Royal Society of Chemistry 2022 |