
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexible organic frameworks sequester neuromuscular blocking agents in vitro and reverse neuromuscular block in vivo†
Yan
Wu
 a,
Yue-Yang
Liu
a,
Hong-Kun
Liu
a,
Shang-Bo
Yu
b,
Furong
Lin
b,
Wei
Zhou
*a,
Hui
Wang
a,
Dan-Wei
Zhang
*a,
Zhan-Ting
Li
*a and
Da
Ma
*c
a,
Yue-Yang
Liu
a,
Hong-Kun
Liu
a,
Shang-Bo
Yu
b,
Furong
Lin
b,
Wei
Zhou
*a,
Hui
Wang
a,
Dan-Wei
Zhang
*a,
Zhan-Ting
Li
*a and
Da
Ma
*c
aDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200438, China. E-mail: zhouw@fudan.edu.cn; zhangdw@fudan.edu.cn; ztli@fudan.edu.cn
bKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China
cSchool of Pharmaceutical and Materials Engineering & Institute for Advanced Studies, Taizhou University, 1139 Shifu Avenue, Jiaojiang, Zhejiang 318000, China. E-mail: dama@fudan.edu.cn
First published on 20th July 2022
Abstract
Supramolecular sequestration and reversal of neuromuscular block (NMB) have great clinical applications. Water-soluble flexible organic frameworks (FOFs) cross-linked by disulfide bonds are designed and prepared. Different linker lengths are introduced to FOFs to give them varied pore sizes. FOFs are anionic nanoscale polymers and capable of encapsulating cationic neuromuscular blocking agents (NMBAs), including rocuronium (Roc), vecuronium (Vec), pancuronium (Panc) and cisatracurium (Cis). A host–guest study confirms that FOFs bind NMBAs in water. The multivalency interaction between FOFs and NMBAs is able to sequester NMBAs, and prevent them from escaping. These FOFs are non-toxic and biocompatible. Animal studies show that FOFs are effective for the reversal of NMB induced by Roc, Vec and Cis, which shorten the time to a train-of-four ratio of 0.9 by 2.6, 3.8 and 5.7-fold compared to a placebo, respectively.
Introduction
The sequestration of pharmaceutical agents, drugs and toxins has great clinical applications.1–3 The clinical uses of sequestration include emergency treatments for drugs of abuse,4–6 the prevention of harmful side effects of pharmaceuticals,7,8 or the reversal of neuromuscular block (NMB).9–11Supramolecular sequestration often serves as a pharmacokinetic approach, and involves molecular recognition.12–14 One important clinical example of a supramolecular sequestration agent is sugammadex, a γ-cyclodextrin derivative, which binds neuromuscular blocking agents (NMBAs) and reverses their NMB.15–17 Other molecular containers, including cucurbit[n]uril-family hosts and water-soluble pillar[n]arenes have also been reported to sequester NMBAs.18–21 Nevertheless, molecular containers lack the structural tunability to be optimized for the binding of NMBAs and other sequestration targets. By contrast, framework materials may be fine-tuned for binding and sequestration by simply changing the building blocks and cross-linkers.22,23
Herein, we explore the sequestration of NMBAs by framework materials. Metal–organic frameworks (MOFs),24–26 covalent–organic frameworks (COFs)27,28 and supramolecular–organic frameworks (SOFs)29–31 are important framework materials. To sequester NMBAs, we use a new type of framework, flexible organic frameworks (FOFs), which are biocompatible, stable and capable of encapsulating pharmaceutical or biological guests.32–34 For this purpose, FOFs cross-linked by disulfide bonds are prepared and used. The resulting water-soluble anionic FOFs are capable of encapsulating NMBAs through hydrophobic and electrostatic interactions. Considering the original intention of modular design, different core moieties were introduced which realized the change in pore size of FOFs and the size of hydrophobic surfaces. These FOFs are confirmed to effectively sequester NMBAs in vitro and reverse the NMB in vivo.
Results and discussion
Synthesis of precursors T1–T3
The synthetic routes of precursors T1–T3 are outlined in Schemes S1–S3,† respectively. The linker moiety Boc-Cys(Trt)-Asp-(OMe)2 was synthesized from two essential amino acid derivatives by the conventional EDCI/HOBt condensation reaction. Triphenylmethane is a sulfhydryl protective group sensitive to medium-strong acid. Formic acid was used to remove the Boc protecting group to yield Cys(Trt)-Asp-(OMe)2. The EDCI/HOBt condensation reaction failed to achieve high yield conversion in the next amidation step. Therefore, TCPM was converted to acyl chloride, followed by the reaction with amine. T1-(Trt)4-(OMe)8 was obtained in high yield by column chromatography and recrystallization (yield 56%). The protective group of sulfhydryl was removed by TFA. To protect the sulfhydryl group from oxidation in air, the Schlenk technique was used to maintain an anaerobic environment during the ester hydrolysis reaction to obtain T1. Detailed synthetic procedures are available in the ESI.†For precursors T2 and T3, acrylate and benzoate fragments were introduced into core moieties through the common coupling reaction, and then the modified derivatives of core moieties were obtained through ester hydrolysis.35,36 The following module splicing, deprotection and ester hydrolysis reactions were operated using the procedures described for T1.
Construction and characterization of FOFs
As shown in Scheme 1, compounds T1-3 were used as precursors to prepare FOF-SS1-3. Precursors T1-3 shared the same core moiety of tetraphenylmethane and flexible linkers of cysteine–aspartic acid dipeptide, which were conjugated with a C–C single bond, ethylene group or p-phenyl group. The three different conjugation groups gave FOF-SS1-3 different pore sizes and recognition cavities. The porous polymeric FOFs were constructed by dynamic covalent disulfide cross-linking with H2O2 oxidation under alkaline conditions. The resulting FOFs are three-dimensional porous structures based on precursors T1-3 and cross-linked by disulfide bonds. Two monocationic NMBAs (rocuronium or Roc and vecuronium or Vec) and two dicationic NMBAs (cisatracurium or Cis and pancuronium or Panc) were chosen for the study.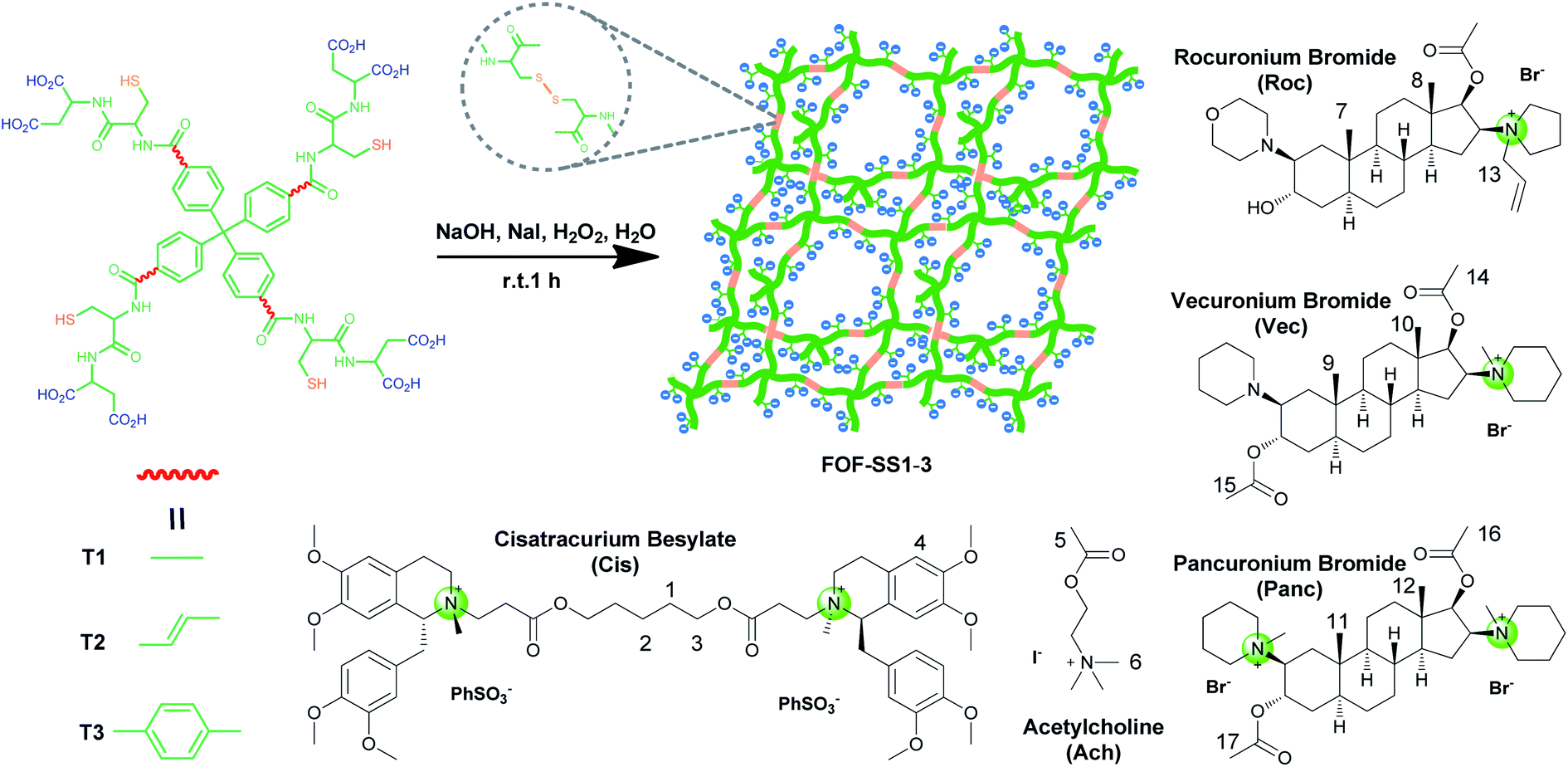 | ||
| Scheme 1 Synthetic scheme for FOF-SS1-3 and chemical structures of NMBAs and acetylcholine. | ||
We used 1H NMR spectroscopy to study the formation of FOF-SS1-3. Briefly, precursors T1-3 (5 mM) were dispersed in D2O. Precursors were neutralized with a stoichiometric amount of NaOH and oxidized by four equivalents of H2O2. Sodium iodide played a role as a catalyst in the oxidation process. This condition could ensure a rapid and clean oxidation reaction.371H NMR spectra showed the emergence of a new set of resonances shortly after the oxidant was added, which remained unchanged after that (Fig. 1a, S10 and S11†). This observation indicated that the cross-linking reaction was rapidly completed. The originally sharp and clearly split peaks became wider and more complex, which proved the formation of a new material. After incubation for 24 h at 310 K, the 1H NMR spectra remained the same as the initial state, confirming that an equilibrium had been quickly reached. To confirm the formation of disulfide bonds, the products of the FOF-SS1-3 cross-linking reaction were acidulated by an excess amount of hydrochloric acid, and the precipitate was collected and dried under high vacuum. As shown in Fig. 1b–d, the proton resonance of sulfhydryl groups almost completely disappeared on the 1H NMR spectra of acidulated FOF-SS1-3, which indicated the cross-linking of sulfhydryl groups and the formation of disulfide bonds.
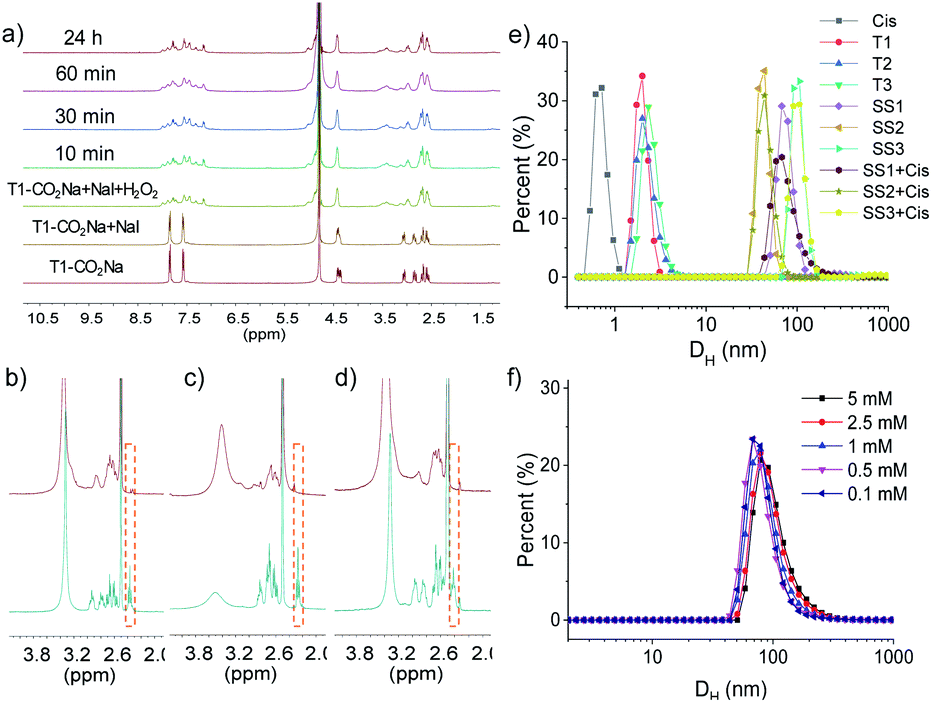 | ||
| Fig. 1 (a) 1H NMR spectra (400 MHz, D2O, 298 K) recorded for the formation of FOF-SS1 after the oxidant was added. 1H NMR spectra (400 MHz, DMSO-d6, 298 K) recorded for (b) T1 and acidulated FOF-SS1, (c) T2 and acidulated FOF-SS2, and (d) T3 and acidulated FOF-SS3. (e) Size distribution of Cis, T1-3, FOF-SS1-3 and FOF-SS1-3 + Cis in water. (f) Size distribution of FOF-SS1 at different concentrations in water (calculated based on [T1]). | ||
Next, the formation of FOFs was investigated by dynamic light scattering (DLS) in water. Briefly, neutralized precursors T1-3 (1 mM) were incubated in an aqueous solution of H2O2 (4 mM), and the size distribution was monitored by DLS. As shown in Fig. 1e, before H2O2 was added, precursors T1-3 alone demonstrated a hydrodynamic diameter (DH) of 2.0–2.3 nm. After incubation for one hour, the DH of the reaction solution increased to 68 nm, 44 nm and 106 nm for precursors T1-3, respectively, confirming the cross-linking and formation of FOF-SS1-3. When FOF-SS1-3 were diluted by 50 times, the hydrodynamic diameter did not show a noticeable change, suggesting that the cross-linking of FOFs prevented them from dissolution or swelling (Fig. 1f and S15†). Combined with the above stability test in 1H NMR, the rapid formation and stability of the compound were verified which ensured repeatability of material preparation. When equivalent Cis was added, the hydrodynamic diameter of FOFs did not show a noticeable change, indicating that guest molecules did not have a significant impact on the size of FOFs. The size distribution of Cis (<1 nm) was not observed, which indicated that Cis was drilled into the cavities of FOFs rather than dispersed in the solution. All three FOFs were easily dispersed to form homogeneous and clear solutions in water up to 50 mg mL−1 due to the solubility enhancement of carboxylic acid groups. The carboxylic acid-rich structure also explained the negatively charged ζ potential, with that of −44.2 mV, −44.9 mV and −42.8 mV for FOF-SS1-3, respectively.
Supramolecular interaction between FOFs and NMBAs in vitro
The pore sizes of FOF-SS1-3 were calculated to be approximately 3.0–4.9 nm, spacious enough to accommodate NMBAs. FOFs are abundant with negatively-charged carboxylic acid groups, which render them capable of binding cationic NMBAs. We used 1H NMR spectroscopy to study the binding in water. As shown in Fig. 2a–c, when mixed with FOF-SS3, methylene protons (H1–H3) of the alkyl chains and the aromatic proton (H4) in Cis underwent significant (0.25–0.3 ppm) upfield shifts.38 In addition, other protons also moved upfield slightly, which suggested the shielding nature of FOF cavities. The same upfield shifted resonances were observed for Cis when mixed with FOF-SS1-2 (Fig. S16 and S17†). Similarly, when FOF-SS3 and steroidal NMBAs (Roc, Vec and Panc) were mixed, resonances of steroidal protons (H7–H12) shifted dramatically upfield (Fig. S18–S20†).18 Therefore, FOFs are capable of encapsulating NMBAs.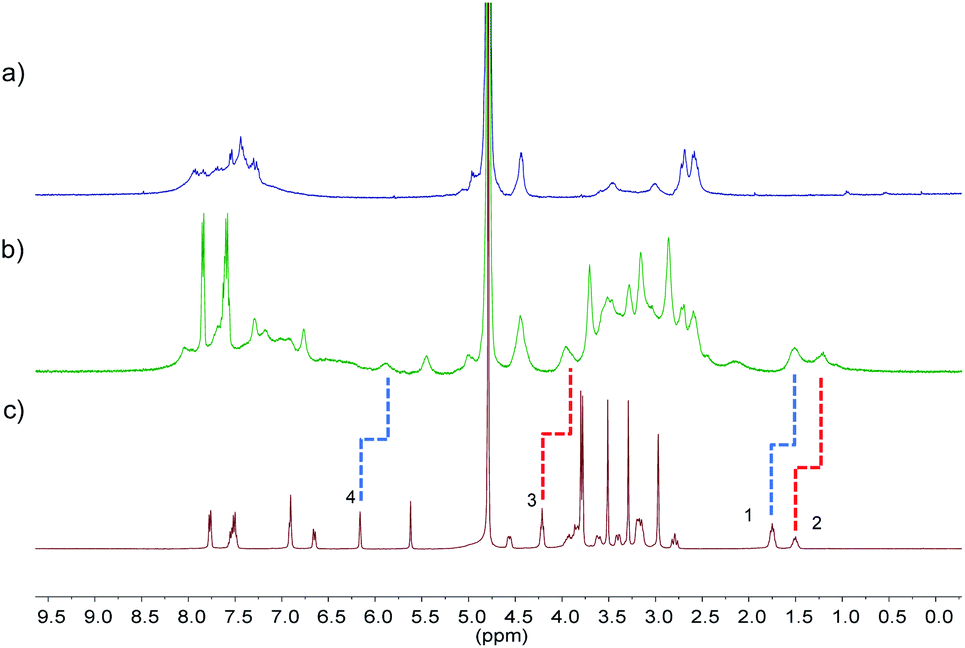 | ||
| Fig. 2 1H NMR spectra (400 MHz, D2O, 298 K) recorded for (a) FOF-SS3 (2 mM, calculated based on [T3]), (b) a mixture of FOF-SS3 (2 mM) and Cis (2 mM), and (c) Cis (2 mM). | ||
A fluorescence titration experiment was employed to study the interaction between Cis and FOFs. Fig. 3a shows that with the gradual addition of Cis into a solution of FOF-SS3 with a fixed concentration, the emission intensity at 752 nm decreased significantly. The emission intensity-Cis concentration curve reached a plateau when the concentration approached 50 μM. This observation indicated that Cis entered the cavity of the FOF and greatly reduced the fluorescence emission intensity, which could be explained by the electrostatic interaction between FOF-SS3 and Cis.
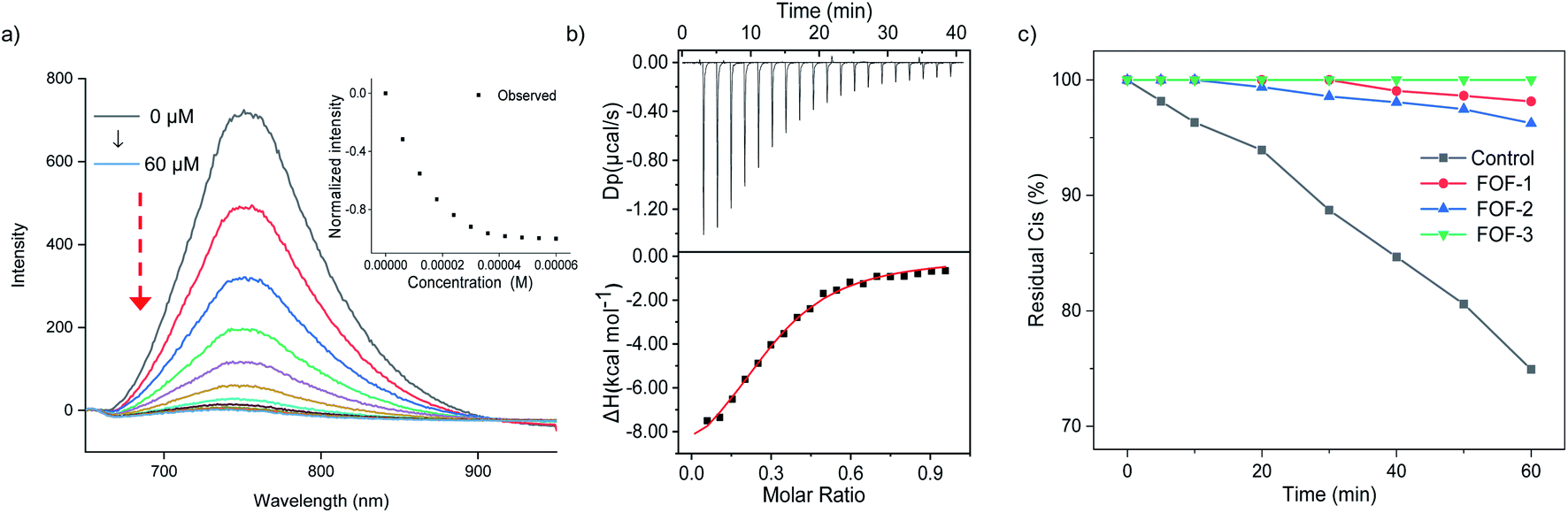 | ||
| Fig. 3 (a) Fluorescence titration of FOF-SS3 (50 μM in PBS) by adding Cis (0–60 μM), λex = 325 nm and λem = 752 nm. (b) Plot of differential power (μcal s−1) vs. time from the titration of FOF-SS3 (0.1 mM) and with Cis (0.8 mM) in PBS (pH 7.4) and plot of the ΔH vs. molar ratio. The red line represents the best non-linear fit of the data Ka = (1.04 ± 0.11) × 105 M−1. (c) The time-dependent change of residual Cis with FOFs in a dialysis bag. [FOF-SS1] = [FOF-SS2] = [FOF-SS3] = 10 mg mL−1and [Cis] = 1 mg mL−1. | ||
To quantitatively analyze the binding strength, isothermal titration calorimetry (ITC) was employed to determine the binding constants of FOFs and NMBAs. The titration curves were well fitted with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. As shown in Fig. 3b, S21–S23.† The value of Ka for the complex of FOF-SS1-3 and Cis was determined to be (1.56 ± 0.65) × 104 M−1, (3.11 ± 0.41) × 104 M−1 and (1.04 ± 0.11) × 105 M−1, respectively. The values of Ka for the complexes of FOFs and other NMBAs were determined by ITC and are summarized in Table 1. The binding strength between FOFs and all the four NMBAs was comparable, indicating that the electrostatic interaction between anionic carboxylate and cationic NMBAs was the key factor in determining the binding strength. The binding strength of FOF-SS3 towards NMBAs was slightly higher compared to that of the other two FOFs, showing that the introduction of a benzene moiety into the FOF helped enhance hydrophobicity and improve binding strength. Acetylcholine (ACh) is a neurotransmitter involved in the NMB process. 1H NMR spectroscopy was employed to compare the interactions between FOFs and Ach or Cis (Fig. S24†). When equivalent stoichiometric FOFs and Ach were mixed, significant (0.26–0.30 ppm) upfield shifts of H5 and H6 in Ach were observed. However, as Cis was added, even excessive ACh did not show stronger competitiveness. The signal of ACh was consistent with that of pure Ach, while that of Cis still moved upfield. The competitive assays showed that the binding towards NMBAs was significantly stronger compared to that of ACh.
:1 binding model. As shown in Fig. 3b, S21–S23.† The value of Ka for the complex of FOF-SS1-3 and Cis was determined to be (1.56 ± 0.65) × 104 M−1, (3.11 ± 0.41) × 104 M−1 and (1.04 ± 0.11) × 105 M−1, respectively. The values of Ka for the complexes of FOFs and other NMBAs were determined by ITC and are summarized in Table 1. The binding strength between FOFs and all the four NMBAs was comparable, indicating that the electrostatic interaction between anionic carboxylate and cationic NMBAs was the key factor in determining the binding strength. The binding strength of FOF-SS3 towards NMBAs was slightly higher compared to that of the other two FOFs, showing that the introduction of a benzene moiety into the FOF helped enhance hydrophobicity and improve binding strength. Acetylcholine (ACh) is a neurotransmitter involved in the NMB process. 1H NMR spectroscopy was employed to compare the interactions between FOFs and Ach or Cis (Fig. S24†). When equivalent stoichiometric FOFs and Ach were mixed, significant (0.26–0.30 ppm) upfield shifts of H5 and H6 in Ach were observed. However, as Cis was added, even excessive ACh did not show stronger competitiveness. The signal of ACh was consistent with that of pure Ach, while that of Cis still moved upfield. The competitive assays showed that the binding towards NMBAs was significantly stronger compared to that of ACh.
| NMBAs | FOF-SS1 | FOF-SS2 | FOF-SS3 |
|---|---|---|---|
| Cis | (1.56 ± 0.65) × 104 | (3.11 ± 0.41) × 104 | (1.04 ± 0.11) × 105 |
| Roc | (1.39 ± 0.86) × 104 | (1.30 ± 0.37) × 104 | (2.78 ± 0.50) × 104 |
| Vec | (1.33 ± 0.47) × 104 | (1.79 ± 0.25) × 104 | (5.36 ± 0.41) × 104 |
| Panc | (2.56 ± 1.16) × 104 | (1.84 ± 0.69) × 104 | (6.95 ± 0.52) × 104 |
Dialysis experiments were conducted to evaluate the capability of FOF-SS1-3 to encapsulate Cis. Briefly, a solution of FOF-SS1-3 (10 mg mL−1) and Cis (1 mg mL−1) in phosphate buffered saline (PBS, pH 7.4) was kept in a dialysis bag (MWCO 1000), and dialyzed against PBS. A solution of Cis alone was used as the control group. The dialysis rate of Cis escaping from the dialysis bag was determined by high-performance liquid chromatography (HPLC), and plotted against time. A concentration-absorption standard curve of Cis was plotted which represented absolute linearity (R2 = 0.9996, Fig. S25†). As shown in Fig. 3c, while approximately 30% of Cis escaped from the dialysis bag containing a solution of Cis within one hour, FOF-SS1-3 reduced the escaping rate to no more than 5%. FOF-SS3 demonstrated the best retaining ability due to its highest binding strength.
Biocompatibility of FOFs in vitro
Cytotoxicity and hemolysis assays were used to verify the biocompatibility of FOFs. The cytotoxicity of FOF-SS1-3 for L02 and H9C2 cells was evaluated using CCK-8 assay. The results showed that the cells maintain a high survival rate (>85%) even at the highest concentration of FOF-SS1-3 (512 μg mL−1). There was no noticeable sign of apoptosis, indicating the low cytotoxicity (Fig. 4a–c). To carry out the hemolysis assay, healthy erythrocytes were transferred into centrifuge tubes and saline with different concentrations of FOF-SS1-3. The absorbance of each sample was recorded at 545nm using a microplate reader to determine the erythrocyte cell rupture ratio. As shown in Fig. 4d–f, the hemolysis rates for all the three FOFs were below 5%, which proved that they did not induce hemolysis.39
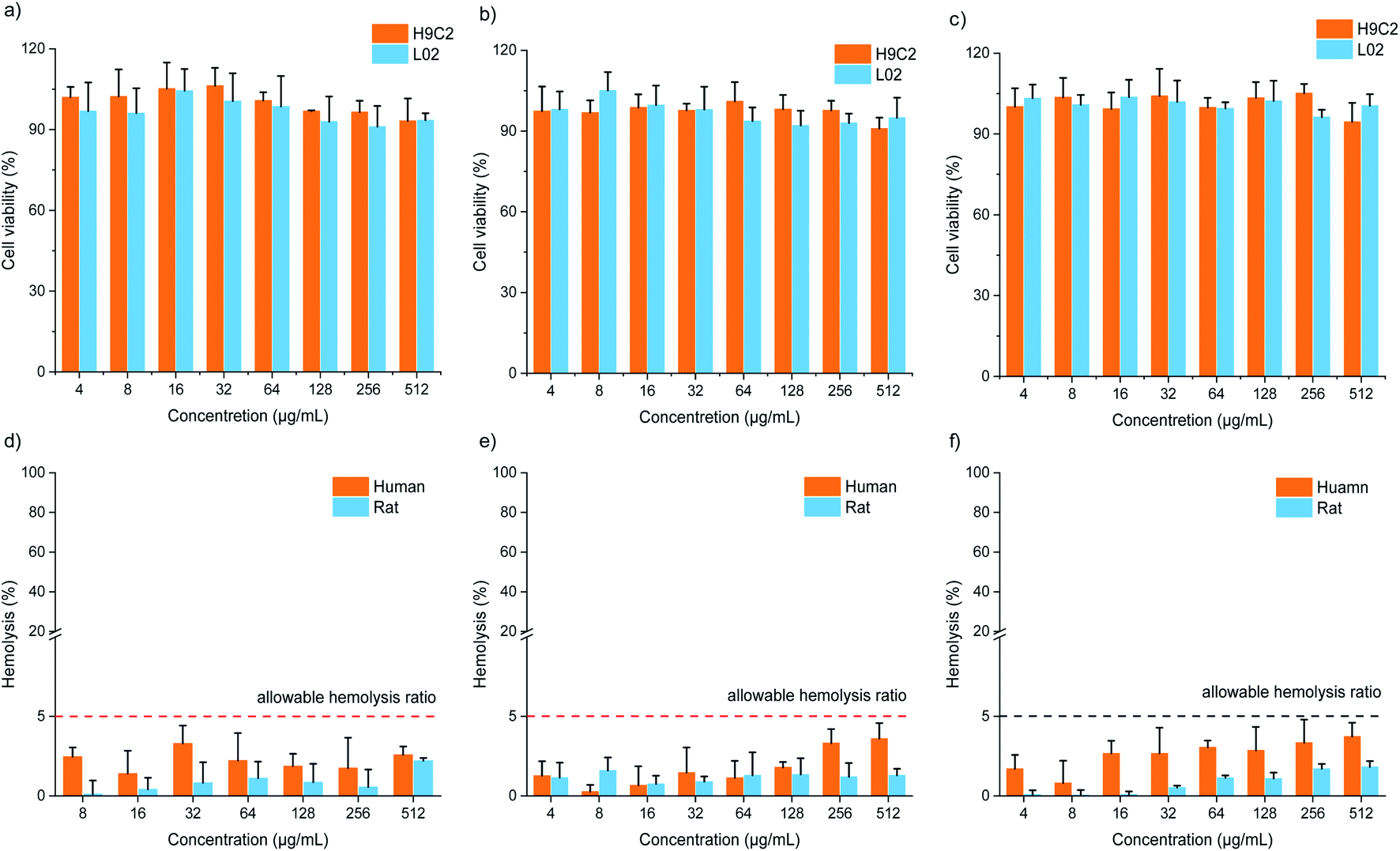 | ||
| Fig. 4 Cell viability values (%) of H9C2 and L02 cells estimated by CCK-8 versus incubation concentrations of (a) FOF-SS1, (b) FOF-SS2, and (c) FOF-SS3. The hemolytic ratio (%) of human and rat erythrocytes versus incubation concentrations of (d) FOF-SS1, (e) FOF-SS2, and (f) FOF-SS3. | ||
Experiment for reversal of NMBAs in vivo
Lastly, we tested the ability of FOF-SS1-3 to reverse NMB in vivo. For this purpose, rats (n = 6) were anesthetized with isoflurane and instrumented with an intravenous line, an arterial line and electrodes to stimulate the femoral nerve. The twitch response of the quadriceps muscle was measured by the acceleration sensor of the Algo TOF-Watch monitor with continuous stimulation at 10 mA current. The monitor was calibrated to ensure a stable response to stimulation, and then switched to TOF mode with continuous stimulation. As the anesthesia started, mechanical ventilation was used to maintain the breath, and Cis was administered by intravenous injection (0.6 mg kg−1, two-fold ED90).18 Thirty seconds later, the quadriceps muscles were completely relaxed (T1 and TOF to 0). Rats were administrated with placebo (0.5 mL), neostigmine bromide (0.06–0.24 mg kg−1) or FOF-SS1-3 (40–120 mg kg−1).As shown in Fig. 5, FOF-SS1-3 can significantly accelerate the recovery of neuromuscular transmission (TOF to 90%) compared with a placebo and neostigmine (placebo: 774 ± 127 s; neostigmine: 210 ± 19 s; FOF-SS1: 202 ± 42 s; FOF-SS2: 187 ± 36 s; FOF-SS3: 115 ± 17 s). The clinical drug neostigmine could shorten the recovery time of NMB induced by Cis compared with a placebo, which was based on the strategy of inhibiting the hydrolysis of acetylcholine.40 Herein we adopted the strategy of sequestering NMBAs to accelerate the recovery of nerve transmission. FOF-SS1-3 could shorten the time by 2.8, 3.1 and 5.7-fold compared to the spontaneous recovery time at the maximum dose. FOF-SS3 showed the highest reversal efficiency, which was consistent with its highest Ka value based on ITC. The recovery time was further shortened than that of neostigmine. Simultaneously, we could observe that with the increasing dosage of FOFs, it showed a significantly improved antagonistic effect against NMB. For FOF-SS3, it reached a plateau at 60 mg kg−1 while FOF-SS1-2 may achieve it at a higher dose.
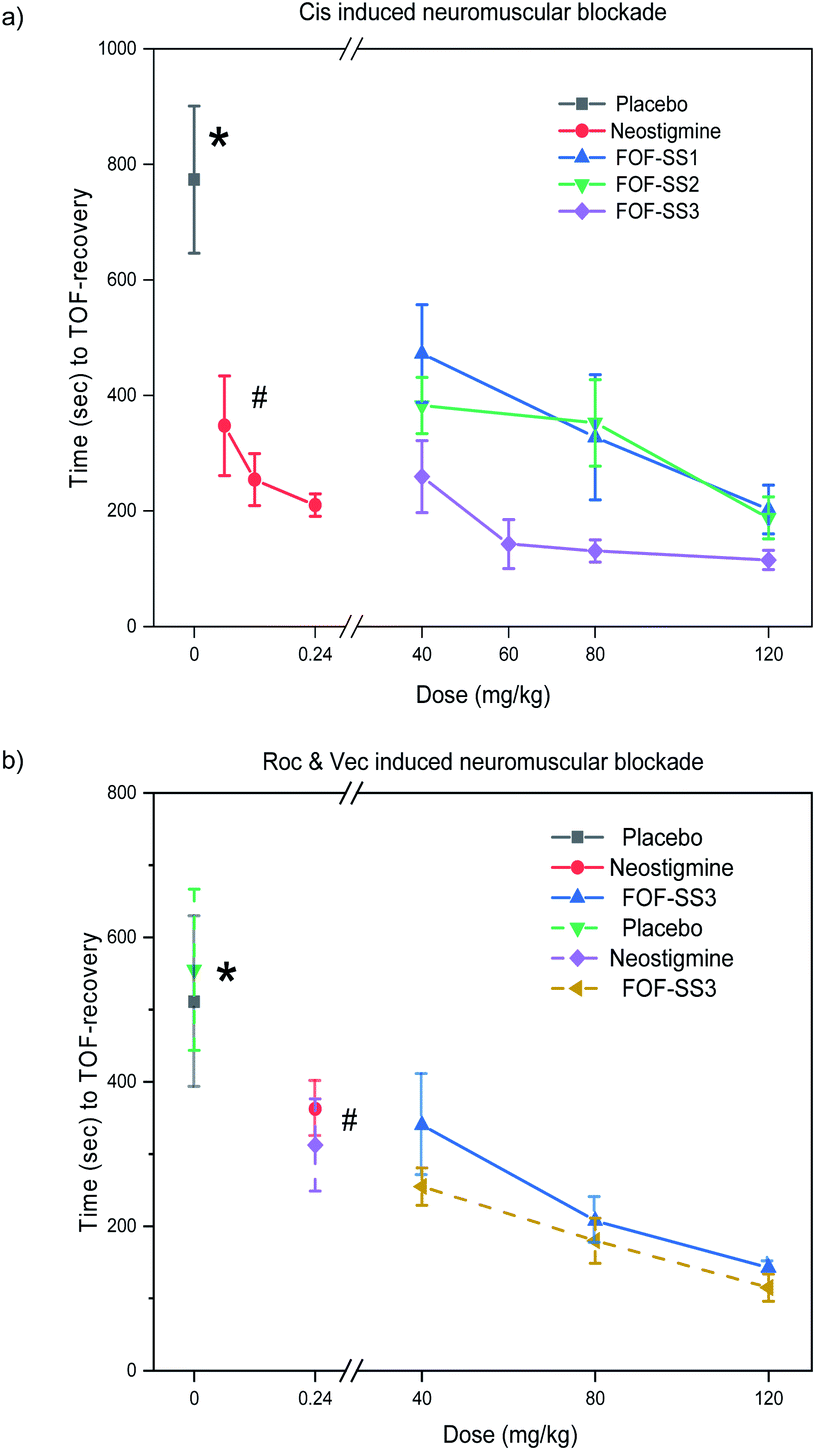 | ||
| Fig. 5 Time to train-of-four (TOF) ratio of 0.9 recovery from: (a) Cis, (b) Roc (solid line) and Vec (dotted line) induced NMB after administration of FOFS, neostigmine or placebo. FOF-SS3 accelerated recovery significantly compared with a placebo (*P < 0.0001). | ||
Encouraged by the good performance in reversing Cis induced NMB, we further explored the potential application of FOF-SS3 in reversing NMB induced by Roc or Vec. FOF-SS3 significantly accelerated the reversal of NMB induced by Roc (placebo: 510 ± 118 s; neostigmine: 362 ± 38 s; FOF-SS3: 142 ± 7.5 s) and Vec (placebo: 555 ± 112 s; neostigmine: 313 ± 64 s; FOF-SS3: 115 ± 19 s). We found that neostigmine can slightly shorten the recovery time while FOF-SS3 could further shorten the time to a train-of-four ratio of 0.9 by 2.6 and 3.8-fold compared to placebo. FOF-SS3 could antagonize benzylisoquinolinium and aminosteroid NMBAs, which may act as a broad-spectrum reversal agent, while the commercial drug sugammadex was only used for aminosteroid NMBAs.
Conclusions
In summary, we constructed three negatively charged and disulfide cross-linked FOFs by oxidative coupling of sulfhydryl groups. FOFs are capable of encapsulating NMBAs due to the synergy between electrostatic and hydrophobic interactions. These FOFs are nontoxic and biocompatible. Animal studies confirm that FOFs are able to efficiently reverse NMB in vivo. This study paves the way to explore other clinical applications of supramolecular sequestration by framework materials.Data availability
Experimental data are available from the authors upon reasonable request.Author contributions
Y. Wu performed the experiments and wrote the manuscript. Y. Y. Liu, H. K. Liu and S. B. Yu guided the cell cytotoxicity experiment and dialysis experiment. F. Lin, H. Wang, and D. W. Zhang participated in data analysis. W. Zhou and D. Ma provided mentorship for the in vivo experiments. D. Ma and Z. T. Li conceptualized the study and revised the manuscript. All authors were involved in the preparation of the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We thank the National Natural Science Foundation of China (No. 21890732, 21890730 and 21921003) for financial support.References
- C. L. Deng, S. L. Murkli and L. Issacs, Chem. Soc. Rev., 2020, 49, 7516–7532 RSC.
- S. M. Bromfield, E. Wilde and D. K. Smith, Chem. Soc. Rev., 2013, 42, 9184–9195 RSC.
- Y. C. Pan, Y. X. Yue, X. Y. Hu, H. B. Li and D. S. Guo, Adv. Mater., 2021, 33, 2104310 CrossRef CAS PubMed.
- P. T. Bremer, A. Kimishima, J. E. Schlosburg, B. Zhou, K. C. Collins and K. D. Janda, Angew. Chem., Int. Ed., 2016, 55, 3772–3775 CrossRef CAS PubMed.
- A. Dahan, L. Aarts and T. W. Smith, Anesthesiology, 2010, 112, 226–238 CrossRef PubMed.
- S. Ganapati, S. D. Grabitz, S. Murkli, F. Scheffenbichler, M. I. Rudolph, P. Y. Zavalij, M. Eikermann and L. Isaacs, ChemBioChem, 2017, 18, 1583–1588 CrossRef CAS PubMed.
- Q. Huang, H. Zhao, M. Shui, D. S. Guo and R. Wang, Chem. Sci., 2020, 11, 9623–9629 RSC.
- T. Mecca, G. M. L. Consoli, C. Geraci, R. L. Spina and F. Cunsolo, Org. Biomol. Chem., 2006, 4, 3763–3768 RSC.
- U. Hoffmann, M. Grosse-Sundrup, K. Eikermann-Haerter, S. Zaremba, C. Ayata, B. Zhang, D. Ma, L. Isaacs and M. Eikermann, Anesthesiology, 2013, 119, 317–325 CrossRef CAS PubMed.
- D. N. Shurpik, O. A. Mostovaya, D. A. Sevastyanov, O. A. Lenina, A. S. Sapunova, A. D. Voloshina, K. A. Petrov, I. V. Kovyazina, P. J. Cragg and I. I. Stoikov, Org. Biomol. Chem., 2019, 17, 9951–9959 RSC.
- H. Yin, D. Bardelang and R. Wang, Trends Chem., 2021, 3, 1–4 CrossRef CAS.
- K. Wang, D. S. Guo, H. Q. Zhang, D. Li, X. L. Zheng and Y. Liu, J. Med. Chem., 2009, 52, 6402–6412 CrossRef CAS PubMed.
- H. Chen, J. Y. W. Chan, S. Li, J. J. Liu, I. W. Wyman, S. M. Y. Lee, D. H. Macartney and R. Wang, RSC Adv., 2015, 5, 63745–63752 RSC.
- Y. M. Zhang, X. Xu, Q. Yu, Y. H. Liu, Y. H. Zhang, L. X. Chen and Y. Liu, J. Med. Chem., 2017, 60, 3266–3274 CrossRef CAS PubMed.
- A. Bom, M. Bradley, K. Cameron, J. K. Clark, J. Egmond, H. Feilden, E. J. MacLean, A. W. Muir, R. Palin, D. C. Rees and M. Q. Zhang, Angew. Chem., Int. Ed., 2002, 41, 265–270 CrossRef CAS.
- J. M. Adam, D. J. Bennett, A. Bom, J. K. Clark, H. Feilden, E. J. Hutchinson, R. Palin, A. Prosser, D. C. Rees, G. M. Rosair, D. Stevenson, G. J. Tarver and M. Q. Zhang, J. Med. Chem., 2002, 45, 1806–1816 CrossRef CAS PubMed.
- W. T. Nicholson, J. Sprung and C. J. Jankowski, Pharmacotherapy, 2007, 27, 1181–1188 CrossRef CAS PubMed.
- D. Ma, B. Zhang, U. Hoffmann, M. G. Sundrup, M. Eikermann and L. Isaacs, Angew. Chem., Int. Ed., 2012, 51, 11358–11362 CrossRef CAS PubMed.
- F. Haerter, J. C. P. Simons, U. Foerster, I. M. Duarte, D. Diaz-Gil, S. Ganapati, K. Eikermann-Haerter, C. Ayata, B. Zhang, M. Blobner, L. Isaacs and M. Eikermann, Anesthesiology, 2015, 123, 1337–1349 CrossRef CAS PubMed.
- W. Xue, P. Y. Zavalij and L. Isaacs, Angew. Chem., Int. Ed., 2020, 59, 13313–13319 CrossRef CAS PubMed.
- X. Zhang, Q. Cheng, L. Li, L. Shangguan, C. Li, S. Li, F. Huang, J. Zhang and R. Wang, Theranostics, 2019, 9, 3107–3121 CrossRef CAS PubMed.
- S. B. Yu, F. Lin, J. Tian, J. Yu, D. W. Zhang and Z. T. Li, Chem. Soc. Rev., 2022, 51, 434–449 RSC.
- J. Tian, H. Wang, D. W. Zhang, Y. Liu and Z. T. Li, Nat. Sci. Rev., 2017, 4, 426–436 CrossRef CAS.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS PubMed.
- J. Li, C. Zhang, S. Gong, X. Li, M. Yu, C. Qian, H. Qiao and M. Sun, Acta Biomater., 2019, 94, 435–446 CrossRef CAS PubMed.
- X. He, Y. Yu and Y. Li, RSC Adv., 2018, 8, 41976–41985 RSC.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef CAS PubMed.
- Y. C. Zhang, P. Y. Zeng, Z. Q. Ma, Z. Y. Xu, Z. K. Wang, B. Guo, F. Yang and Z. T. Li, Drug Delivery, 2022, 29, 128–137 CrossRef CAS PubMed.
- B. Yang, X. D. Zhang, J. Li, J. Tian, Y. P. Wu, F. X. Yu, R. Wang, H. Wang, D. W. Zhang, Y. Liu, L. Zhou and Z. T. Li, CCS Chem., 2019, 1, 156–165 CAS.
- J. Tian, B. Huang, Z. Cui, P. Wang, S. Chen, G. Yang and W. Zhang, Acta Biomater., 2021, 130, 447–459 CrossRef CAS PubMed.
- J. L. Lin, Z. K. Wang, Z. Y. Xu, L. Wei, Y. C. Zhang, H. Wang, D. W. Zhang, W. Zhou, Y. B. Zhang, Y. Liu and Z. T. Li, J. Am. Chem. Soc., 2020, 142, 3577–3582 CrossRef CAS PubMed.
- Z. Y. Xu, H. K. Liu, Y. Wu, Y. C. Zhang, W. Zhou, H. Wang, D. W. Zhang, D. Ma and Z. T. Li, ACS Appl. Bio. Mater., 2021, 4, 4591–4597 CrossRef CAS PubMed.
- Z. K. Wang, J. L. Lin, Y. C. Zhang, C. W. Yang, Y. K. Zhao, Z. W. Leng, H. Wang, D. W. Zhang, J. Zhu and Z. T. Li, Mater. Chem. Front., 2021, 5, 869–875 RSC.
- L. Felix, U. Sezer, M. Arndt and M. Mayor, Eur. J. Org. Chem., 2014, 2014, 6884–6895 CrossRef CAS.
- D. Liu, Z. Xie, L. Ma and W. Lin, Inorg. Chem., 2010, 49, 9107–9109 CrossRef CAS PubMed.
- M. Kirihara, Y. Asai, S. Ogawa, T. Noguchi, A. Hatano and Y. Hiraib, Synthesis, 2007, 21, 3286–3289 CrossRef.
- N. Mistry, A. D. Roberts, G. E. Tranter, P. Francis, I. Barylski, I. M. Ismail, J. K. Nicholson and J. C. Lindon, Anal. Chem., 1999, 71, 2838–2843 CrossRef CAS.
- R. Zhao, T. Ma, F. Cui, Y. Tian and G. Zhu, Adv. Sci., 2020, 7, 2001899 CrossRef CAS PubMed.
- J. Luo, S. Chen, S. Min and L. Peng, Ther. Clin. Risk Manag., 2018, 14, 2397–2406 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02456j |
| This journal is © The Royal Society of Chemistry 2022 |