
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation assisted binding and cleavage of dinitrogen by uranium complexes†
Nadir
Jori
 a,
Thayalan
Rajeshkumar
c,
Rosario
Scopelliti
a,
Ivica
a,
Thayalan
Rajeshkumar
c,
Rosario
Scopelliti
a,
Ivica
![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) ivković
b,
Andrzej
Sienkiewicz
de,
Laurent
Maron
c and
Marinella
Mazzanti
*a
ivković
b,
Andrzej
Sienkiewicz
de,
Laurent
Maron
c and
Marinella
Mazzanti
*a
aInsititut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratory for Quantum Magnetism, Institute of Physics, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, 1015, Switzerland
cLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, Cedex 4, 31077 Toulouse, France
dLaboratory for Quantum Magnetism, Institute of Physics, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
eADSresonances Sàrl, Route de Genève 60B, 1028 Préverenges, Switzerland
First published on 14th July 2022
Abstract
The role of alkali promoters in N2 cleavage by metal complexes remains poorly understood despite its relevance to the industrial production of ammonia from N2. Here we report a series of alkali bound-oxo-bridged diuranium(III) complexes that provide a unique example of decreasing N2 binding affinity with increasing cation size (from K to Cs). N2 binding was found to be irreversible in the presence of K. A N2 complex could be isolated in the solid state in the presence of the Rb cation and crystallographically characterized, but N2 binding was found to be reversible under vacuum. In the presence of the Cs cation N2 binding could not be detected at 1 atm. Electrochemical and Computational studies suggest that the decrease in N2 binding affinity is due to steric rather than electronic effects. We also find that weak N2 binding in ambient conditions does not prevent alkali assisted N2 cleavage to nitride from occurring. More importantly, we present the first example of cesium assisted N2 cleavage leading to the isolation of a N2 derived multimetallic U/Cs bis-nitride. The nitrides readily react with protons and CO to yield ammonia, cyanate and cyanide.
Introduction
Uranium compounds are attracting increasing interest in catalysis and in the activation of small molecules such as N2 and CO2.1 Inorganic uranium compounds were also reported in early studies to be efficient catalysts for N2 conversion to ammonia,2 but iron-based catalysts were adopted in the Haber–Bosch process. As a result, the dinitrogen chemistry of molecular uranium compounds remains less developed1j,m,3 than d-block metals, and only three examples of dinitrogen cleavage were reported so far (complexes A–C in Chart 1) where multimetallic cooperativity between uranium and s- or d-block metals is thought to play an important role.4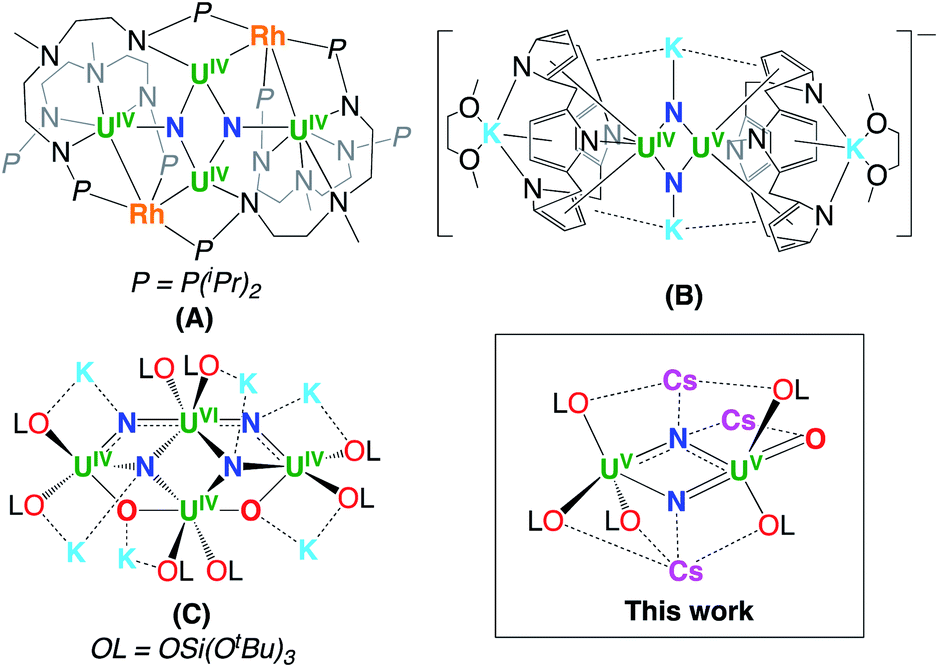 | ||
| Chart 1 The eamples of dinitrogen cleavage by uranium complexes. | ||
The role of multimetallic cooperativity and of alkali ions in dinitrogen binding and cleavage by mono- and multimetallic complexes has attracted large attention1j,3j,5 due to its relevance to the iron-catalysed Haber–Bosch process for the industrial production of ammonia. Most catalysts developed for N2 reduction to ammonia use potassium as an electronic promoter because it has showed higher efficiency than the other alkali ions.5g,6 However, the role of alkali ions in dinitrogen cleavage remains poorly understood as most studies have focused on investigating the effects of N2-bound alkali ions on the extent of dinitrogen activation in molecular dinitrogen complexes of transition metals5g,7a,b with rare examples of studies involving f elements being reported.8,9 Fewer studies were directed to investigate the effect of alkali cations on N2 binding affinity or N2 cleavage and they were mostly limited to d-block metals and iron in particular.5c,e,j,10 Examples of cation assisted dinitrogen cleavage by d-block transition metals were reported for Li, Na K and Rb.10b–d In contrast, no example of dinitrogen cleavage promoted by Cs has been reported despite its higher reducing power. The role of the nature of the reducing alkali ions in the dinitrogen reduction has been investigated5f,g,11 for molecular multimetallic complexes of iron but cleavage of N2 was never observed in the presence of cesium most likely due to kinetic effects favouring the formation of multimetallic intermediates unable to promote N2 cleavage.5c,e,f,11a
The few multimetallic uranium–alkali ion systems reported so far that effect the four-electron reduction of N2,1j,m,3h or even its complete cleavage to nitride,4 all contain the potassium cation. It should be noted that although the number of reported uranium nitride complexes is rapidly increasing12,13 only three examples of uranium nitrides have been obtained from N2 reduction (Chart 1).
Here we report the first example of cesium assisted N2 cleavage which is effected by a multimetallic uranium–cesium complex. Moreover, we isolated a series of multimetallic complexes that are analogues of the previously reported oxide bridged diuranium(III)–K complex [K2{UIII(OSi(OtBu)3)3}2(µ-O)]3h presenting different alkali ions and demonstrated the important effects of the nature of the alkali ion on dinitrogen binding affinity and cleavage.
Results and discussion
U(III)/U(III) oxide-bridged complexes with different alkali metal counterions
We recently reported3h the well-defined multimetallic oxide bridged diuranium(III)–K complex [K2{UIII(OSi(OtBu)3)3}2(µ-O)] K2-2 that effects the four-electron reduction of dinitrogen to yield the diuranium(V) complex [K2{UV(OSi(OtBu)3)3}2(µ-O)(µ-N2)], K2-N2 which was further reduced by excess KC8 to yield a multimetallic nitride cluster (C, Scheme 1) via a putative bis-nitride intermediate (“K4-(N)2”) that could not be structurally characterized.4c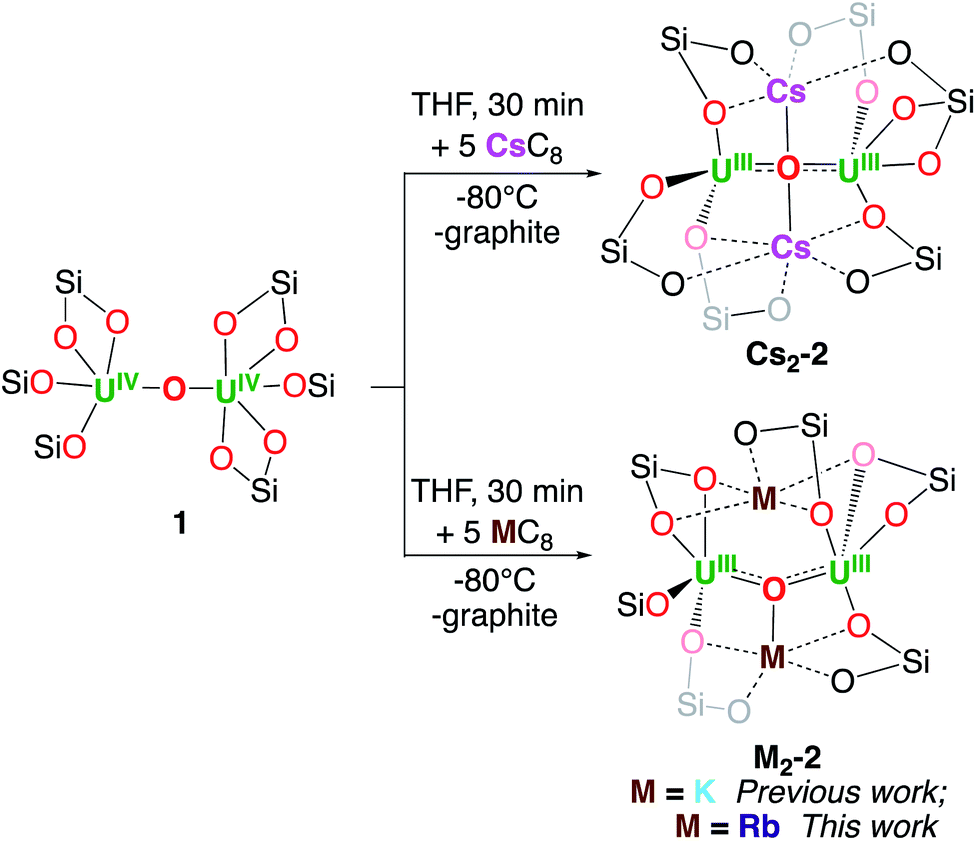 | ||
| Scheme 1 Synthesis of M2-2. | ||
Since K2-2 was obtained by reduction with KC8 of the diuranium(IV) complex [{UIV(OSi(OtBu)3)3}2(µ-O)], 1, this system was identified as an ideal candidate to investigate the effect of the nature of alkali ions on dinitrogen cleavage by uranium complexes. Gratifyingly, the analogous dinuclear uranium(III) complexes [M2{UIII(OSi(OtBu)3)3}2(µ-O)], (M = Rb, Rb2-2; M = Cs, Cs2-2), were obtained by reduction of complex 1 with 5 equivalents of RbC8 or CsC8 respectively in THF at −80 °C. The multimetallic U(III) complexes [M2{UIII(OSi(OtBu)3)3}2(µ-O)], (M = Rb, Cs; Rb2-2 and Cs2-2) (Scheme 1), were isolated from a cold (−40 °C) hexane and toluene solution with yields of 60 and 70% respectively.
The M2-2 complexes showed low thermal stability in THF solution, with a faster decomposition observed for K2-2 (complete decomposition over the course of 12 h)3h compared to Rb2-2 (60% remaining after 24 h) and Cs2-2 (80% remaining after 24 h). The stability of these complexes is greatly increased in THF solution at −40 °C, with 84% of Cs2-2 after 21 days and 62% of Rb2-2 after 14 days. Moreover, the complex Cs2-2 shows a remarkable stability in toluene solution at room temperature (no decomposition is observed up to 17 days), compared with Rb2-2 which decomposes completely over the course of 5 days and K2-2 which decomposes over the course of 24 h in the same conditions.3h The latter two decompose to a mixture of [M2{UIV(OSi(OtBu)3)3}2(µ-O)2], M2-4 and other unidentified species. A few crystals of Rb2-4 could be isolated and characterized by X-ray diffraction (Fig. S59†) and 1H NMR spectroscopy (Fig. S14†).
The synthesis of the Na and Li analogues was also pursued but, since the MC8 reducing material is inaccessible, different synthetic strategies were followed without success. Notably, the reduction of 1 with Na0 mirror led only to partial reduction and to the isolation of the mixed-valent complex [Na{UIII(OSi(OtBu)3)3}{UIV(OSi(OtBu)3)3}(µ-O)], Na-3 in 30% yield (Fig. S58†).
The 1H NMR spectra of Rb2-2 and Cs2-2 both show for the siloxide ligands a broad signal in THF at −40 °C and one sharp signal in toluene at −40 °C. Only one sharp signal is observed in the 1H NMR spectra of Rb2-2 and Cs2-2 measured in toluene or THF at 25 °C suggesting fluxional behaviour in solution of the siloxide ligands. Similar 1H NMR spectra have been reported for the K2-2 complex. Overall, the binding of different cations results in a small shift of the signals assigned to the siloxide ligands (Fig. S15,† in a range of −0.08 ppm to −0.28 ppm in toluene solution at −40 °C). The sequential addition of 2 equiv. of 2.2.2-cryptand to a THF solution of K2-2 at −40 °C, results in the displacement of the resonance assigned to the siloxide ligands (Fig. S58†). These studies indicate that the cations remain bound to the siloxides both in toluene and in THF solutions.
The solid-state molecular structure of Rb2-2 (Fig. 1, middle) shows an ion-paired complex with two U(III) ions bridged by an oxo ligand, which also binds one Rb+ cation (at a Rb–O distance of 3.007(6) Å) located in the pockets formed by four oxygen atoms of the siloxide ligands. A second Rb+ cation is coordinated by five oxygen atoms of the siloxide ligands (2.899(6)–3.146(6) Å) but is located at a non-bonding Rb–O distance (3.696(6) Å).
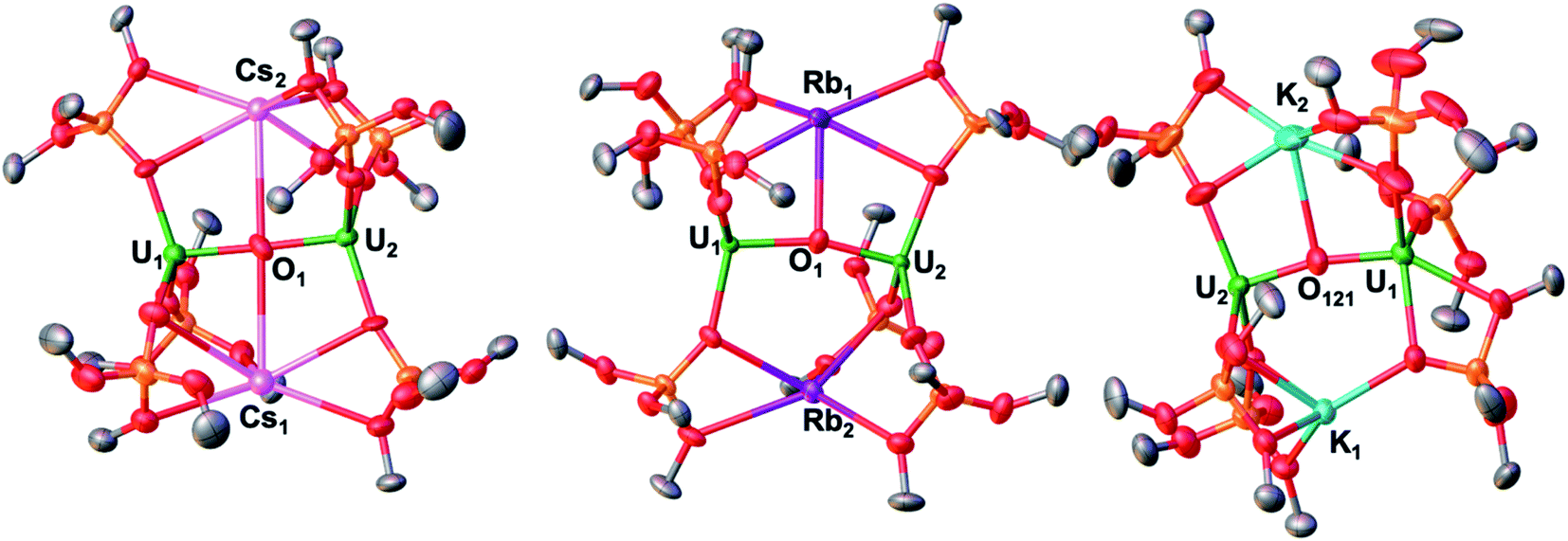 | ||
| Fig. 1 Solid-state molecular structure of Cs2-2 (left) Rb2-2 (middle) and K2-2 (redrawn from ref. 3h) with 50% probability ellipsoids. Color code: uranium (green), potassium (light blue) rubidium (purple), cesium (pink), oxygen (red), carbon (grey), silicon (orange). Hydrogen atoms and tBu groups were omitted for clarity. | ||
The binding mode found in Rb2-2 is similar to that reported for K2-2 (Fig. 1, right) with an oxo bound K+ at 2.913(4). Å and an unbound K+ at 3.392(4) Å. The K+ ions are bound by four and five siloxide oxygen atoms, respectively, at distances in the range 2.643(3)–3.008(4) Å, shorter than those found for the Rb-siloxides in Rb2-2.
The values of the UIII–(µ-O) distances (2.100(5) and 2.135(5) Å) are similar to those reported for 1 (2.085(1) and 2.137(1) Å) and compare well to the values reported for K2-2,3h and the only two other U(III) bridging oxo complexes previously reported.14 The two uranium atoms are held in close proximity at a U–U distance of 4.1972(8) Å similar to that found in K2-2 (4.262(1) Å). The U–(µ-O)–U core is bent (164.7(3)°), as was also reported for K2-2 (167.4(2)°).
The solid-state molecular structure of Cs2-2 (Fig. 1, left) displays a similar ion-paired dinuclear U(III) complex bridged by an oxo group at a U–U distance of 4.247(1) Å as K2-2 and Rb2-2, but with some differences: first the binding of the oxo group to the U(III) centers in Cs2-2 is more symmetric than in K2-2 and in Rb2-2 with two very similar UIII–(µ-O) distances (2.137(7) and 2.126(7) Å); second the U–(µ-O)–U core is almost linear (177.9(4)°) while it is bent in K2-2 and in Rb2-2. Moreover, in Cs2-2 both Cs+ cations are coordinated to the oxo group (3.434(8) and 3.336(8) Å) (Table 1).
| Complex | U–U | U1–(µ-O) | U2–(µ-O) | U1–(µ-O)–U2 | M1–(µ-O) | M2–(µ-O) |
|---|---|---|---|---|---|---|
| K2-2 | 4.262(1) | 2.178(3) | 2.120(3) | 167(4) | 3.392(4) | 2.913(4) |
| Rb2-2 | 4.1972(8) | 2.135(5) | 2.100(5) | 164.7(3) | 3.007(6) | 3.696(6) |
| Cs2-2 | 4.247(1) | 2.137(7) | 2.126(7) | 177.9(4) | 3.336(8) | 3.434(8) |
Overall, the binding of the cations K, Rb and Cs in the three complexes is quite similar, but we were interested in investigating how the observed structural differences would affect their magnetic properties and their reactivity.
Variable temperature SQUID magnetic data were measured for the complexes Rb2-2 and Cs2-2 and were compared with the previously reported magnetic data for K2-2.3h Data were measured only up to 250 K due to the low stability of the complexes above 250 K. The measured values of the magnetic moment per ion at 250 K are 2.1 µB for K2-2, 2.77 µB for Rb2-2 and 2.51 µB for Cs2-2 and decrease to a low value of 0.9 µB for K2-2, 0.7 µB for Rb2-2 and 0.4 µB for Cs2-2 at 2 K. Low values of χT at low temperature have been previously reported previously for other U(III) complexes.15 The complex Cs2-2 shows a significantly different behaviour of the magnetic susceptibility measured in function of the temperature, as can be clearly observed in the plot of the magnetic susceptibility (χ) versus T (Fig. S65†). The χ versus T plot for Cs2-2 indicates the unambiguous presence of antiferromagnetic coupling between the U(III) centers, with a maximum of the χ at 10 K. In contrast, the plot of χ versus T of the K2-2 and Rb2-2 complexes shows the magnetic behaviour associated with two magnetically independent U(III) ions.16 These results suggest that the presence of two Cs atoms in the core of the molecule allows a magnetic communication between the metal centers which is most likely correlated to a more covalent U–O–U bond in agreement with its linear geometry.
Binuclear complexes of uranium(III) are rare and only very few examples of magnetic communication between U(III) centers were reported including the diuranium(III) nitride complex [K3{UIII(OSi(OtBu)3)3}2(µ-N)], K3UNU complex1j which showed antiferromagnetic coupling with a maximum at a T of 23 K.3h Stronger antiferromagnetic coupling with the highest value of χ at 110 K was reported by Cummins and Diaconescu for an arene-bridged U(III) dimer.15a,17
Reactivity of M2-2 with N2
In order to identify the role of the cation in N2 binding and reduction we investigated the reaction of Rb2-2 and Cs2-2 with N2 and compared it with that previously reported for K2-2 (Scheme 2 and S44†).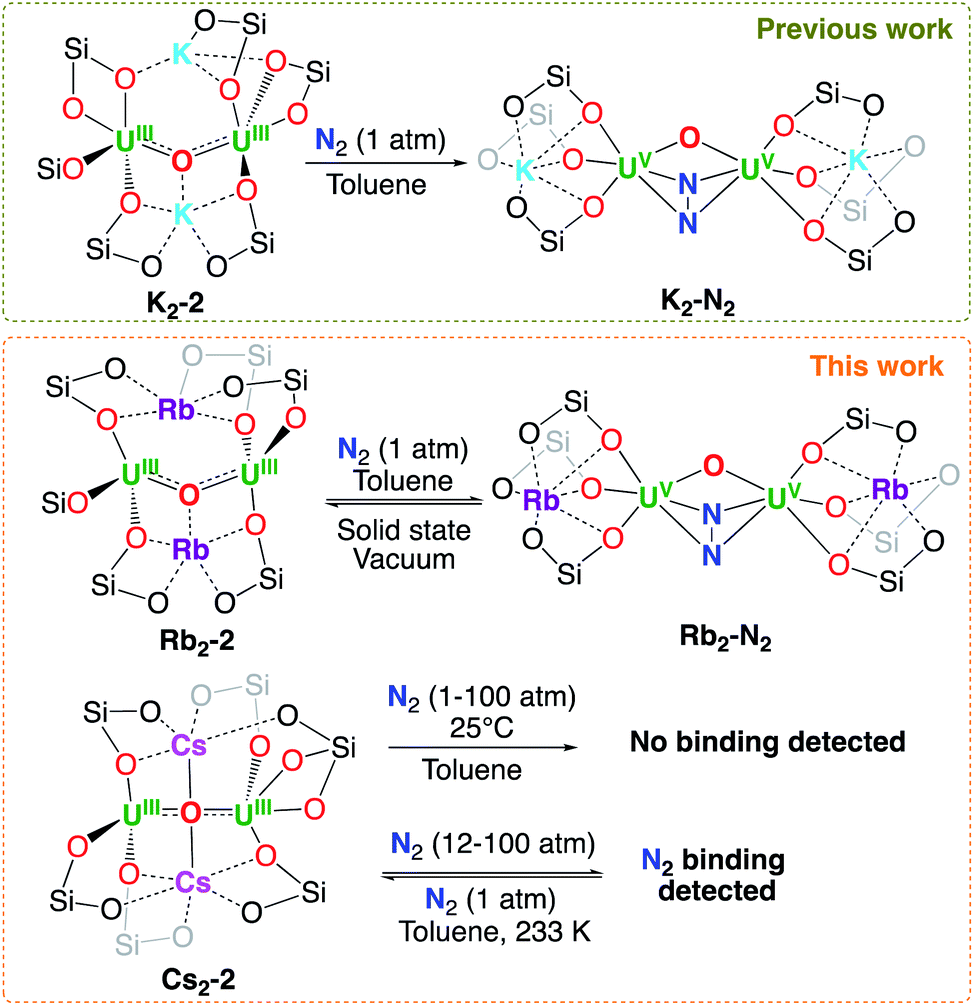 | ||
| Scheme 2 N2 reactivity of M2-2. | ||
We previously showed that K2-2 reacts rapidly with N2 in ambient conditions effecting its four-electron reduction and yielding the diuranium(V)–N2 complex [K2{UV(OSi(OtBu)3)3}2(µ-O)(µ-N2)], K2-N2.
Complex K2-N2 was shown to be stable even under dynamic vacuum although addition of acid or H2 led to N2 release. Similarly, when exposing a dark red toluene solution of Rb2-2 to 1 atm of N2, at 25 °C its colour changed suddenly to dark brown and the 1H NMR spectrum showed the disappearance of the signals of the Rb2-2 and the appearance a new signal at a chemical shift of −2.27 ppm (Fig. S18†) that was assigned to the N2 complex [Rb2{UV(OSi(OtBu)3)3}2(µ-O)(µ-N2)], Rb2-N2 (Scheme 2).
However, the removal of N2 from the solution led to the loss of uranium bound dinitrogen and the formation of the Rb2-2 complex as shown by 1H NMR studies (Fig. S20†) demonstrating that the binding of Rb2-N2 occurs at ambient pressure and temperature but is reversible under vacuum.
Dark brown crystals of [Rb2{UV(OSi(OtBu)3)3}2(µ-O)(µ-N2)], Rb2-N2, were grown from a concentrated toluene solution under N2 atmosphere in 53% yield (Scheme 2). However, leaving the isolated solid under dynamic vacuum resulted in dinitrogen loss and in mixtures of Rb2-2 and Rb2-N2 complexes (Fig. S19†). Addition of acid or H2 (Fig. S21†) to Rb2-N2 also resulted only in N2 loss, as confirmed by the comparison of the 1H NMR spectrum with the reaction of Rb2-2 with H2, which gave the same set of resonances. This was observed previously for the formation of a bis-hydride from K2-N2.3h
In the crystal unit cell of Rb2-N2 (Fig. 2) there are two crystallographically independent molecules. In both molecules an oxo group (U1–O–U2 = 104(1)° and U3–O–U4 = 103.7(7)°) and a side-on bound hydrazido moiety (N24−) (U1–N2A–U2 = 103.3(2)° and U1–N1A–U2 = 100.5(10)° and U3–N3A–U4 = 102.9(12)° and U1–N1A–U2 = 99.4(5)°) bridge two uranium(V) centers with a short U–U distance of 3.4083(1) Å. As found in the previously reported structure of K2-N2, the alkali metal cations are not N2-bound, but are located in the pockets formed by the siloxide moieties.
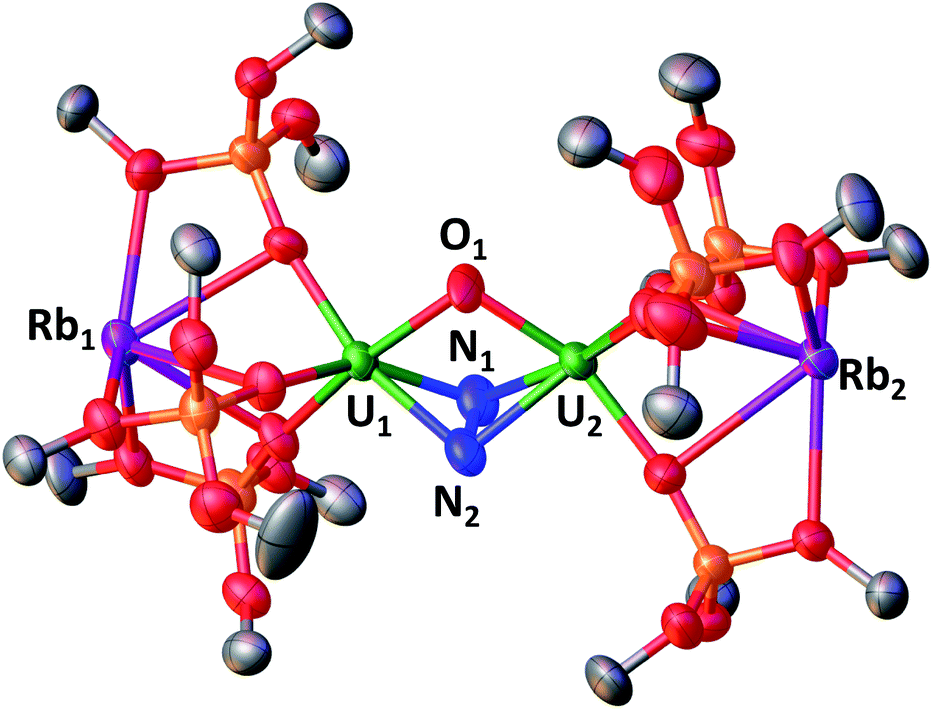 | ||
| Fig. 2 Solid-state molecular structure of Rb2-N2 (50% probability ellipsoids). Color code: uranium (green), rubidium (purple), oxygen (red), carbon (grey), nitrogen (blue), silicon (orange). Hydrogen atoms and tBu groups were omitted for clarity. | ||
The U2(µ-η2-N2) moiety in Rb2-N2 has U–N distances between 2.14(3) Å and 2.3(1) Å, comparable to those found in K2-N2 and in the previously reported diuranium(V) hydrazido complex [K3{UV(OSi(OtBu)3)3}2(µ-N)(µ-N2)], K3UNU.1j The N–N bond length in Rb2-N2 (1.41(1) Å) is comparable to that observed in hydrazine, H2NNH2 (1.47 Å), and falls in the range of values (1.377–1.548 Å) reported for hydrazido complexes of Zr(IV), U(IV) and U(V).1h,m,3h,18 The structural parameters point to a formulation of the complex as a [Rb2{UV(OSi(OtBu)3)3}2(µ-O)(µ-N2)] which is supported by the DFT studies (see infra) and EPR data. Notably, low-temperature (6 K) continuous-wave (CW) X-band EPR of 20 mM toluene solutions of Rb2-N2 measured as toluene/hexane glass (Fig. S82†) revealed the presence of a broad (∼3000 G) and poorly resolved EPR signal, with slightly better distinguished features in the low magnetic field region having the g-factor value of 3.31, which is consistent with a 5f1 electronic configuration. Similar g-values have been observed in other uranium(V) complexes.12m,13e
The structural and EPR data suggest a similar degree of activation of the bound N2 occurring in Rb2-N2 and K2-N2, but release of N2 occurs for Rb2-N2 under vacuum.
The lower stability of the N2 complex Rb2-N2 compared to K2-N2 under vacuum indicates a decreased binding constant of the N2 complex despite the structural similarity of the two U(III) complexes Rb2-2 and K2-2 and their N2 complexes K2-N2 and Rb2-N2 (see structural description above). It should also be noted that although the alkali ion does not bind directly the N2 in the solid-state structure of the final N2 complex, it plays an important role in N2 binding.
Finally, 1H NMR studies show that when exposing solid Cs2-2 or a toluene solution to 1 atm of N2 at 25 °C or lower temperatures (−40 °C or −80 °C) no reaction is observed. The addition of higher pressures of N2 (10 to 100 bar) at 25 °C to a dark red toluene solution of Cs2-2 did not yield any new species, as confirmed by 1H NMR studies (Fig. S41†). However, exposing a dark red solution of Cs2-2 to higher pressures (12–100 atm) of N2 at −40 °C resulted in the partial to total consumption of the U(III) starting material and the formation of a new species as confirmed by 1H NMR studies showing one broad signal for the siloxide ligands similarly to what found for the K2-N2 and Rb2-N2 complexes, (Fig. S42–S44†). The binding of dinitrogen is reversible when removing the excess pressure at −40 °C, forming Cs2-2 over the course of 16 h (Fig. S43†). When increasing the temperature at 100 atm formation of Cs2-2 is also observed together with unidentified decomposition products. Favourable binding of N2 at low temperature has been previously observed for titanium and lanthanide complexes.19
These results show the important effect of the coordinated cation in the binding of dinitrogen by the dinuclear oxo bridged diuranium(III) complexes [M2{[UIII(OSi(OtBu)3)3}2(µ-O)], M2-2. In particular, an important decrease in N2 binding affinity is observed from K+ to Rb+ to Cs+ that can be correlated with the increased size and decreased Lewis acidity of the cation. The electronic effects of the bound cation on the reactivity of the diuranium(III) complex are not straightforward since an increased Lewis acidity of the cation should result in a less electron-donating siloxide ligand and as a result a less reducing uranium center which was confirmed by cyclic voltammetry studies (see infra).
Steric effects arising from the presence of a larger cation binding the siloxide could also play a role. Notably, the larger size of the Cs+ compared to K+ and the binding of two Cs+ to the bridging oxide results in an overall arrangement that renders more difficult or even prevents the access to the U centers, as can be seen for the space filling diagrams of the M2-2 complexes (Fig. S56†) and by computational data (Fig. 5).
Although the alkali ions do not bind dinitrogen in the final M2-N2 complexes, cation binding to the reduced U(III)–O–U(III) complex affects significantly the ability of the metal centers to bind and reduce N2. Since differences in N2 binding arise from the different binding of alkali cations to the oxo linkers, we believe these results bear some relevance to the Haber–Bosch process since the Mittasch catalyst is based on metal oxides.
Indeed, besides the steric effects of the molecular siloxide envelope, this study hints that the binding of larger ions such as Cs to the M–O–M fragment is likely to reduce the N2 binding constant in metal oxides.
Electrochemical studies
To fully characterize and compare the reduction power of the M2-2 complexes, cyclic voltammetry data were measured under argon atmosphere for these complexes and for complex 1 in ∼0.1 M THF solution of [Bu4N][BPh4] and are presented in Fig. 3. All redox potentials are referenced against the [(C5H5)2Fe]+/0 redox couple.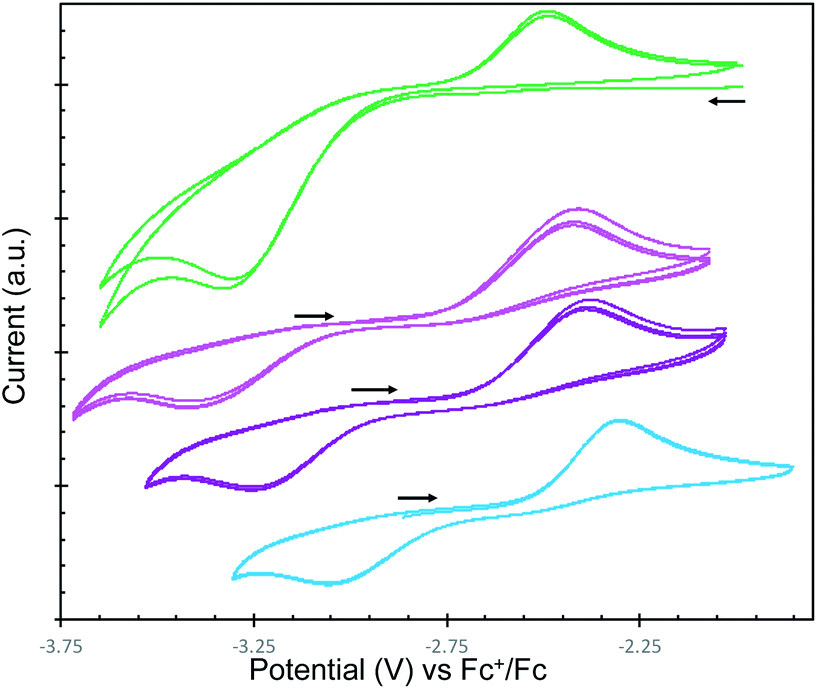 | ||
| Fig. 3 [−3.8 V; −1.85 V] region of cyclic voltammogram of THF solutions of complexes 1 (3.0 mM, green) and M2-2 (3.5 mM, M = Cs, pink), (3.0 mM, M = Rb, purple), (3.0 mM, M = K, light blue) recorded in 0.1 M [NBu4][BPh4] under Ar at 25 °C, at a scan rate of 100 mV s−1, referenced against [Fe(C5H5)2]+/0. | ||
The voltammogram of complex 1 shows a distinctive irreversible reduction event at Epc = −3.32 V associated with the irreversible oxidation process at Epa = −2.48 V. This irreversible redox event can be associated to the U(III)/U(IV) couple, since the oxidation at Epa = −2.48 V is not observed in the voltammogram of 1 when it is swept initially from −1.98 V towards the positive region.
The same irreversible redox events were observed in the voltammograms of the family of complexes M2-2. Interestingly, a distinctive oxidation event (Epc = −2.3, −2.37 and −2.41 V for M = K, Rb and Cs, respectively) is observed always when swept towards positive region, which shifts towards negative values when decreasing the Lewis acidity of M. The respective reduction event (Epc = −3.07, −3.23 and −3.4 V for M = K, Rb and Cs, respectively) is only observed after initial oxidation of the complexes and a negative shift is also observed when decreasing the Lewis acidity of M (Table 2).
| Compound | E pc (V) | E pa (V) | ΔE (V) |
|---|---|---|---|
| 1 | −3.32 | −2.48 | 0.84 |
| Cs2-2 | −3.4 | −2.41 | 0.99 |
| Rb2-2 | −3.23 | −2.37 | 0.86 |
| K2-2 | −3.07 | −2.3 | 0.77 |
These results show that the reducing potential of M2-2 increases with the decrease in the Lewis acidity of the alkali metal counterion, with the highest reducing power for the series observed for Cs2-2 (Epc = −3.4 V and Epa = −2.41 V).
The values of the redox potentials assigned to the redox couple U(IV)/U(III) in M2-2 are significantly more negative than previously reported values for the same redox couple in a family of cyclopentadienyl U(III) complexes (in the range between −1.04 V and −1.54 V).20 These results show that siloxide supported diuranium(III) oxides are highly reducing, but also indicate that the difference in reactivity towards dinitrogen observed for the M2-2 complexes cannot be related to differences in redox potential of the uranium centers. Notably the most reducing complex Cs2-2 is showing the lowest reactivity with N2.
Dinitrogen cleavage and functionalization by Rb2-2 and Cs2-2
We recently reported that the reduction of the N2 complex K2-N2 with 2 equiv. of KC8 leads to the formation of a new species with a higher degree of activation of the bound N2. On the basis of the structure of the tetranitride complex isolated from the reduction of K2-N2 with excess KC8 and on the basis of computational and reactivity studies this species was proposed to be a bis-nitride complex but its molecular structure was not elucidated.4cTo gain further insight into the role of the alkali ions in the cleavage of N2 to nitrides we explored the reduction of Rb2-N2 with 2 equiv. RbC8 and of Rb2-2 and Cs2-2 with 2 equiv. RbC8 and CsC8 respectively under N2.
1H NMR studies (Fig. S34 and S35†) showed that the reduction of Rb2-N2 with 2 equiv. of RbC8 at −40 °C under 1 atmosphere N2 or the reduction of Rb2-2 with 2 equiv. RbC8 under 1 atmosphere N2 lead to the formation of a new species which displays 1H NMR signals similar to those reported for the putative bis-nitride intermediate obtained from the 2 electron reduction of K2-N2 (Fig. S36–S39†).4c Unfortunately also in this case the putative nitride could not be isolated from the reaction mixture. However, the addition of excess acid to the reaction mixture between Rb2-2 and 2 equiv. of RbC8 after removing the graphite yielded NH4Cl in 85% yield (1.7 equiv., 100% conversion corresponding to 2 equiv. of NH4Cl) suggesting that cleavage of the bound N2 has occurred.
Despite the fact that we were not able to observe N2 binding by Cs2-2 at −40 °C and 1 atm N2 by 1H NMR spectroscopy we could not completely rule out the possibility of weak N2 binding not observable by NMR.19c,21
Gratifyingly the reduction of Cs2-2 with 2 equiv. of CsC8 at −40 °C under 1 atmosphere of N2 revealed the complete consumption of the dinuclear U(III) starting material and the formation of a new major species (Fig. S22 and S23†). Golden-yellow crystals of [Cs3{UV(OSi(OtBu)3)3}(µ-N)2{UV(OSi(OtBu)3)2(κ-O)}][CsOSi(OtBu)3] Cs4-(N)2 (Fig. 4) were grown in 50% yield by leaving a concentrated toluene solution of Cs4-(N)2 at −40 °C over the course of two days (Scheme 3). 1H NMR studies indicated that the same reaction performed under an Ar atmosphere, did not lead to new reduction products but only to formation of small traces of decomposition (Fig. S28†).
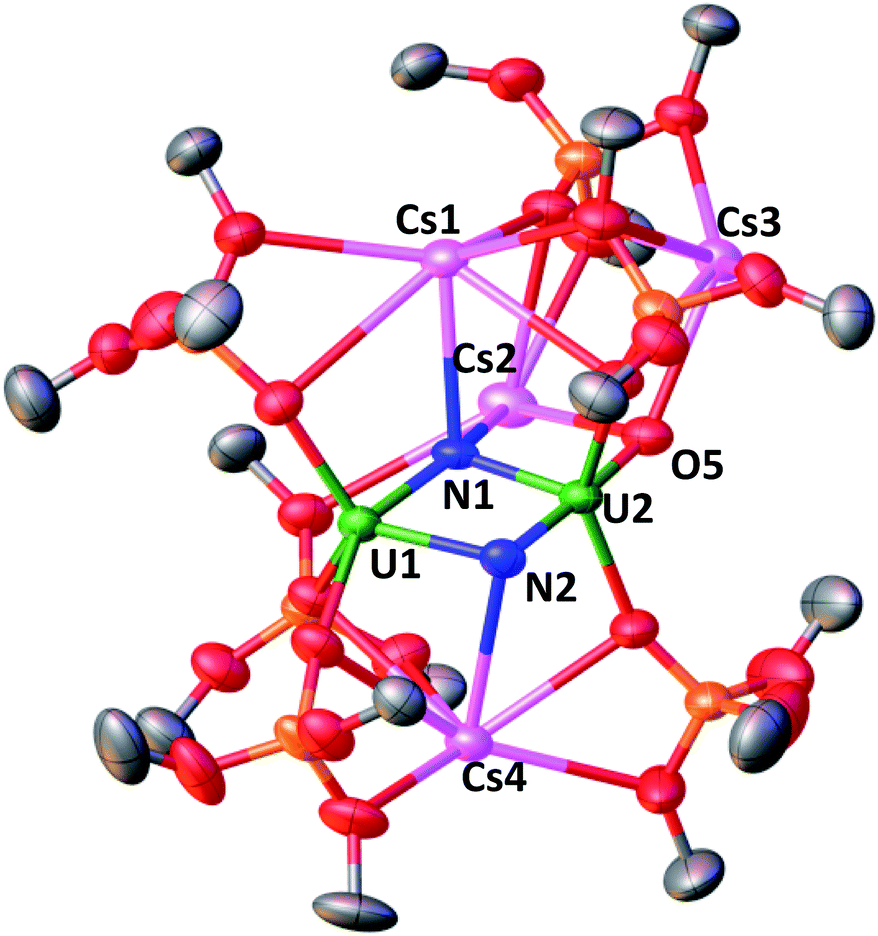 | ||
| Fig. 4 Solid-state molecular structure of Cs4-(N)2 (50% probability ellipsoids). Color code: uranium (green), cesium (pink), oxygen (red), carbon (grey), nitrogen (blue), silicon (orange). Hydrogen atoms and tBu groups were omitted for clarity. | ||
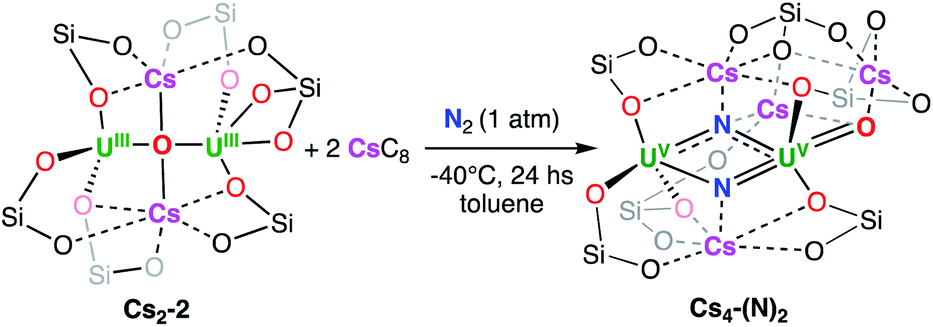 | ||
| Scheme 3 Reduction of Cs2-2 under N2. | ||
These results suggest that reversible binding of N2 by Cs2-2 must occur to some extent in toluene solution also at −40 °C and 1 atm although it was not possible to detect it by 1H NMR spectroscopy.
Cs4-(N)2 is unstable in toluene solution at 25 °C, decomposing completely over the course of 12 h (Fig. S26†). The decomposition is slower at −40 °C, with 75% of Cs4-(N)2 still present in solution after 2 weeks (Fig. S27†).
The molecular structure of complex Cs4-(N)2 (Fig. 4) shows the presence of two independent dimeric complexes where two uranium ions and three cesium ions are bridged by two nitrides (the µ4-nitride N1 bridges two U centers and two Cs and the µ3-nitride N2 bridges two U and one Cs). The two U(V) centers are both pentacoordinated with a distorted square pyramidal geometry and are held together by the bridging nitrides and the siloxide framework at a short U–U distance of 3.337(2) Å. U1 is coordinated by two oxygen atoms from the siloxide ligands, the two nitrides and a terminal oxo group, while U2 is coordinated by three oxygen atoms from the siloxide ligands and two nitrides.
Asymmetric U–N bond distances are found in complex Cs4-(N)2. In particular, a very short U–N distance (U2–N2 = 1.85(1) Å) and a longer U–N distance (U1–N2 = 2.34(1) Å) is found for the (µ3-nitride) which is trans to the terminal oxo group, which suggest that N2 binds U2 with a multiple bond and U1 with a single bond. The (µ4-nitride) bridges the two U(V) centers in a more symmetric fashion (U–N1: 2.09(1) Å and 2.13(1) Å). The latter distances compare well to those found in the previously reported dinuclear U(IV)/U(V) (U–N: 2.076(6) and 2.099(5) Å),4a and the U(V)/U(V) bis-nitrides (U–N: 2.101(6) and 2.022(5) Å) complexes.13a
The terminal oxo O5 binds two cesium atoms resulting in a U1–O5 distance (1.856(4) Å) longer than the one observed in the previously reported U(V) terminal oxo [U(O){N(Si(Me3))2}3] (U–O distance 1.817(1) Å).22 The complex Cs4-(N)2 presents a unique and second of its kind nitride-substituted analogue of the uranyl(V) ion. The distances observed in the trans oxo-nitrido moiety [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UVN] found in Cs4-(N)2 (U2–O5: 1.856(4) Å; U2–N2 = 1.85(1) Å) are significantly longer than those found in the only other known trans oxo-nitrido U(VI) complex reported by Hayton and coworkers12c (U–O = 1.797(7) Å; U–N = 1.818(9) Å).
UVN] found in Cs4-(N)2 (U2–O5: 1.856(4) Å; U2–N2 = 1.85(1) Å) are significantly longer than those found in the only other known trans oxo-nitrido U(VI) complex reported by Hayton and coworkers12c (U–O = 1.797(7) Å; U–N = 1.818(9) Å).
An additional siloxide ligand is held in the complex through the binding of three Cs cations. The shift of the bridging oxo to terminal oxo is likely to be responsible for the cleavage of the U2–OSi(OtBu)3 bond.
1H NMR studies show that most of the Cs-bound siloxide ligand remains associated in toluene solution but dissociation was observed in THF (Fig. S31–S33†).
Variable temperature SQUID magnetic data were measured in the temperature range 2 K–250 K for Cs4-(N)2 (due to the low stability of the complex at room temperature) and compared with the previously reported putative “K4-(N)2” intermediate. The measured magnetic data of Cs4-(N)2 are similar to those measured in situ for the putative “K4-(N)2”4c with a magnetic moment per ion of µ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) = 1.51 µB at 250 K for “K4-(N)2” and of µ = 1.57 µB at 250 K for Cs4-(N)2 and µ = 0.33 µB for Cs4-(N)2 and µ = 0.27 µB for “K4-(N)2” at 2 K, indicative of the presence of U(V) cations, and a χ versus T behaviour in agreement with the presence of two magnetically independent ions (Fig. S66†), similarly to what was found for “K4-(N)2”.
= 1.51 µB at 250 K for “K4-(N)2” and of µ = 1.57 µB at 250 K for Cs4-(N)2 and µ = 0.33 µB for Cs4-(N)2 and µ = 0.27 µB for “K4-(N)2” at 2 K, indicative of the presence of U(V) cations, and a χ versus T behaviour in agreement with the presence of two magnetically independent ions (Fig. S66†), similarly to what was found for “K4-(N)2”.
1H NMR studies showed that the in situ reduction of the diuranium(IV) complex 1 with 4 equiv. of CsC8 in toluene at −40 °C carried out under N2 resulted in the formation of the bis-nitride complex Cs4-(N)2 as the major species after 24 hours (Fig. S53†) via the Cs2-2 intermediate (Fig. S54†). Previous studies had shown that reduction of 1 with 4 equiv. of KC8 in toluene at −40 °C resulted in the formation of multiple products due to different redox reactions occurring at the same time.4c
The reactivity of the nitrides in Cs4-(N)2 with electrophiles (H+ and CO) was then probed.
The addition of excess HCl(Et2O) to isolated Cs4-(N)2 yielded NH4Cl in 100% yield (Fig. S30†). The addition of excess acid to the reaction mixture between Cs2-2 and 2 equiv. of CsC8 under N2 after removing the graphite yielded NH4Cl in 83% yield (1.66 equiv., 100% conversion corresponding to 2 equiv. of NH4Cl).
These results show that the bis-nitride formed from N2 reaction reacts readily with protons to give ammonia.
The addition of excess 13CO (5 equiv.) to Cs4-(N)2 resulted in the formation of the diuranium(IV) bis-oxo complex [Cs2{[UIV(OSi(OtBu)3)3]2(µ-O)2}], Cs2-4 (Scheme 4) in 91% yield as shown by 1H NMR studies (Fig. S48†) and single crystal X-ray diffraction (Fig. S60†). Moreover, the 13C NMR spectrum of the reaction mixture quenched in D2O shows the presence of Cs13CN and CsN13CO in 1:1 ratio with overall yield of 100%.
 | ||
| Scheme 4 Reactivity of Cs4-(N)2. | ||
The formation of the bis-oxo and Cs13CN indicates that one bridging nitride promotes the cleavage and deoxygenation of carbon monoxide to afford a N–C triple bond and a bridging oxo group. A second molecule of CO effects the reductive carbonylation of the second nitride to yield a diuranium(IV) bis-oxo complex, Cs2-4 and isocyanate. Rare examples of nitride functionalization by CO23 including CO cleavage by uranium nitrides have been reported previously.24 Notably similar reactivity with CO was observed for the putative diuranium(V) nitride analogue “K4-(N)2”4c and for the previously reported diuranium(V) bis-nitride prepared by reaction of U(III) with alkali azides.13a
Computational studies
To get some insights on the effect of the different alkali atoms, DFT calculations (B3PW91) were carried out. Complexes K2-2, Rb2-2 and Cs2-2 were first optimized. Interestingly, for Cs2-2, it has been possible to obtain both a ferromagnetic and antiferromagnetic (AF) coupling, that were found to be at the same energy (0.2 kcal mol−1 difference with AF slightly lower in energy), whereas only a ferromagnetic coupling was obtained for the other complexes. The bonding was analysed in these three complexes and a UO double bond is found (see ESI† for details). The UO double bonds are found in the three complexes to be strongly polarized toward O (93% in K2-2, 90% in Rb2-2 and 83% in Cs2-2). Thus, the UO bonds are slightly more covalent in the latter complex in line with the linear structure of the oxo. Then, the binding of N2 was investigated in the three cases and three stable structures K2-N2, Rb2-N2 and Cs2-N2 were obtained. The binding energy of N2 was thus computed and is found to decrease (−19.4 kcal mol−1 for K, −5.1 kcal mol−1 for Rb and finally −0.1 kcal mol−1 for Cs) for increasing values of the cation atomic number Z and decreased charge density on the alkali ion. These values are in line with the experiments since the binding is irreversible for K, reversible for Rb in ambient conditions and binding occurs for Cs only at high pressure and low temperature.
This difference of N2 binding is attributed to steric effects since no clear electronic differences in the three systems K2-2, Rb2-2 and Cs2-2 are found (e.g. the U charge is 1.62 and the K one is 0.96 K2-2, 1.65 for U and 0.96 for Rb in Rb2-2 and 1.59 for U and 1.02 for Cs in Cs2-2, see ESI† for a complete report of the density analysis).
It is interesting to note that the charge at U is the lowest in Cs2-2 where the charge of the alkali atom is the largest, indicating that U is more basic. This is in line with the electrochemistry where U is slightly more reducing in Cs2-2 than in K2-2. Indeed, the N2 binding in Cs2-N2 would clearly induce a steric clash in the complex while it is pronounced in the two other cases. This is highlighted in Fig. 5 where a “big atom” representation of the optimized structures of K2-2, Rb2-2 and Cs2-2 (top) as well as K2-N2, Rb2-N2 and Cs2-N2 (bottom) is provided (in this representation each atom is drawn according to its atomic radius). In all cases, the bound dinitrogen molecule is found to be N24− in line with the presence of two U(V) (see unpaired electron density in ESI†).
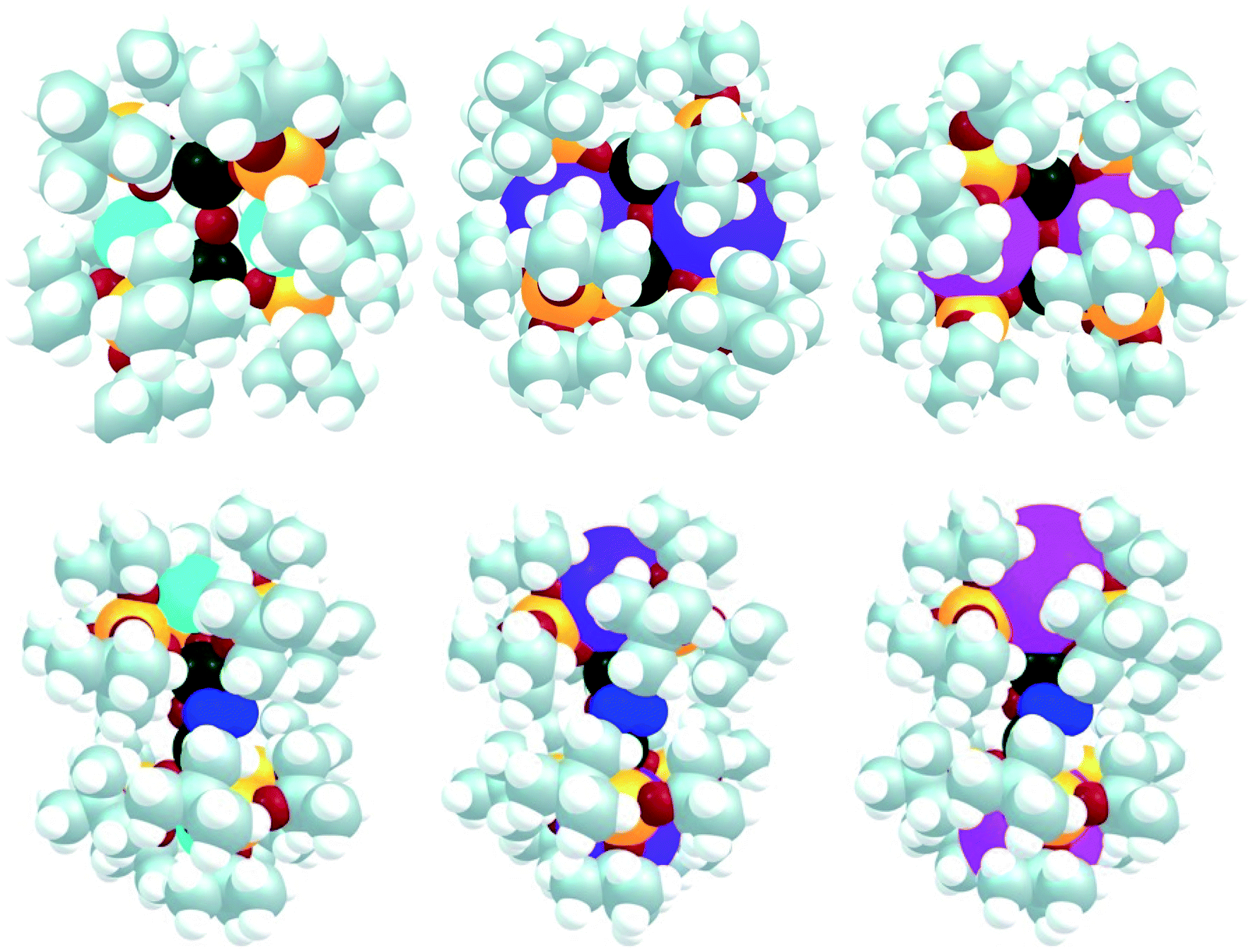 | ||
| Fig. 5 Big atom representation of K2-2, Rb2-2 and Cs2-2 (top) as well as K2-N2, Rb2-N2 and Cs2-N2 (bottom). | ||
Finally, the full reduction of N2, that is the formation of complexes M4-(N)2 with M = K, Rb, Cs, was investigated computationally. A stable structure was optimized in the three cases and the enthalpy of formation of M4-(N)2 from M2-2 is ranging from −83.1 kcal mol−1 (M = Rb) to −91.9 kcal mol−1 (M = K). This is in line with the experiment and the slow conversion (24 hours) for Cs2-2 is due to the very low binding affinity of N2 (Fig. 6).
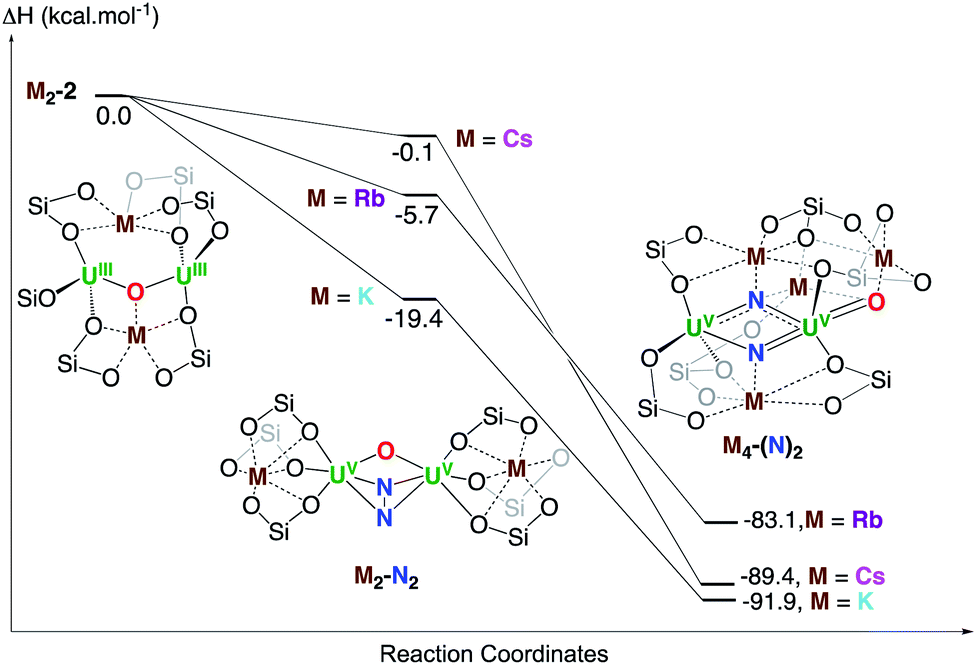 | ||
| Fig. 6 Computed enthalpy profile in kcal mol−1 at room temperature for the formation of K4-(N)2, Rb4-(N)2 and Cs4-(N)2 from the reduction of N2 by K2-N2, Rb2-N2 and Cs2-N2 respectively. | ||
The bonding in Cs4-(N)2 (Fig. S7†) was analysed using the Natural Bonding Analysis (NBO). The presence of an almost linear N–U–O moiety (172°) is of particular interest in Cs4-(N)2. The bonding in this fragment is very similar to what found for uranyl ion with a set a U–O triple bond and a set of U–N triple bond. The three U–O bonds (1σ + 2π) are strongly polarized toward O(81%) and involve an overlap between df orbitals on U (47% 6d + 53% 5f)and sp on O. A relatively similar bonding mode is observed for the U–N triple bond. However, the bond is slightly less polarized toward N (71% vs. 81% for O). These bonds also imply overlap between df hybrid orbital on U (roughly 50–50) and sp orbital on N. The fact that these bonds are strongly polarized explains the relatively low WBI found (0.31 for UN and 0.14 for UO). The second nitride is found to mainly bind to the second uranium with also a triple bond.
Conclusions
Here we compared the structural, redox and magnetic properties of a unique series of structurally analogous multimetallic complexes of low valent uranium where an oxide bridge connects two U(III) centers and two alkali ions of different nature [M2{UIII(OSi(OtBu)3)3}2(µ-O)] (M = K, K2-2; Rb, Rb2-2; M = Cs, Cs2-2). Overall, the binding of the cations K, Rb and Cs in the three complexes is quite similar, with the main differences being the coordination of two Cs+ to the bridging oxide compared to only one K+ or Rb+ and the resulting symmetric and linear U–O–U arrangement in Cs2-2 compared to an asymmetric and bent U–O–U in the K and Rb analogues. These differences result in different magnetic properties with a weak antiferromagnetic coupling observed between the U(III) ions in Cs2-2 while the two U(III) behave as independent paramagnets in K2-2 and Rb2-2. Cyclic voltammetry measurements show an increasing reducing power for the U(III) ions with the increasing ionic radii of the alkali ion and subsequent reduced charge density (K < Rb < Cs). The three complexes show a very different binding ability towards dinitrogen which decreases with the increasing size of the alkali ion and is therefore not correlated to the difference in redox potential. Notably the K2-2 complex binds irreversibly N2 in ambient conditions (1 atm and 25 °C) while N2 binding is reversible for Rb2-2 in the same conditions and only occurs at high pressures (100 atm) and low temperatures (−40 °C) for Cs2-2. DFT analysis indicated that N2 binding by Cs2-2 is hindered by steric effects, but the structures computed for the M2-N2 species show a similar degree of activation of bound N2 for all cations. Remarkably, although N2 binding could not be detected in ambient conditions, reduction of Cs2-2 under N2 led to the first example of cesium assisted N2 cleavage by a metallic complex. The molecular structure of the N2 cleavage product Cs4-(N)2 presents two Cs-bound nitrides binding two U(V) in a diamond-shaped arrangement while the bridging oxide has switched to a terminal binding mode. Furthermore, the two nitrides are readily and quantitatively functionalized by protons and CO. This study provided a relevant molecular model for N2 binding in metal oxides and suggests that structural effects dominate the effect of alkali ions in N2 binding. We also showed that strong binding is not required for further reduction of N2 to nitride which can also be effected by cesium cations.Data availability
All data were given in ESI and CCDC.†Author contributions
N. J. carried out all the synthetic experiments and analyzed the experimental data. R. S. carried out the X-ray single crystal structure analyses. I. Z. collected the variable-temperature magnetic data. A. S. recorded and analyzed the EPR data. T. R. and L.M. carried out the computational studies. M. M. originated the central idea, coordinated the work, and analyzed the experimental data. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 178793 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Farzaneh Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses. We thank Dr David Savary in the group of Prof. Paul Dyson for its help with the high-pressure experiments. We thank Roxane Moinat for carrying out the elemental analyses. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.References
- (a) I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed; (b) S. C. Bart and K. Meyer, in Structure and Bonding, 2008, vol. 127, pp. 119–176 Search PubMed; (c) M. S. Eisen, Top. Organomet. Chem., 2010, 31, 157–184 CrossRef CAS; (d) O. Cooper, C. Camp, J. Pecaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed; (e) I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed; (f) H. S. La Pierre and K. Meyer, in Progress in Inorganic Chemistry, ed. K. D. Karlin, 2014, vol. 58, pp. 303–415 Search PubMed; (g) D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317–321 CrossRef CAS PubMed; (h) M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS; (i) P. L. Arnold and Z. R. Turner, Nat. Rev. Chem., 2017, 1, 0002 CrossRef CAS; (j) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed; (k) H. Liu, T. Ghatak and M. S. Eisen, Chem. Commun., 2017, 53, 11278–11297 RSC; (l) L. Barluzzi, M. Falcone and M. Mazzanti, Chem. Commun., 2019, 55, 13031–13047 RSC; (m) P. L. Arnold, T. Ochiai, F. Y. T. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS PubMed; (n) M. A. Boreen and J. Arnold, J. Chem. Soc., Dalton Trans., 2020, 49, 15124–15138 RSC; (o) D. R. Hartline and K. Meyer, JACS Au, 2021, 1, 698–709 CrossRef CAS PubMed.
- (a) F. Haber, Ammonia, German Pat., DE229126, 1909 Search PubMed; (b) F. Haber, Angew. Chem., 1914, 27, 473–477 CrossRef CAS.
- (a) A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836–5837 CrossRef CAS; (b) P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS; (c) G. F. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352–9353 CrossRef PubMed; (d) W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264–14265 CrossRef CAS PubMed; (e) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed; (f) S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS; (g) M. D. Walter, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS; (h) M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed; (i) E. Lu, B. E. Atkinson, A. J. Wooles, J. T. Boronski, L. R. Doyle, F. Tuna, J. D. Cryer, P. J. Cobb, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, N. Kaltsoyannis and S. T. Liddle, Nat. Chem., 2019, 11, 806–811 CrossRef CAS PubMed; (j) D. Singh, W. R. Buratto, J. F. Torres and L. J. Murray, Chem. Rev., 2020, 120, 5517–5581 CrossRef CAS PubMed.
- (a) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed. Engl., 2002, 41, 3433–3436 CrossRef CAS; (b) X. Q. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Q. Zhu, J. Am. Chem. Soc., 2020, 142, 15004–15011 CrossRef CAS PubMed; (c) N. Jori, L. Barluzzi, I. Douair, L. Maron, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2021, 143, 11225–11234 CrossRef CAS PubMed.
- (a) C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef CAS PubMed; (b) S. Gambarotta and J. Scott, Angew. Chem., Int. Ed. Engl., 2004, 43, 5298–5308 CrossRef CAS PubMed; (c) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780–783 CrossRef CAS PubMed; (d) T. Shima, S. W. Hu, G. Luo, X. H. Kang, Y. Luo and Z. M. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed; (e) K. P. Chiang, S. M. Bellows, W. W. Brennessel and P. L. Holland, Chem. Sci., 2014, 5, 267–274 RSC; (f) K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807–16816 CrossRef CAS PubMed; (g) G. P. Connor and P. L. Holland, Catal. Today, 2017, 286, 21–40 CrossRef CAS PubMed; (h) L. R. Doyle, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2019, 58, 6674–6677 CrossRef CAS PubMed; (i) M. J. Dorantes, J. T. Moore, E. Bill, B. Mienert and C. C. Lu, Chem. Commun., 2020, 56, 11030–11033 RSC; (j) S. J. K. Forrest, B. Schluschass, E. Y. Yuzik-Klimova and S. Schneider, Chem. Rev., 2021, 121, 6522–6587 CrossRef CAS PubMed; (k) F. Masero, M. A. Perrin, S. Dey and V. Mougel, Chem.–Eur. J., 2021, 27, 3892–3928 CrossRef CAS PubMed.
- (a) R. Krabetz and C. Peters, Angew. Chem., Int. Ed. Engl., 1965, 4, 341–347 CrossRef; (b) H. P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- (a) P. L. Holland, J. Chem. Soc., Dalton Trans., 2010, 39, 5415–5425 RSC; (b) J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS PubMed.
- W. J. Evans, M. Fang, G. Zucchi, F. Furche, J. W. Ziller, R. M. Hoekstra and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11195–11202 CrossRef CAS PubMed.
- M. Fang, D. S. Lee, J. W. Ziller, R. J. Doedens, J. E. Bates, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 3784–3787 CrossRef CAS PubMed.
- (a) R. Ferguson, E. Solari, C. Floriani, D. Osella, M. Ravera, N. Re, N. ChiesiVilla and C. Rizzoli, J. Am. Chem. Soc., 1997, 119, 10104–10115 CrossRef CAS; (b) G. K. B. Clentsmith, V. M. E. Bates, P. B. Hitchcock and F. G. N. Cloke, J. Am. Chem. Soc., 1999, 121, 10444–10445 CrossRef CAS; (c) A. Caselli, E. Solari, R. Scopelliti, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, J. Am. Chem. Soc., 2000, 122, 3652–3670 CrossRef CAS; (d) H. Kawaguchi and T. Matsuo, Angew. Chem., Int. Ed. Engl., 2002, 41, 2792–2794 CrossRef CAS; (e) G. Ung and J. C. Peters, Angew. Chem., Int. Ed. Engl., 2015, 54, 532–535 CAS; (f) T. D. Lohrey, R. G. Bergman and J. Arnold, J. Chem. Soc., Dalton Trans., 2019, 48, 17936–17944 RSC; (g) S. Suzuki, Y. Ishida, H. Kameo, S. Sakaki and H. Kawaguchi, Angew. Chem., Int. Ed. Engl., 2020, 59, 13444–13450 CrossRef CAS PubMed.
- (a) S. F. McWilliams and P. L. Holland, Acc. Chem. Res., 2015, 48, 2059–2065 CrossRef CAS PubMed; (b) K. C. MacLeod, F. S. Menges, S. F. McWilliams, S. M. Craig, B. Mercado, M. A. Johnson and P. L. Holland, J. Am. Chem. Soc., 2016, 138, 11185–11191 CrossRef CAS PubMed.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed; (b) G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2008, 47, 3040–3042 CrossRef CAS PubMed; (c) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed; (d) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed; (e) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed; (f) T. K. Todorova, L. Gagliardi, J. R. Walensky, K. A. Miller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 12397–12403 CrossRef CAS PubMed; (g) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (h) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (i) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed; (j) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266, 2–15 CrossRef; (k) L. Maria, I. C. Santos, V. R. Sousa and J. Marcalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed; (l) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed; (m) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed; (n) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC; (o) K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M. H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed; (p) L. Barluzzi, N. Jori, T. Y. He, T. Rajeshkumar, R. Scopelliti, L. Maron, P. Oyala, T. Agapie and M. Mazzanti, Chem. Commun., 2022, 58, 4655–4658 RSC.
- (a) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (b) J. Z. Du, D. M. King, L. Chatelain, E. L. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC; (c) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC; (d) L. Barluzzi, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 19047–19051 CrossRef CAS PubMed; (e) M. A. Boreen, G. D. Rao, D. G. Villarreal, F. A. Watt, R. D. Britt, S. Hohloch and J. Arnold, Chem. Commun., 2020, 56, 4535–4538 RSC; (f) L. Chatelain, E. Louyriac, I. Douair, E. L. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS PubMed; (g) C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS PubMed; (h) M. Yadav, A. J. Metta-Magana and S. Fortier, Chem. Sci., 2020, 11, 2381–2387 RSC; (i) L. Barluzzi, F. C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096–8104 RSC; (j) J. Z. Du, J. A. Seed, V. E. J. Berryman, N. Kaltsoyannis, R. W. Adams, D. Lee and S. T. Liddle, Nat. Commun., 2021, 12, 5649 CrossRef CAS PubMed.
- (a) W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS; (b) D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2021, 60, 3737–3744 CrossRef CAS PubMed.
- (a) B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens, Jr., L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef; (b) K. R. Meihaus, S. G. Minasian, W. W. Lukens, S. A. Kozimor, D. K. Shuh, T. Tyliszczak and J. R. Long, J. Am. Chem. Soc., 2014, 136, 6056–6068 CrossRef CAS PubMed; (c) L. C. J. Pereira, C. Camp, J. T. Coutinho, L. Chatelain, P. Maldivi, M. Almeida and M. Mazzanti, Inorg. Chem., 2014, 53, 11809–11811 CrossRef CAS PubMed.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- (a) M. D. Fryzuk, T. S. Haddad and S. J. Rettig, J. Am. Chem. Soc., 1990, 112, 8185–8186 CrossRef CAS; (b) J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed; (c) Y. Ohki and M. D. Fryzuk, Angew. Chem., Int. Ed. Engl., 2007, 46, 3180–3183 CrossRef CAS PubMed.
- (a) R. D. Sanner, D. M. Duggan, T. C. McKenzie, R. E. Marsh and J. E. Bercaw, J. Am. Chem. Soc., 1976, 98, 8358–8365 CrossRef CAS; (b) W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef CAS; (c) T. E. Hanna, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 14688–14689 CrossRef CAS PubMed.
- J. C. Wedal, J. M. Barlow, J. W. Ziller, J. Y. Yang and W. J. Evans, Chem. Sci., 2021, 12, 8501–8511 RSC.
- T. Suzuki, K. Fujimoto, Y. Takemoto, Y. Wasada-Tsutsui, T. Ozawa, T. Inomata, M. D. Fryzuk and H. Masuda, ACS Catal., 2018, 8, 3011–3015 CrossRef CAS.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- (a) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 446–447 CrossRef CAS PubMed; (b) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed; (c) P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2014, 53, 10412–10415 CrossRef CAS PubMed; (d) J. A. Buss, C. Cheng and T. Agapie, Angew. Chem., Int. Ed. Engl., 2018, 57, 9670–9674 CrossRef CAS PubMed.
- M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2016, 55, 12290–12294 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2154518–2154525. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02530b |
| This journal is © The Royal Society of Chemistry 2022 |