
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zintl cluster supported low coordinate Rh(I) centers for catalytic H/D exchange between H2 and D2†
Oliver P. E.
Townrow
 a,
Simon B.
Duckett
b,
Andrew S.
Weller
*b and
Jose M.
Goicoechea
*a
a,
Simon B.
Duckett
b,
Andrew S.
Weller
*b and
Jose M.
Goicoechea
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: jose.goicoechea@chem.ox.ac.uk
bDepartment of Chemistry, University of York, YO10 5DD, UK. E-mail: andrew.weller@york.ac.uk
First published on 9th June 2022
Abstract
Ligand exchange reactions of [Rh(COD){η4-Ge9(Hyp)3}] with L-type nucleophiles such as PMe3, PPh3, IMe4 (IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene) or [W(Cp)2H2] result in the displacement of the COD ligand to afford clusters with coordinatively unsaturated trigonal pyramidal rhodium(I) centers [Rh(L){η3-Ge9(Hyp)3}]. These species can be readily protonated allowing access to cationic rhodium–hydride complexes, e.g. [RhH(PPh3){η3-Ge9(Hyp)3}]+. These clusters act as catalysts in H/D exchange between H2 and D2 and alkene isomerisation, thereby illustrating that metal-functionalized Zintl clusters are active in both H–H and C–H bond activation processes. The mechanism of H/D exchange was probed using parahydrogen induced polarization experiments.
Introduction
Transition-metal/main-group element alloys (TMMGAs) are an interesting class of materials that combine late transition metals (e.g. nickel, platinum or palladium) with low melting point post-transition metals such as gallium, indium, tin, lead or bismuth. In their molten state, these alloys have been employed as catalysts in a number of challenging processes that involve light alkane valorization, e.g. methane pyrolysis to form graphitic carbon and hydrogen, dry reforming of methane, or propane dehydrogenation.1–7 These melts offer access to “solvated” late transition metal atoms accounting for their impressive catalytic performance, however in situ monitoring of reaction profiles/mechanisms remains a challenge due to the harsh conditions employed (e.g. operating temperatures of 1040 °C) and the lack of suitable spectroscopic probes. This led us to turn our attention to the synthesis of molecular TMMGAs, compounds in which a late transition-metal–ligand fragment is supported on a main-group cluster platform. For this purpose, we have explored the use of Zintl clusters, anionic clusters of the main-group elements, that can be readily functionalized with transition-metals. In principle, these compounds can act as molecular models for TMMGAs. However, poor solubility in non-polar solvents, low yielding syntheses, and a tendency for oxidative coupling have long hindered their application in homogeneous catalysis.The silylated Zintl ion [Ge9(Hyp)3]− (Hyp = Si(SiMe3)3) is an attractive platform for the design of molecular TMMGAs due to its increased solubility in hydrocarbon solvents.8,9 Recently we demonstrated that the rhodium(I) cluster [Rh(COD){η4-Ge9(Hyp)3}] can be employed as a catalyst for the hydrogenation of cyclic alkenes.10 This is the first example of a Zintl cluster being used in catalysis, and a proof-of-concept study illustrating that this class of compound may be used to mimic the impressive reactivity of TMMGAs. Furthermore, the [Ge9(Hyp)3]− cage can readily isomerize to adapt to the steric and electronic requirements of the transition metal.10 These molecular dynamics have the potential to play a pivotal role in catalysis, where geometric responses to substrate and product binding can provide lower energy pathways in elementary catalytic steps. Herein we demonstrate that, in addition to being active hydrogenation catalysts, metal-functionalized Zintl clusters are also active catalysts for H/D exchange between H2 and D2, and alkene isomerization. While both of these catalytic bond transformations are common in organometallic chemistry they are unknown in Zintl ion chemistry.11
Results and discussion
We have previously shown that reaction of [Rh(COD){η4-Ge9(Hyp)3}] with the bidentate, κ2, ligand dppe (1,2-bis(diphenylphosphino)ethane) results in both the displacement of the COD ligand and cluster isomerization, so that the rhodium centre moves from occupying at a 4-connected vertex position in the cluster to a 5-connected vertex position. The rhodium(I) center is formally 18-electron in both.10 This prompted us to explore whether the COD ligand could be displaced by monodentate L-type ligands to afford a coordinatively unsaturated rhodium(I) metal center that has a formal 16-valence electron count. Consequently, the addition of one equivalent of PMe3 to [Rh(COD){η4-Ge9(Hyp)3}] led to the reaction shown in Scheme 1. The two hypersilyl environments initially present in the 1H NMR spectrum of [Rh(COD){η4-Ge9(Hyp)3}] disappeared giving rise to a single resonance (0.50 ppm). In the 31P{1H} NMR spectrum a doublet resonance was observed at −13.5 ppm, (1JP–Rh = 210.3 Hz). This new species, [Rh(PMe3){η3-Ge9(Hyp)3}] (1a), and free COD are the only two products observable by 1H NMR spectroscopy. The single hypersilyl resonance observed at room temperature points to a fluxional process that makes these groups equivalent on the NMR timescale, as also noted for [Rh(dppe){η5-Ge9(Hyp)3}].10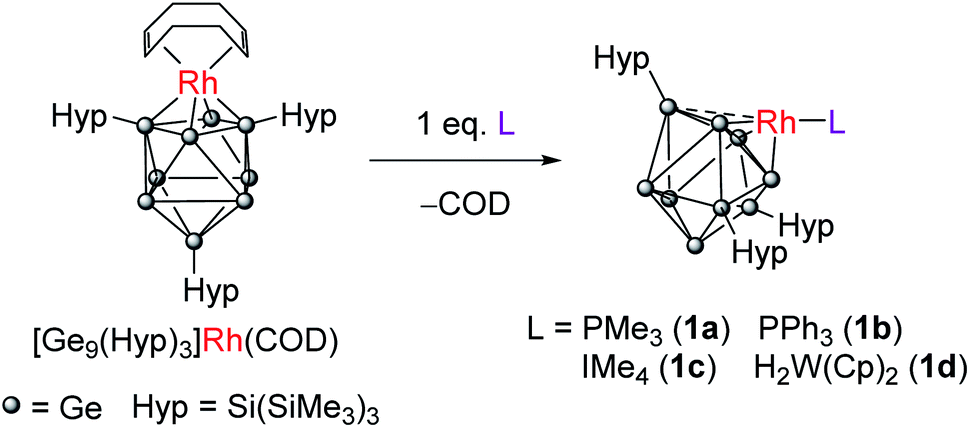 | ||
| Scheme 1 Synthesis of 1a–1d from reaction of [Rh(COD){η4-Ge9(Hyp)3}] with Lewis bases. | ||
The related clusters [Rh(PPh3){η3-Ge9(Hyp)3}] (1b), [Rh(IMe4){η3-Ge9(Hyp)3}] (1c), and [Rh{η3-Ge9(Hyp)3}(μ-H)2W(Cp)2] (1d) were synthesized in a similar manner, from the reaction of [Rh(COD){η4-Ge9(Hyp)3}] with one equivalent of the L-type ligands PPh3, IMe4 (IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene) or [W(Cp)2H2],12–15 respectively. Clusters 1b–1d exhibit similar NMR spectra to 1a (see ESI† for full details), and are thus also fluxional at room temperature. In the case of 1d, the 1H NMR spectrum exhibits a doublet resonance (integrating to two protons) at −16.15 ppm (1JRh–H = 26.2 Hz) that exhibits satellites due to 183W coupling (1JW–H is 76.0 Hz), signaling Rh–H–W bridging hydrides. These data are comparable to those found for two known [Rh](μ-H)2W(Cp)2 complexes reported in the literature, only one of which has been structurally authenticated, i.e. [Rh(PPh3)2(μ-H)2W(Cp)2][PF6].16–18
Crystallization of 1a, 1c and 1d from saturated n-hexane or n-pentane solutions allowed for structural characterization of the new clusters by single-crystal X-ray diffraction. All three structures exhibit coordinatively unsaturated rhodium(I) centers, bound to the [Ge9(Hyp)3]− cluster in an η3 mode (Fig. 1), in which the ligand (e.g. PMe3) sits trans to a silylated vertex. This gives the cluster overall, non-crystallographic, Cs symmetry. The τ4 values for 1a, 1c and 1d are 0.79, 0.83 and 0.85, respectively,‡ in line with the value for an ideal C3v coordination geometry (0.85).19 Despite there being an apparent vacant coordination site, no structural or spectroscopic evidence was observed for an interaction between the most proximal hypersilyl substituent with the metal center (e.g. the closest Rh⋯C interatomic distances are greater than 3.65 Å).20 The structures of 1a and 1d each feature a single crystallographically unique cluster in the asymmetric unit which exhibits positional disorder. This disorder is best accounted for by two different orientations of the {RhL} fragment that are related by rotation with respect to the (static) [Ge9(Hyp)3]− cluster. By contrast, the structure of 1c reveals two crystallographically unique clusters in the asymmetric unit with near identical bond metrics. The rhodium centers in all the complexes bind to the cluster through three short Rh–Ge bonds (1a: 2.394(1)–2.433(1) Å; 1c: 2.397(1)–2.418(1) Å; 1d: 2.409(2)–2.429(1) Å), and a slightly longer Rh⋯Ge contact with the nearest silylated germanium atom (1a: 2.517(7) Å; 1c: 2.667(1) Å; 1d: 2.644(2) Å). The Rh–L distances for 1a and 1c are similar to those reported in related rhodium−phosphine,21 and rhodium–carbene complexes.22 In the case of 1d, the Rh⋯W distance of 2.852(1) Å is notably longer than that in [Rh(PPh3)2(μ-H)2W(Cp)2][PF6] (2.721(3) Å),16 which we hypothesize is a consequence of the steric demands of the hypersilyl substituents of the [Ge9(Hyp3)]− cage. From an electron-counting perspective, 1a–1d can be viewed as hypercloso-like, and therefore similar to [Rh(CO)3(B9H9)]+.23 The effect of displacing a four-electron donor ligand (COD) by a two electron donor (e.g. PMe3) is to reduce the overall cluster electron count, and this is consequently accompanied by a structural distortion. The reverse effect has previously been seen when hypercloso-[Ru(PEt3)2(C2B7H9)] converts to closo-[Ru(PEt3)3(C2B7H9)] on addition of PEt3.24,25 Significant rhodium d-orbital participation in cluster bonding can be observed for 1b on inspection of frontier orbitals (see ESI†).
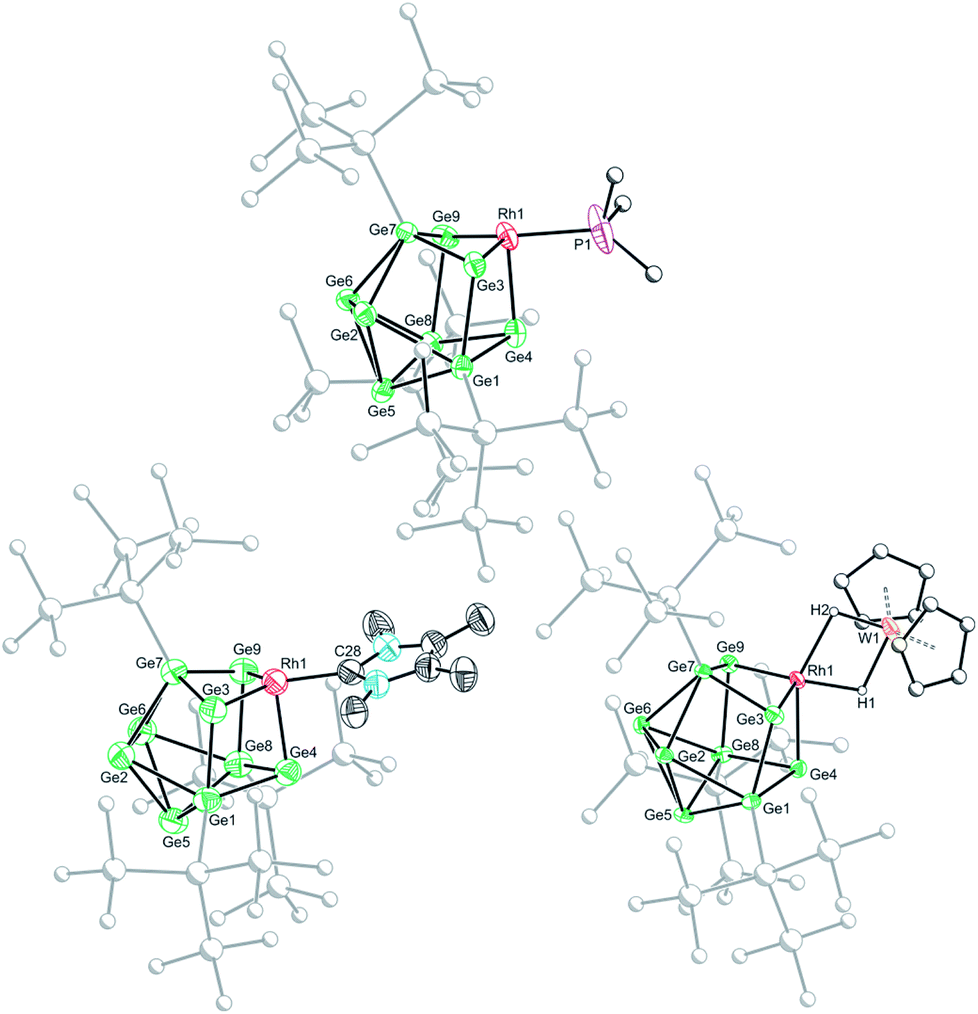 | ||
| Fig. 1 Molecular structures of 1a (top), 1c (bottom left) and 1d (bottom right). Anisotropic displacement ellipsoids are set at 50% probability. Hydrogen atoms have been omitted for clarity. Only the major disordered component is shown. | ||
Based on these crystallographically-determined structures, one would expect two chemically inequivalent hypersilyl substituents to be observed in the solution phase 1H NMR spectra of 1a–1d. The fact that only one 1H resonance is observed suggests that, in solution, a fluxional process is operating that makes the hypersilyl groups equivalent. This process was modelled using density functional theory (DFT) calculations for 1b, and shown to proceed via a Cs to C3v transition (Fig. 2) whereby the rhodium center adopts a tetrahedral coordination geometry and coordinates to a triangular face of the [Ge9(Hyp)3]− cage. This process involves cleavage of the Rh1⋯Ge7 contact (as per the numbering scheme in Fig. 1) and the contraction of the distance between Ge3 and Ge9 by ca. 0.7 Å. Consistent with the observed fluxionality, that is not frozen out at −80 °C, the optimized geometries for the Cs and C3v symmetric clusters were found to be within 2.4 kcal mol−1 of one another, with a computed transition state barrier of 4.4 kcal mol−1. The positional disorder observed in the single-crystal X-ray diffraction structures of 1a and 1d is consistent with the superposition of two of these Cs symmetric isomers in the lattice.
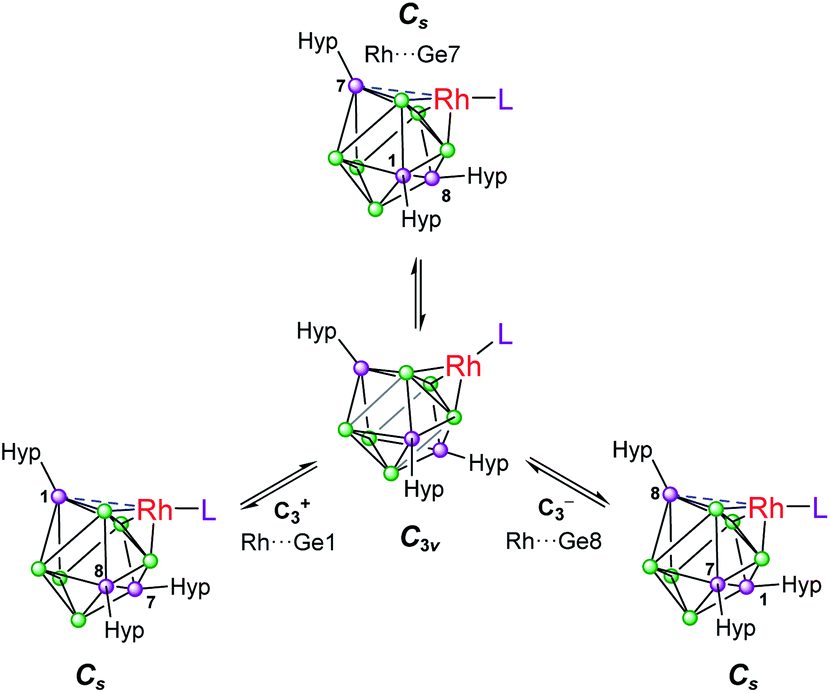 | ||
| Fig. 2 Fluxional behavior of 1b. Numbering of silylated vertices as per Fig. 1. | ||
Trigonal pyramidal rhodium(I) compounds are rare, and, in some cases, have been shown to be weakly electrophilic.26–28 For example, Grützmacher has shown that [Rh(trop2SiMe)(C2H4)] (trop2SiMe = bis(5H-dibenzo[a,d]cyclohepten-5-yl)methyl-silane) will reversibly bind THF to afford a trigonal bipyramidal compound. However, this reactivity is finely balanced, as other nucleophiles, such as PPh3, will instead displace the coordinated alkene in the equatorial site rather than bind in the axial position.26 Reaction of 1a–1d with alkenes indicate that these clusters are not particularly strong electrophiles, as no adduct is observed to be formed to the detection limit of 1H NMR spectroscopy. Indeed, inspection of the computed frontier orbitals reveals an energetically accessible HOMO (with a significant rhodium d orbital contribution) suggesting that these complexes are more nucleophilic in character. Thus their reactivity is predicted to mimic that of more traditional organometallic nucleophiles such as Cp*Rh(PMe3)2.29
We probed this chemically by reaction of 1b and 1d with Brookhart's acid [H(OEt2)2][BArF4] (ArF = 3,5-(CF3)2C6H3).30 Reaction of 1b with 1 equivalent of this acid converts the rhodium(I) center to its conjugate rhodium(III) hydride, [RhH(PPh3){η3-Ge9(Hyp)3}][BArF4] (2b[BArF4]; Scheme 2). The 1H NMR spectrum now shows two new broad singlet resonances at 0.07 and 0.33 ppm (in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio) corresponding to its hypersilyl substituents on a now static Zintl cage,§ as well as a new doublet of doublets, integrating to one proton, at −1.48 ppm (1JH–Rh = 12.6 Hz, 2JH–P = 3.4 Hz) assigned to Rh–H. 1H{31P} NMR spectroscopy measurements allowed for the unequivocal assignment of the coupling constants (see ESI†). Similarly, 1d can also be protonated to afford the rhodium(III) trihydride, 2d[BArF4]. On inspection of the 1H NMR spectrum of 2d[BArF4], the hypersilyl groups present as two inequivalent peaks, and there is a new doublet resonance (−0.03 ppm; 1JRh–H = 12.2 Hz) indicating a terminal hydride. The Rh–H–W bridging hydride resonance also shifts from −16.15 ppm (1JRh–H = 26.2 Hz, 1JW–H = 76.0 Hz) to −15.41 ppm (1JRh–H = 27.8 Hz, 1JW–H = 83.3 Hz).
:2 ratio) corresponding to its hypersilyl substituents on a now static Zintl cage,§ as well as a new doublet of doublets, integrating to one proton, at −1.48 ppm (1JH–Rh = 12.6 Hz, 2JH–P = 3.4 Hz) assigned to Rh–H. 1H{31P} NMR spectroscopy measurements allowed for the unequivocal assignment of the coupling constants (see ESI†). Similarly, 1d can also be protonated to afford the rhodium(III) trihydride, 2d[BArF4]. On inspection of the 1H NMR spectrum of 2d[BArF4], the hypersilyl groups present as two inequivalent peaks, and there is a new doublet resonance (−0.03 ppm; 1JRh–H = 12.2 Hz) indicating a terminal hydride. The Rh–H–W bridging hydride resonance also shifts from −16.15 ppm (1JRh–H = 26.2 Hz, 1JW–H = 76.0 Hz) to −15.41 ppm (1JRh–H = 27.8 Hz, 1JW–H = 83.3 Hz).
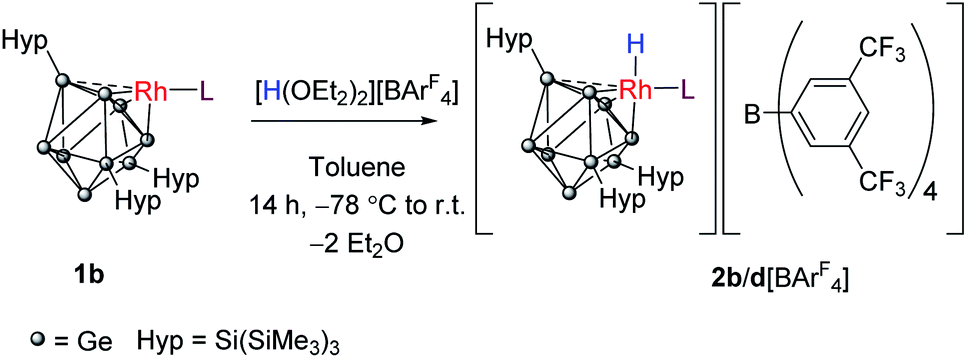 | ||
| Scheme 2 Synthesis of 2b[BArF4] and 2d[BArF4]. | ||
Crystals of 2b[BArF4]·1.5hex (Fig. 3) and 2d[BArF4]·Et2O suitable for single crystal X-ray diffraction were grown from concentrated n-hexane and diethyl ether solutions, respectively. The cationic cluster 2d exhibits positional disorder (two different cluster orientations related by rotation in a 2:8 ratio), consequently only the bond metric data for 2b[BArF4]·1.5hex will be discussed in detail. The crystal structure reveals a single crystallographically unique cationic cluster in the unit cell, [RhH(PPh3){η3-Ge9(Hyp)3}]+, accompanied by a [BArF4]− counterion and solvent of crystallization. The cluster core of 2b is comparable to those of 1a and 1c, with the notable exception that the rhodium metal center now adopts a trigonal bipyramidal geometry with a (located) hydride in an axial position, fully consistent with protonation of the HOMO in 1b. On protonation, the formal oxidation state of the rhodium center changes from +1 to +3. This is consistent with the moderate contraction (0.03 Å) of the Rh–Ge distances, 2.365(1) to 2.397(1) Å that is observed, relative to those observed for 1a and 1c (cf. 2.397(1) to 2.418(1) Å for 1c). As with 1a and 1c, a close Rh⋯Ge contact to the nearest silylated germanium center is seen (2.677(1) Å).
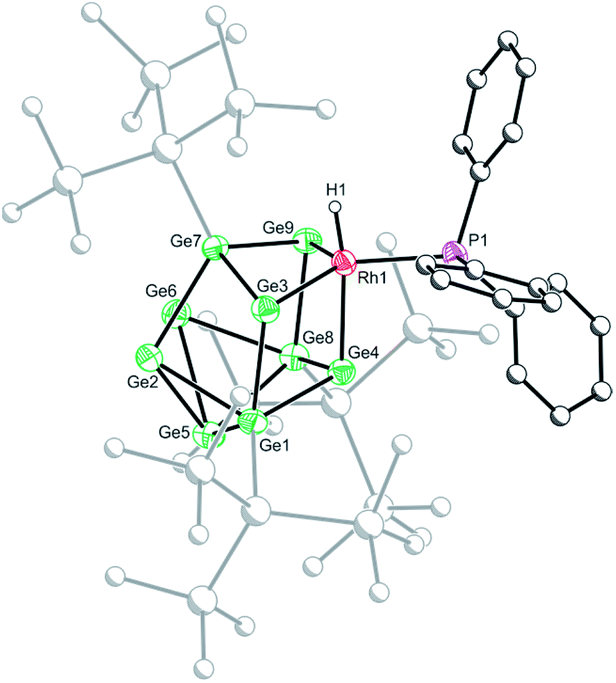 | ||
| Fig. 3 Molecular structure of the cationic moiety in 2b[BArF4]·1.5hex. Anisotropic displacement ellipsoids are set at 50% probability. C–H hydrogen atoms have been omitted for clarity. | ||
Having probed the electronic structure of 1b and 1d through protonation studies, we turned our attention to see if they might be viable catalysts. Given the presence of an apparent vacant coordination site, we chose to explore the reactivity of these compounds towards dihydrogen activation, a ubiquitous process in organometallic chemistry that often occurs at Rh(I) metal centers.11
Placing a C6D6 solution of 1b under an atmosphere of H2 resulted in no change to the 1H NMR spectrum. Similarly, no changes were observed to the NMR spectra of 1b if placed under an atmosphere of D2. While this observation seemed at odds with the presence of an apparent vacant coordination site on the rhodium(I) center on the cluster, motifs that are well known to oxidatively add H2, we show later H2 addition is rapid, reversible, and endergonic. We thus turned our attention the dihydride bridged compound 1d to indirectly probe the possibility of reversible H2 addition to 1b through reaction with D2.31,32
A C6D6 solution of 1d was placed under an atmosphere of D2 (Scheme 3). Almost immediately, the 1H signal for the hydrides (−16.15 ppm) started to decrease in intensity and signals for dissolved H2 and HD (4.43 ppm, 1:1:1 triplet, 1JH–D = 42.8 Hz) were observed. After approximately 3 hours, two overlapping hydride resonances were clearly visible that are assigned to 1d and its isotopologue [Rh{Ge9(Hyp)3}(μ-H)(μ-D)W(Cp)2}]. Complete loss of the bridging 1H signals was observed after 16 hours as [Rh{Ge9(Hyp)3}(μ-D)2W(Cp)2], d2-1d, now predominates. The presence of the bridging Rh–D–W isotopologue was confirmed by 2H NMR spectroscopy, by a resonance observed at −16.06 ppm (1JW–D = 12.0 Hz; 1JRh–D = 4.1 Hz). Replacing the D2 atmosphere with H2 resulted in complete regeneration of 1d and the observation of dissolved HD, over the course of 24 hours. For context, this reactivity does not occur when [W(Cp)2H2] is placed under D2, although [Rh(PPh3)2(μ-H)2W(Cp)2)][PF6] does undergo H/D exchange in D2O/d6-acetone.16
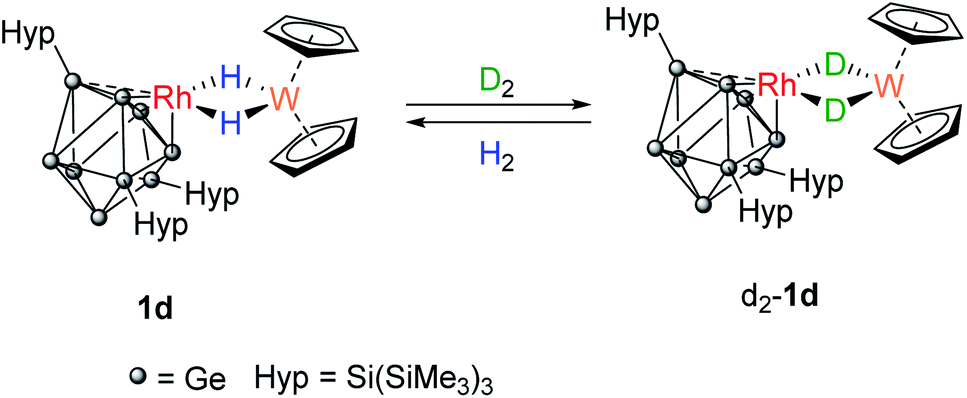 | ||
| Scheme 3 Reversible H2/D2 exchange at 1d proceeds via the Rh(μ-H)(μ-D)W form (not shown). | ||
The reversible nature of H/D exchange at 1d prompted us to re-evaluate the reaction of 1b with H2. We reasoned that perhaps H–H bond activation does indeed take place when the cluster is placed under an H2 atmosphere, but that this process is endergonic, rapid and reversible. In order to probe this, 1b was placed under a mixture of H2 and D2. The immediate formation of HD(dissolved) was observed on time of mixing, indicating fast H2 and D2 activation on the NMR timescale, with 1b acting a catalyst for H/D exchange. This reaction was unaffected by addition of mercury to the reaction mixture, suggesting a homogeneous process.33 To further investigate the H/D exchange process, parahydrogen (p-H2) induced polarization experiments were conducted by reacting 1b and 1d with p-H2. Due to the hyperpolarization effects arising from a bias in spin relaxation pathways, such experiments can be used to observe short-lived and low concentration intermediates in reactions involving p-H2.34–38 While these experiments did not allow us to directly observe any rhodium−hydride intermediates, two important observations were made allowing us to infer that rapid and reversible H–H bond activation takes place on reaction of 1b or 1d with H2. The first of these is that when solutions of 1b or 1d are exposed to NMR silent p-H2 (3 bar) in an airtight NMR tube, instant formation of NMR detectable ortho-dihydrogen (o-H2) is observed. The second important observation is that when a dilute solution of 1b (0.0005 mmol ml−1) was cooled to 263 K in the NMR spectrometer, removed from the magnet and shaken under p-H2 (3 bar), on returning to the NMR spectrometer a signal with Partially Negative Lineshape (PNL) was observed at 4.56 ppm. This enhancement has a lifetime of around 5 s at this temperature, after which, only o-H2 was observed. The observation of a PNL effect at 263 K, whist no hydride species are seen, suggests a very rapid and reversible transfer of p-H2 onto the cluster (most likely the rhodium center) in which the two spins become distinct. This would be consistent with the endergonic formation of either a dihydrogen, Rh(H2), or a dihydride, Rh(H)2, motif.39,40 Both are common intermediates in hydrogenation reactions mediated by organometallic complexes.11 At room temperature p-H2 destruction to form o-H2 proceeds so rapidly, by reversible formation of a dihydride complex, that the PNL effect is quenched.
What remains undetermined at this stage is the mechanism of H/D exchange by 1b. Oxidative addition of H2/D2 at the rhodium(I) center is supported by the studies using p-H2. As this would result in an 18-valence electron rhodium complex, association of a second molecule of H2/D2 would necessitate a change in the hapticity of the [Ge9(Hyp)3]− cage (for example from η3 to η1),41or phosphine dissociation. The resulting dihydride/σ-dihydrogen compound could undergo H/D exchange via a σ-CAM type mechanism.42,43 DFT calculations, however, show that cluster isomerization from η3 to η1 is unfavorable (42.4 kcal mol−1), as is phosphine dissociation (32.8 kcal mol−1). An alternative is a proton-catalyzed mechanism, as previously invoked by Brookhart and co-workers for the oxidative-addition of H2 by the d8 iridium(I) complex [Ir(PONOP)(CH3)] (PONOP = 2,6-bis(di-tert-butylphosphinito) pyridine).44 Here H/D exchange would occur by initial reversible protonation of 1b by adventitious water to form trace amounts of formally 16-electron 2b[OH], which would then undergo H/D exchange. To explore such reactivity, 2b[BArF4] was exposed to a H2/D2 mixture. While these studies were hampered by the low solubility of 2b[BArF4] and its propensity to precipitate out of solution, they showed that C6D6 solutions of 2b[BArF4] generate HD when exposed to a mixture of H2 and D2. Moreover, such H/D scrambling occurs on a similar timescale to that observed when using 1b (i.e. approx. 15 minutes). Thus, it is possible that adventitious moisture in the solvent or gas mixture could give rise to trace amounts of 2b[BArF4] on dissolving 1b and that this species is, in fact, the active catalyst. Arguing against this hypothesis is that addition of proton sponge to 1b H2/D2 mixture did not suppresses H/D exchange, as might be anticipated for a proton-transfer mechanism. However, we cannot discount the formation of trace [OH]− under these conditions that may act to deprotonate intermediate dihydrogen or hydride complex to form 2b.45 While the precise details of the mechanism remain to be resolved, clear is that rapid H/D exchange does occur, a first for a Zintl cluster.
The oxidative addition of H2 to 1b, and the ability to subsequently bind an additional ligand, is further demonstrated by isotope scrambling experiments when 1b is used to catalyze the deuteration of 1-hexene. Addition of 1-hexene to 1b (2 mol%) under a D2 atmosphere (1 bar, 16 hours) resulted in the formation of a mixture of the deuterated alkenes dn-1-hexene (major) and dn-2-hexene (minor), as well as dn-hexane, as measured by 1H and 2H NMR spectroscopy. The dn-1-hexene has deuterium incorporated into both geminal positions (∼75% D total) as well as the vicinal position (∼90% D) of the alkene. HD(dissolved) is also observed as pictured in Scheme 4A. Recharging with D2 results in only dn-hexane being observed after a further 16 hours, in which deuterium has been incorporated into the 1-, 2- and 3-positions. These observations suggest the formation of di-deuteride intermediate (consistent with both H2/D2 exchange and p-H2 experiments), followed by reversible coordination of 1-hexene, and reversible insertion into either of the alkene positions of 1-hexene, followed by a rate-determining reductive elimination of hexane, Scheme 4B. The observation of 2-hexene and d-incorporation into positions 1, 2 and 3 of the final product, hexane, indicates a slower isomerization process also occurs, likely via non-degenerative β-elimination from a 2° alkyl-hydride intermediate. As for H2/D2 exchange we cannot discount that catalysis occurs by a proton-catalyzed mechanism, via an (undetected) analogue of mono-hydride 2b[BArF4].
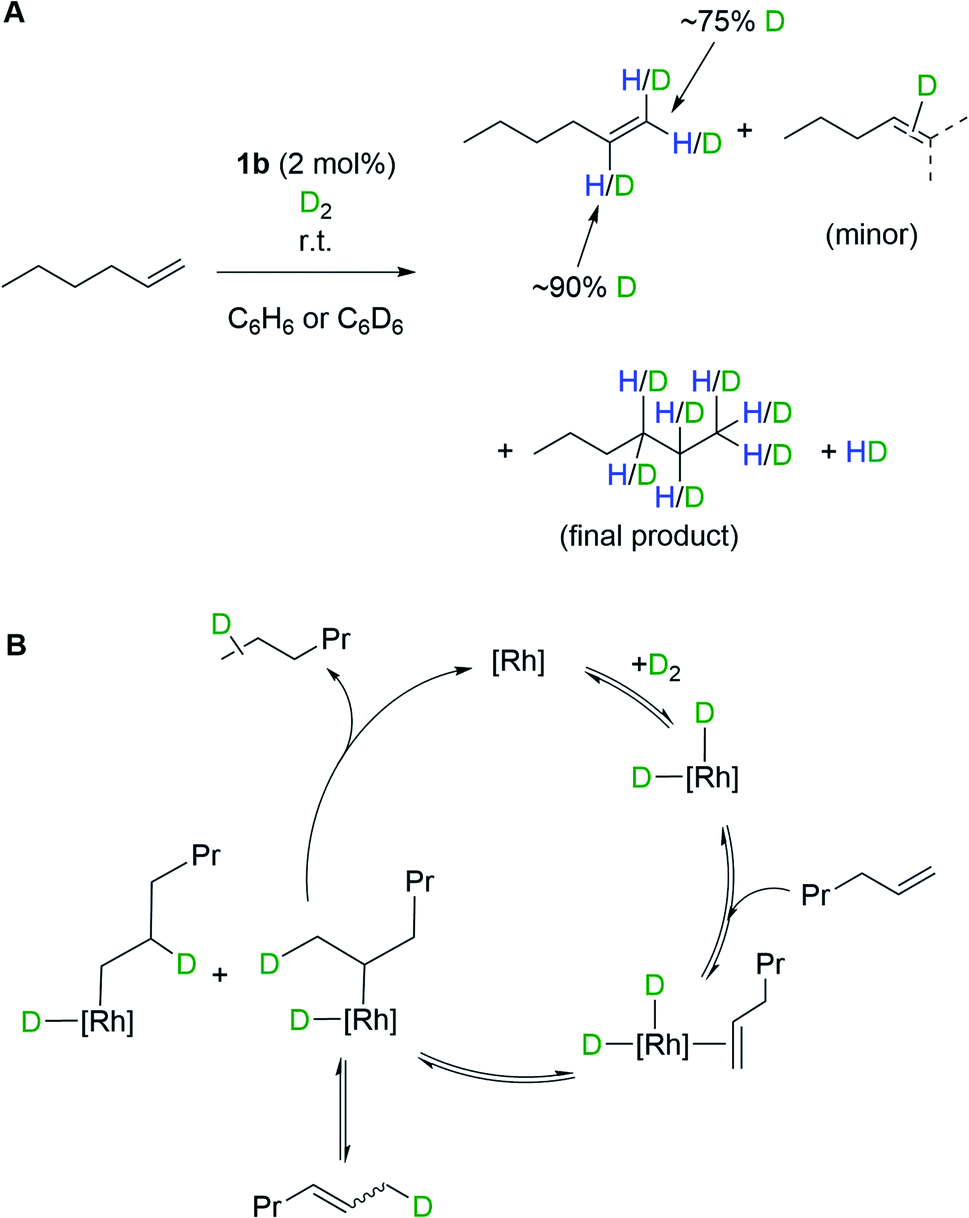 | ||
| Scheme 4 (A) H/D exchange into 1-hexene, as catalyzed by complex 1b. (B) Proposed mechanism. | ||
Conclusions
To conclude, we have shown that the cluster [Rh(COD){Ge9(Hyp)3}] can be modified resulting in coordinatively unsaturated rhodium(I) containing Zintl clusters. Protonation results in a Rh-hydride, while rapid, catalytic H/D exchange occurs between H2 and D2 in the presence of such clusters. The organometallic chemistry of rhodium-phosphine-hydrides is well established – particularly with supporting cyclopentadienyl ligands – and has been critical in the development of our collective understanding of important concepts in the field, such as structure/property relationships, reactivity patterns and catalysis.46 However, the “inorganometallic” chemistry of analogous species with Zintl ions as supporting ligands is essentially unexplored.47,48 This contribution shows that molecular TMMGAs offer similar rich structural and bond activation chemistry that suggests further study into their uses in catalysis is warranted.Experimental
Materials and general procedures
All reactions and product manipulations were carried out under an inert atmosphere of argon or dinitrogen using standard Schlenk-line or glovebox techniques (MBraun UNIlab glovebox maintained at < 0.1 ppm H2O and < 0.1 ppm O2). n-pentane (pent; Sigma-Aldrich, HPLC grade, ≥99%), n-hexane (hex; Sigma-Aldrich, HPLC grade, ≥97%), benzene (Rathburn, HPLC grade, 99.8%), and toluene (Sigma-Aldrich, HPLC grade, 99.8%), were purified using an MBraun SPS-800 solvent system. C6D6 (Sigma-Aldrich, 99.5%) and C7D8 (Sigma-Aldrich, 99.5%) were distilled over sodium metal/benzophenone. All dry solvents were stored under argon in gas-tight ampoules over activated 3 Å molecular sieves. K4Ge9 was synthesized by heating a mixture of the elements (potassium, 99.95%, Alfa Aesar; germanium powder −100 mesh, 99.999%, Alfa Aesar) at 900 °C for 2 days in sealed niobium containers jacketed in evacuated fused-silica ampules according to previously reported synthetic procedures. [Rh(COD)Cl]2,49 K{Ge9[Si(SiMe3)3]3},50 [Rh(COD){Ge9(Hyp3)3}],10 IMe4 (IMe4 = 1,3,4,5-tetramethylimidazol-2-ylidene),51 Na[B(3,5-C6H3(CF3)2)4],52 and [H(OEt2)2][B(3,5-C6H3(CF3)2)4]30 were prepared according to literature procedures. Triphenylphosphine (PPh3; Sigma Aldrich, 99%), trimethylphosphine (PMe3; Sigma Aldrich, 97%) chlorotris(trimethylsilyl)silane (HypCl; TCI, 97%) rhodium(III) chloride hydrate (Precious Metals Online), 1,5-cyclooctadiene (Sigma-Aldrich, >99%), and proton sponge (Sigma-Aldrich, 99%) were used as received without further purification. Parahydrogen was sourced as previously described.37NMR samples were prepared inside an inert atmosphere glovebox in NMR tubes fitted with a gas-tight valve. 1H NMR spectra were recorded at either 499.9 MHz or 400.1 MHz on a Bruker AVIII 500 or a Bruker AVIII 400 NMR spectrometer, respectively. 13C{1H} NMR spectra were recorded at either 151 MHz, 125.8 MHz or 100.6 MHz on a Bruker NEO 600 with broadband helium cryoprobe, a Bruker AVII 500 fitted with a cryoprobe or a Bruker AVIII 400 NMR spectrometer, respectively. 31P{1H} NMR spectra were recorded at 202.4 MHz or 162.0 MHz on a Bruker AVIII 500 or a Bruker AVIII 400 NMR spectrometer, respectively. 1H and 13C{1H} NMR spectra are reported relative to TMS and referenced to the most downfield residual solvent resonance. 31P{1H} NMR spectra are externally referenced to an 85% solution of H3PO4 in H2O (δ = 0 ppm).
Elemental analyses were carried out by Elemental Microanalyses Ltd (Devon, UK). Samples (approx. 10 mg), submitted in sealed Pyrex ampoules.
Synthetic procedures
:10 mixture of toluene and n-pentane (∼0.5 ml total volume) and cooling to −40 °C. Attempts to heat the reaction to above 45 °C or stir the reaction longer (∼1 week) resulted in the formation of decomposition products. Elemental analysis calcd for C37H93Ge9RhSi12W (M = 1815.59 g mol−1): C 24.48, H 5.16. Found: C 23.62, H 4.24. 1H NMR (C6D6, 298 K, 400.16 MHz): δ (ppm) −16.15 (d, 1JRh–H = 26.2 Hz, 1JW–H = 76.0 Hz, 2H; RhH2W), 0.59 (s, 81H; CH3), 4.26 (s, 10H; CH). 13C{1H} NMR (C6D6, 298 K, 125.72 MHz): δ (ppm) 3.26 (Si(CH3)3), 77.55 (CH). 29Si NMR (C6D6, 298 K, 99.32 MHz): δ (ppm) −95.21, −9.55.
Data availability
Spectral data, selected crystallographic information and computational data are included in the ESI.†Author contributions
O. P. E. T. carried out all of the synthetic and computational work described in the manuscript, O. P. E. T. and J. M. G. conducted the crystallographic studies, O. P. E. T. and S. B. D. conducted the parahydrogen experiments, and A. S. W. and J. M. G. wrote the manuscript and managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Shell Global Solutions International B.V., the University of Oxford and the EPSRC for financial support of this research (Industrial CASE studentship O. P. E. T.; EP/M024210 ASW). The University of Oxford is also acknowledged for access to Chemical Crystallography facilities.Notes and references
- D. C. Upham, V. Agarwal, A. Khechfe, Z. R. Snodgrass, M. J. Gordon, H. Metiu and E. W. McFarland, Science, 2017, 358, 917–921 CrossRef CAS PubMed
.
- N. Taccardi, M. Grabau, J. Debuschewitz, M. Distaso, M. Brandl, R. Hock, F. Maier, C. Papp, J. Erhard, C. Neiss, W. Peukert, A. Görling, H. P. Steinrück and P. Wasserscheid, Nat. Chem., 2017, 9, 862–867 CrossRef CAS PubMed
- C. Palmer, M. Tarazkar, H. H. Kristoffersen, J. Gelinas, M. J. Gordon, E. W. McFarland and H. Metiu, ACS Catal., 2019, 9, 8337–8345 CrossRef CAS
- C. Palmer, D. C. Upham, S. Smart, M. J. Gordon, H. Metiu and E. W. McFarland, Nat. Catal., 2020, 3, 83–89 CrossRef CAS
- T. Daeneke, K. Khoshmanesh, N. Mahmood, I. A. De Castro, D. Esrafilzadeh, S. J. Barrow, M. D. Dickey and K. Kalantar-Zadeh, Chem. Soc. Rev., 2018, 47, 4073–4111 RSC
- G. Bo, L. Ren, X. Xu, Y. Du and S. Dou, Adv. Phys. X, 2018, 3, 412–442 Search PubMed
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed
- A. Schnepf, Angew. Chem., Int. Ed., 2003, 42, 2624–2625 CrossRef CAS PubMed
- F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS PubMed
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef CAS PubMed
-
J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, Sausalito, USA, 2010 Search PubMed
- R. N. Perutz and B. Procacci, Chem. Rev., 2016, 116, 8506–8544 CrossRef CAS PubMed
- A. Albinati, R. Naegeli, A. Togni and L. M. Venanzi, Organometallics, 1983, 2, 926–928 CrossRef CAS
- A. Hicken, A. J. P. White and M. R. Crimmin, Dalton Trans., 2018, 47, 10595–10600 RSC
- L. Rocchigiani, W. T. Klooster, S. J. Coles, D. L. Hughes, P. Hrobárik and M. Bochmann, Chem. –Eur. J., 2020, 26, 8267–8280 CrossRef CAS
- N. W. Alcock, O. W. Howarth, P. Moore and G. E. Morris, J. Chem. Soc., Chem. Commun., 1979, 1160–1162 RSC
- O. W. Howarth, C. H. McAteer, P. Moore, G. E. Morris and N. W. Alcock, J. Chem. Soc., Dalton Trans., 1982, 541–548 RSC
- H. Brunner and D. Mijolovic, J. Organomet. Chem., 1999, 577, 346–350 CrossRef CAS
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed
- Based on 409 entries in the CCDC (CSD version 5.41; update March 2020). Mean distance: 2.303 Å; variance: 0.003 Å; std. deviation: 0.054 Å; mean deviation: 0.039 Å.
- A. Neveling, G. R. Julius, S. Cronje, C. Esterhuysen and H. G. Raubenheimer, Dalton Trans., 2005, 181–192 RSC
- R. L. Johnston and D. M. P. Mingos, Inorg. Chem., 1986, 25, 3321–3323 CrossRef CAS
- R. T. Baker, Inorg. Chem., 1986, 25, 109–111 CrossRef CAS
- C. W. Jung, R. T. Baker and M. F. Hawthorne, J. Am. Chem. Soc., 2002, 103, 810–816 CrossRef
- D. Ostendorf, C. Landis and H. Grützmacher, Angew. Chem., Int. Ed., 2006, 45, 5169–5173 CrossRef CAS PubMed
- For d8 trigonal pyramidal palladium(II) and platinum(II) complexes see: C. Tsay, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 13975–13977 CrossRef CAS PubMed
- For d9 trigonal pyramidal rhodium(0) complexes see: P. J. Nance, N. B. Thompson, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2019, 58, 6220–6224 CrossRef CAS PubMed
- See for example:
(a) H. Werner, Angew. Chem., Int. Ed. Engl., 1983, 22, 927–949 CrossRef
- M. Brookhart, B. Grant and A. F. Volpe Jr, Organometallics, 1992, 11, 3920–3922 CrossRef CAS
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711–1732 RSC
- J. M. Brown, P. L. Evans and A. R. Lucy, J. Chem. Soc., Perkin Trans., 1987, 2, 1589–1596 RSC
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A Chem., 2003, 198, 317–341 CrossRef CAS
- O. Torres, B. Procacci, M. E. Halse, R. W. Adams, D. Blazina, S. B. Duckett, B. Eguillor, R. A. Green, R. N. Perutz and D. C. Williamson, J. Am. Chem. Soc., 2014, 136, 10124–10131 CrossRef CAS
- M. E. Halse, B. Procacci, S. L. Henshaw, R. N. Perutz and S. B. Duckett, J. Magn. Reson., 2017, 278, 25–38 CrossRef CAS
- D. Blazina, S. B. Duckett, J. P. Dunne and C. Godard, Dalton Trans., 2004, 2601–2609 RSC
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS
- S. B. Duckett and R. E. Mewis, Acc. Chem. Res., 2012, 45, 1247–1257 CrossRef CAS PubMed
- A. S. Kiryutin, G. Sauer, A. V. Yurkovskaya, H. H. Limbach, K. L. Ivanov and G. Buntkowsky, J. Phys. Chem. C, 2017, 121, 9879–9888 CrossRef CAS
- A. Johnson, C. G. Royle, C. N. Brodie, A. J. Martínez-Martínez, S. B. Duckett and A. S. Weller, Inorg. Chem., 2021, 60, 13903–13912 CrossRef CAS PubMed
- For examples of η1-[Ge9R3]− clusters see:
(a) F. S. Geitner, W. Klein, O. Storcheva, T. D. Tilley and T. F. Fässler, Inorg. Chem., 2019, 58, 13293–13298 CrossRef CAS
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed
- R. N. Perutz, S. Sabo-Etienne and A. S. Weller, Angew. Chem., Int. Ed., 2022, 61, e202111462 CrossRef CAS PubMed
- M. Findlater, W. H. Bernskoetter and M. Brookhart, J. Am. Chem. Soc., 2010, 132, 4534–4535 CrossRef CAS PubMed
- R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef CAS
-
C. Elschenbroich in Organometallics, Wiley-VCH, Weinheim, 3rd edn, 2006 Search PubMed
-
T. P. Fehlner, Inorganometallic Chemistry, Springer, Boston, USA, 1992 Search PubMed
- B. Marciniec, C. Pietraszuk, P. Pawluć and H. Maciejewski, Chem. Rev., 2022, 122, 3996–4090 CrossRef CAS
- G. Giordano and R. H. Crabtree, Inorg. Synth., 1979, 19, 218–220 CAS
-
(a) F. Li and S. C. Sevov, Inorg. Chem., 2012, 51, 2706–2708 CrossRef CAS
- M. B. Ansell, D. E. Roberts, F. G. N. Cloke, O. Navarro and J. Spencer, Angew. Chem., Int. Ed., 2015, 54, 5578–5582 CrossRef CAS PubMed
- A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR spectra, crystallographic data and computational results. CCDC 2155445–2155449. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02552c |
| ‡ Note that for 1d the τ4 value is calculated by taking into consideration the Rh–W bond as one of the four bonds. |
| § Protonation blocks fluxionality via a C3v symmetric cluster with a tetrahedrally coordinated rhodium(I) center as previously observed for 1a–1d. |
| This journal is © The Royal Society of Chemistry 2022 |