
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic catalytic aerobic oxidation of C–sp(3)–H bonds under mild conditions using galactose oxidase model compound CuIIL†
Xiao-Hui
Liu
a,
Hai-Yang
Yu
a,
Jia-Ying
Huang
a,
Ji-Hu
Su
c,
Can
Xue
 a,
Xian-Tai
Zhou
*a,
Yao-Rong
He
b,
Qian
He
b,
De-Jing
Xu
a,
Chao
Xiong
b and
Hong-Bing
Ji
*b
a,
Xian-Tai
Zhou
*a,
Yao-Rong
He
b,
Qian
He
b,
De-Jing
Xu
a,
Chao
Xiong
b and
Hong-Bing
Ji
*b
aFine Chemical Industry Research Institute, School of Chemical Engineering and Technology, Sun Yat-sen University, Zhuhai, 519082, China. E-mail: zhouxtai@mail.sysu.edu.cn
bFine Chemical Industry Research Institute, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China. E-mail: jihb@mail.sysu.edu.cn
cCAS Key Laboratory of Microscale Magnetic Resonance, University of Science and Technology of China, Hefei 230026, China. E-mail: sujihu@ustc.edu.cn
First published on 28th July 2022
Abstract
Developing highly efficient catalytic protocols for C–sp(3)–H bond aerobic oxidation under mild conditions is a long-desired goal of chemists. Inspired by nature, a biomimetic approach for the aerobic oxidation of C–sp(3)–H by galactose oxidase model compound CuIIL and NHPI (N-hydroxyphthalimide) was developed. The CuIIL–NHPI system exhibited excellent performance in the oxidation of C–sp(3)–H bonds to ketones, especially for light alkanes. The biomimetic catalytic protocol had a broad substrate scope. Mechanistic studies revealed that the CuI-radical intermediate species generated from the intramolecular redox process of CuIILH2 was critical for O2 activation. Kinetic experiments showed that the activation of NHPI was the rate-determining step. Furthermore, activation of NHPI in the CuIIL–NHPI system was demonstrated by time-resolved EPR results. The persistent PINO (phthalimide-N-oxyl) radical mechanism for the aerobic oxidation of C–sp(3)–H bond was demonstrated.
Introduction
Oxidation is one of the most useful chemical transformations in organic synthesis and the modern chemical industry.1 Oxidation of the C–sp(3)–H bond is extremely challenging due to the high bond energy of the C–H bond.2 Various catalysts have been used for the conversion of hydrocarbons, ranging from heterogeneous3 to homogeneous catalytic systems,4 as well as bio- and enzymatic catalysts.5 In heterogeneous catalytic systems, alkanes are usually first converted to more active alkenes via oxidative dehydrogenation.6 To achieve acceptable conversion and selectivity, these processes are often carried out under harsh conditions (e.g., high temperatures 500–900 °C).7 In the presence of O2 or other peroxide oxidants, C–H bonds could be oxyfunctionalized by transition metals in homogeneous catalytic systems.1a Usually, assistance of light,8 electricity,9 or other additives10 is necessary. Therefore, a more sustainable and environmentally benign oxidative protocol for the chemical conversion of hydrocarbons is desirable.Copper-containing metalloprotein galactose oxidase (GO) is an enzyme which selectively catalyzes the aerobic oxidation of alcohols to aldehydes under mild conditions.11 From the structure, it could be known that GOase contains two one-electron redox centers, a mononuclear copper center and a tyrosyl radical ligand (Tyr 272).12 The Tyr 272 radical ligand in GOase abstracts H from the α-carbon of alcoholate and undergoes intramolecular electron transfer to form aldehyde and a CuI species. Finally, the reduced GOase is converted to its original form by O2 along with the formation of 1 equiv. of H2O2 (Scheme 1A).13
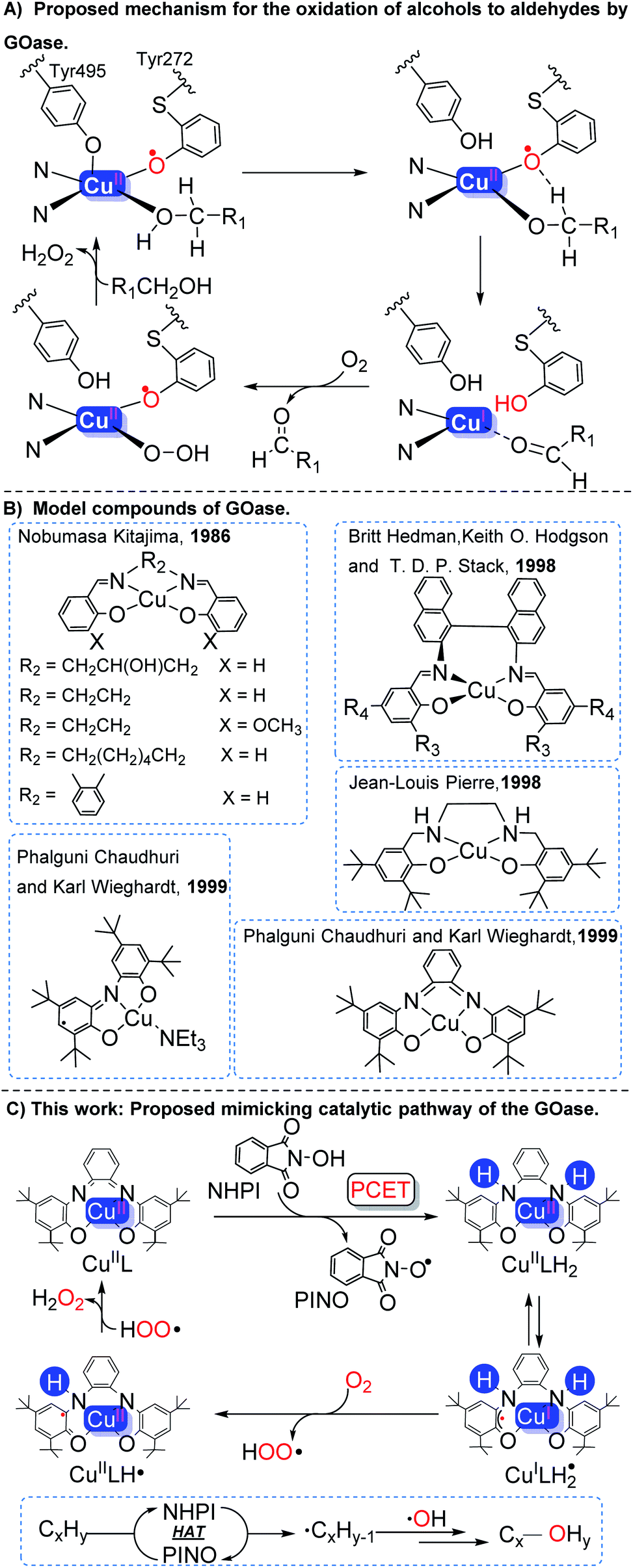 | ||
| Scheme 1 (A) Proposed mechanism for the oxidation of alcohols to aldehydes by GOase. (B) Model compounds of GOase. (C) This work. | ||
In the past, some GOase model compounds have been synthesized by employing redox-active non-innocent ligands (Scheme 1B).14 Kitajima reported a salen type copper(II) complex as a possible GOase model.15 Stack16 and Pierre's17 functional models were developed. Subsequently, a mononuclear CuII–phenoxyl complex was synthesized by Chaudhuri and Wieghardt.13a,18 These model compounds are capable of oxidizing primary/secondary alcohols to the corresponding ketones in the presence of O2. However, GOase has insufficient activity to oxidize hydrocarbons due to the high BDE (bond dissociation energy) of the C–H bond in most hydrocarbons (85–105 kcal mol−1).2b,19
N-Hydroxyphthalimide (NHPI) is an efficient and low-cost N-oxyl precursor capable of producing a highly active phthalimide-N-oxyl (PINO) radical, which is widely used in the aerobic oxidation of hydrocarbons.20 Ishii and co-workers innovatively introduced metal salts and NHPI for the oxidation reaction of isobutane with molecular oxygen (100 °C).21 Jiao and co-workers developed PdCl2/NHPI for C–sp(2)–H hydroxylation (100 °C).22 However, PINO readily self-decomposes at high temperature (>80 °C) and under alkaline conditions.19a,23 In addition, the application of NHPI was restricted because of the long initiation period and short existence time of PINO.20d In response to these challenges, an electrochemical pathway was developed to activate NHPI under mild conditions by Jensen and co-workers.24 Wang described the approach of PINO radical generation through the irradiation of α-Fe2O3 and NHPI using 455 nm light.25 It has also been reported that ionic liquids26 and fluorine-containing solutions27 were utilized to stabilize PINO.
Inspired by the structure of galactose oxidase, the oxidation protocol of the C–sp(3)–H bond was developed, in which the CuIIL catalyst (ligand: N,N′-bis(3,5-di-tert-butyl-2-hydroxyphenyl)-1,2-phenylenediamine)18 and NHPI were introduced. Through the proton coupling electron transfer (PCET) process, two electrons and protons were transferred from NHPI to CuIIL to form CuIILH2 and the PINO radical. The PINO radical can abstract H from the C–H bond of the substrate to produce a high-activity carbon-centered radical through hydrogen atom transformation (HAT). Subsequently, in situ generated CuIILH2 reacts with O2 to produce H2O2 and CuIIL (Scheme 1C). In addition, the detailed mechanism was investigated with the assistance of in situ UV-vis spectroscopy, electron paramagnetic resonance (EPR) spectroscopy, X-ray absorption near-edge structure (XANES), as well as thermodynamic and kinetic analysis combined with density functional theory (DFT).
Results and discussion
We initiated aerobic oxidation of the C–sp(3)–H bond by using isobutane as a model substrate (Table 1). After an extensive survey of reaction conditions, including the amount of catalyst (CuIIL) (Fig. S1†), the amount of NHPI (Fig. S2†) and reaction temperature (Fig. S3†), the optimized conditions were obtained (isobutane 21 mmol, O2 1.0 MPa, 70 °C, CuIIL 0.8 mol%, NHPI 4.0 mol%, CH3CN solvent 20 mL). Under the optimized conditions, isobutane conversion was up to 75%, and the desired hydroxylation product tert-butanol (1) was obtained in 80% selectivity (Table 1, entry 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from the standard conditions | Conv. (%) | Selectivitiesb (%) | ||
| 1 | 2 | 3 | |||
| a Standard conditions: isobutane (21 mmol), CuIIL (0.8 mol%), CH3CN (20 mL), O2 (1.0 MPa), 70 °C, 7 h. b Determined by GC with naphthalene as the internal standard. n.r. = no reaction, “—” means no detection. | |||||
| 1 | None | 75 | 80 | 20 | 0 |
| 2 | Without CuIIL | 4 | 24 | 16 | 60 |
| 3 | Without NHPI | 10 | 39 | 62 | 0 |
| 4 | CuCl2 (0.8 mol%) instead of CuIIL | 45 | 78 | 22 | 0 |
| 5 | CuCl (0.8 mol%) instead of CuIIL | 29 | 72 | 28 | 0 |
| 6 | Dichloroethane as a solvent | 23 | 21 | 24 | 55 |
| 7 | DMF as a solvent | 33 | 74 | 17 | 9 |
| 8 | Air (2.0 MPa) instead of O2 | 68 | 84 | 16 | 0 |
| 9 | Co(OAc)2·4H2O (0.8 mol%) instead of CuIIL | 26 | 60 | 12 | 28 |
| 10 | BHT (10 mol%) as a radical scavenger | n.r. | — | — | — |
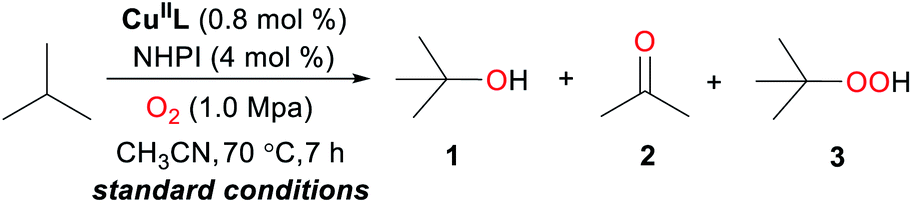
The control experiments indicated that CuIIL and NHPI were indispensable for the success of this oxidation. Especially, the selectivity of tert-butanol was closely related to CuIIL or NHPI (Table 1, entries 2 and 3). When using other Cu-based catalysts such as CuCl2 and CuCl, a lower yield of 1 and conversion of isobutane was observed (Table 1, entries 4 and 5). The results of solvent screening showed that CH3CN was the optimal solvent for this reaction (Table 1, entries 6 and 7). Surprisingly, when the oxidation was conducted in air (2.0 MPa), the selectivity of 1 increased (84%) and the conversion slightly decreased (68%) (Table 1, entry 8). Gratifyingly, after 5 cycles of the reaction, the catalytic system still showed excellent catalytic activity (Fig. S4†). Poor efficiency and selectivity were obtained when using the typical Co(OAc)2/NHPI as the catalyst under the same conditions as previously reported21 (Table 1, entry 9). Addition of butylated hydroxytoluene (BHT, radical scavenger) resulted in oxidation being completely suppressed (Table 1, entry 10).
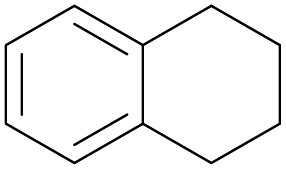
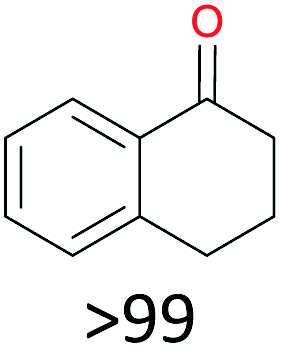
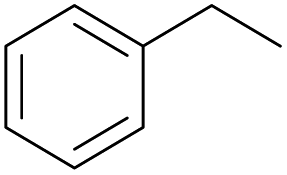
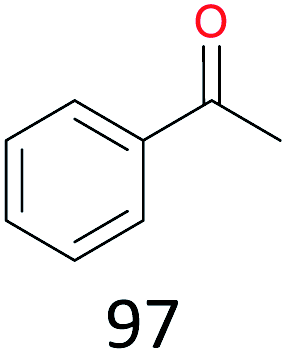
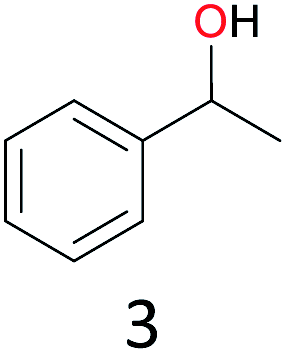
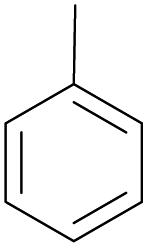
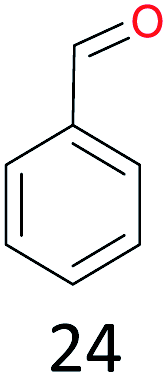
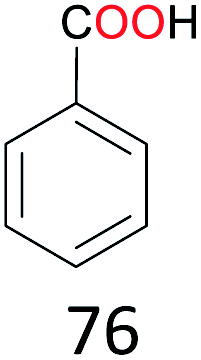
With the optimal reaction conditions in hand, the scope of hydrocarbons was subsequently explored. Indane and 1,2,3,4-tetrahydronaphthalene were oxidized to the corresponding ketones in remarkable conversion (99%) and selectivity (99%) (Table 2, entries 1 and 2). In addition, ethylbenzene was transformed into acetophenone in excellent selectivity (97%) (Table 2, entry 3). Meanwhile, toluene was oxidized to benzaldehyde (24%) and benzoic acid (76%) (Table 2, entry 4).
| En try | Substrate | Conv.b (%) | Selectivitiesb (%) | |
|---|---|---|---|---|
| a Standard conditions: Substrate (20 mmol), CuIIL (0.8 mol%), NHPI (4 mol%), CH3CN (20 mL), O2 (1 MPa), 70 °C, 7 h. b Determined by GC with naphthalene as the internal standard. c NHPI (10 mol%), 100 °C. | ||||
| 1 |
|
99 |
|
|
| 2 |
|
99 |
|
|
| 3 |
|
98 |
|
|
| 4 |
|
48 |
|
|
| 5 |
|
39 |
|
|
| 6 |
|
47 |
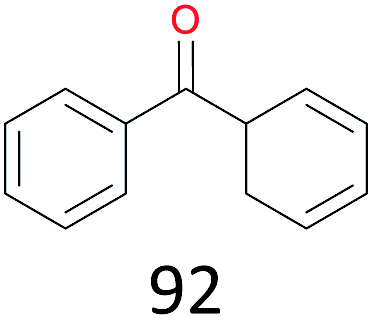
|
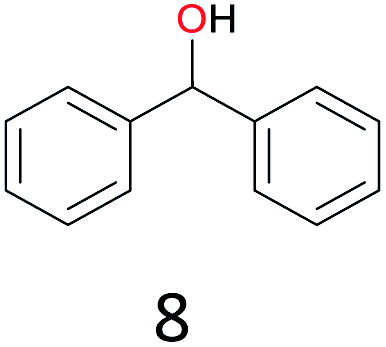
|
| 7 |
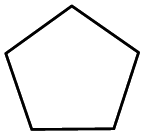
|
37 |
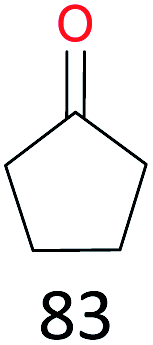
|
|
| 8 |
|
76 |
|
|
|
||||
| 9c |

|
4 |
|
|
| 10c |
|
34 |
|
|
| 11c |
|
66 |
|
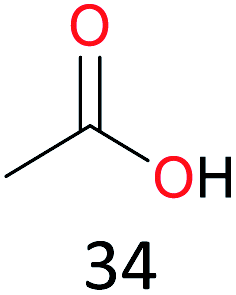
|
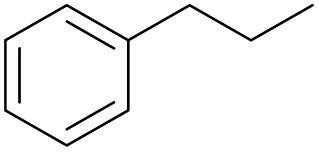
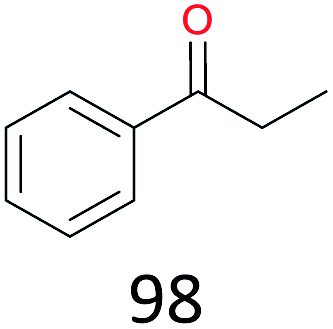
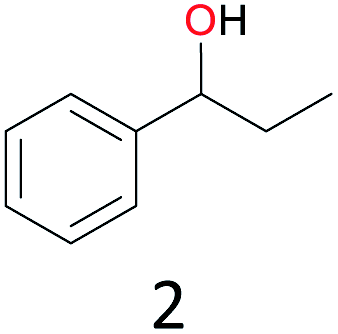
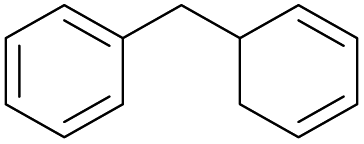
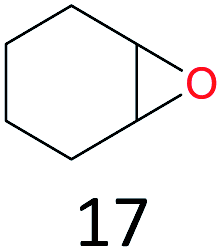
The effect of steric hindrance was observed from the oxidation of propylbenzene and dphenylmethane (Table 2, entries 5 and 6). Cyclopentane was oxidized to cyclopentanone with 83% selectivity and cyclopentanol (17% selectivity) (Table 2, entry 7). For cyclohexene containing both C–sp(3)–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, the total selectivity of C–sp(3)–H bond oxidation to alcohols and ketones was 83%, and the selectivity of epoxide was 17% (Table 2, entry 8). It indicated that the biomimetic catalytic system exhibited high regioselectivity.
C bonds, the total selectivity of C–sp(3)–H bond oxidation to alcohols and ketones was 83%, and the selectivity of epoxide was 17% (Table 2, entry 8). It indicated that the biomimetic catalytic system exhibited high regioselectivity.
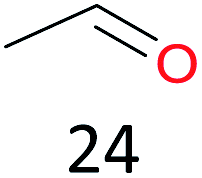
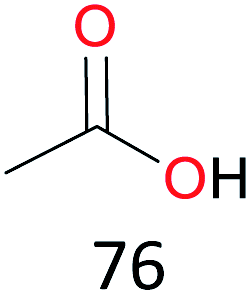
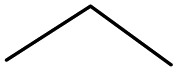
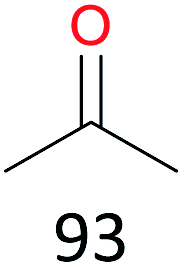
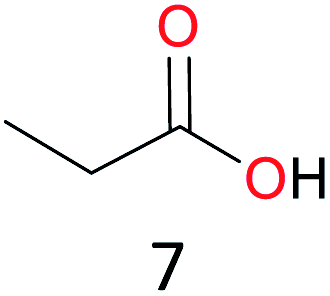
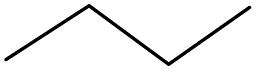
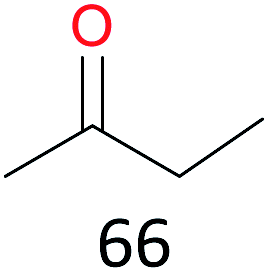
Surprisingly, the quite inert ethane (C–H BDE: 101.1 kcal mol−1)28 could be oxidized to acetaldehyde (24%) and acetic acid (76%) at 100 °C (Table 2, entry 9). Heartened by this result, we also conducted experiments on the oxidation of propane. Propane was oxidized to acetone with satisfactory conversion (34%) and 93% selectivity (Table 2, entry 10). Moreover, n-butane was smoothly oxidized to methyl ethyl ketone (MEK) with 66% conversion and 66% selectivity (Table 2, entry 11).
To gain insight into the reaction pathway, the reaction between CuIIL and NHPI was originally investigated. The X-band EPR spectrum of the nearly square-planar (D4h) CuIIL in CH3CN recorded at 90 K displayed a typical Cu(II) signal with an unpaired electron in the dx2−y2 orbital (Fig. S5†).29 From the optimal geometric configuration of CuIIL (B3LYP/6-31G(d,p)/IEF-PCM (solvent model) level with CH3CN as a solvent), the dx2−y2 orbital of the central Cu(II) atom was perpendicular to the π orbitals in the aromatic ring (Fig. S6†). This orbital orientation minimized the spatial overlap between the orthogonal dx2−y2 orbital of Cu(II) and the ligand's π orbitals, resulting in a difficulty for ligand to metal Cu(II) charge transfer (LMCT) to occur.30
CuIIL has characteristic bands at 435 nm and 1167 nm with a black-green color in CH3CN solution (Fig. 1A, orange line). To determine the type of absorption peak, the electronic UV-vis-NIR absorption spectra of CuIIL were recorded using the TD-B3LYP/6-31G(d,p) method and IEF-PCM solvent model for CH3CN. The obtained absorption spectra, the FMO pictures, and the vertical excitation energies are given in Fig. S6†, S7 and Table S1,† respectively. The obtained peaks at 435 nm (calcd. λ = 529 nm, f = 0.0581) and 1167 nm (calcd. λ = 1022 nm, f = 0.1249) of CuIIL were attributed to β-SOMO (the highest singly occupied molecular orbital)-4 to β-SUMO (the lowest singly unoccupied molecular orbital) and β-SOMO−1 to β-SUMO transitions, respectively. These are mainly intraligand charge-transfer (ILCT) transitions.31 Another intense band of CuIIL in the UV region (<380 nm) was attributed to the π to π* transition within the ligand's aromatic ring (α-SOMO to α-SUMO+2, calcd. λ = 364 nm, f = 0.2001).32 It indicates that it is difficult for an intramolecular redox reaction to occur in planar structured CuIIL.
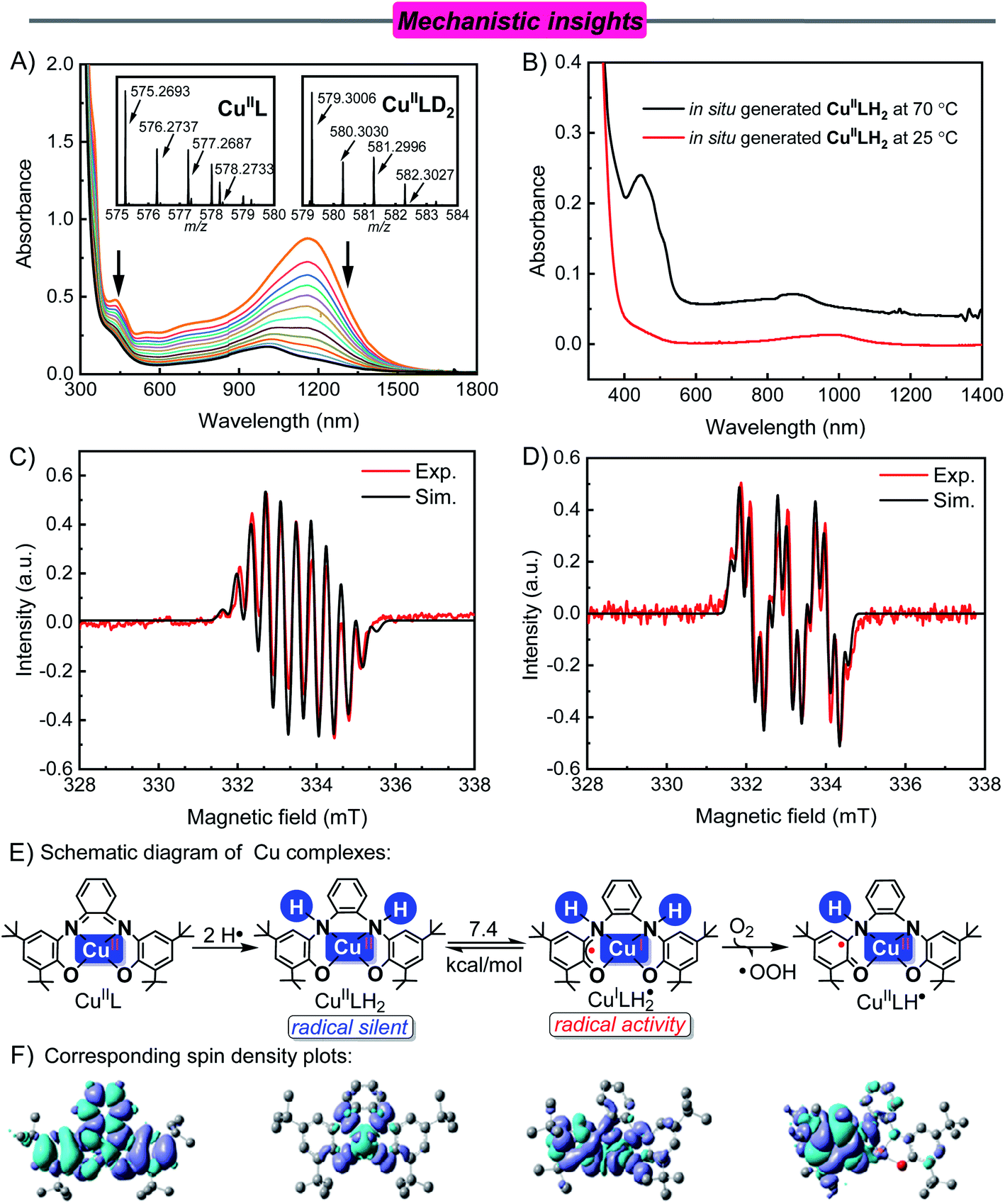 | ||
| Fig. 1 (A) Electronic spectral changes observed in the reaction of CuIIL (0.1 mM) and NHPI (0.2 mM) in CH3CN solvent under an argon atmosphere at 25 °C. Insets show the CSI-MS spectra of CuIIL and CuIILD2. (B) Electronic spectra of in situ generated CuIILH2 at 25 °C (red line) and 70 °C (black line) in CH3CN solvent under an argon atmosphere. (C) Experimental (red) and simulated (black) X-band EPR spectra of CuIIL (0.10 mM) and NHPI (0.20 mM) in CH3CN solvent under an argon atmosphere at 343 K. Frequency: 9.3615 GHz; Microwave power: 2.0 mW; modulation amplitude: 0.8 G. Parameters of simulated hyperfine splitting constants: giso = 2.0043 and AN,N,H,H,H,H = [7.8, 3.5, 3.5, 3.5, 3.5, 3.5 ] G. (D) X-band EPR spectra of in situ generated CuIILH2 under an argon atmosphere followed by reaction with O2 (1.0 MPa) in CH3CN at 70 °C, giso = 2.0043 and AN,H,H,H,H = [9.5, 3.0, 2.0, 2.0, 2.0] G. (E) and (F) Schematic diagram and corresponding spin density plots of the Cu complex. | ||
As shown in the cyclic voltammograms (Fig. S9†), the CuIIL complex in CH3CN showed two reversible oxidations (Eox11/2 = −0.17 V and Eox21/2 = 0.30 V vs. Fc+/0, the same below) and two reversible reductions (Ered11/2 = −0.77 V and Ered21/2 = −1.46 V). For comparison, NHPI has an oxidation potential E = 1.04 V, which means that the single electron transfer (SET) reaction could not occur between NHPI and CuIIL. However, after adding NHPI (0.20 mM) to an argon-saturated CH3CN solution of CuIIL (0.10 mM) at 25 °C, the absorption band at 1167 nm disappeared, and the absorption intensity decreased at 435 nm. A new absorption band appeared at 998 nm, and the color of the solution changed from black-green to yellow (Fig. 1A).
A blue-shift (1167 nm to 998 nm) occurred in the long-wavelength region, which indicated a decrease in the electron density of the conjugated system.33 In addition, the characteristic absorption peak of the resulting solution was consistent with the directly synthesized CuIILH2 and DFT computed UV-vis/NIR spectrum (Fig. S10†). The EPR spectrum of the resulting solution was also consistent with that of the directly synthesized CuIILH2 (Fig. S11†). The presence of CuIILH2 in the resulting solution was also indicated by high resolution mass spectrometry (Fig. S12†). Furthermore, when NHPI-d was used instead of NHPI (see Fig. S13† for the synthesis and characterization of NHPI-d), the CSI-MS (cold-spray ionization mass spectrometry) spectrum of the resulting solution exhibited a prominent ion peak at m/z = 579.3006, whose mass and isotope distribution patterns corresponded to CuIILD2 (calcd m/z = 579.3002) (Fig. 1A, inset). In addition, EPR showed that the PINO radical (AN = 4.6, g = 2.0073) was produced from homolysis of the O–H bond of NHPI (Fig. S14†).20a,34 These results demonstrate unambiguously that CuIIL reacted with the hydrogen donor (NHPI) by H-atom transfers to form CuIILH2.
Compared with 25 °C, the in situ generated CuIILH2 shows a new absorption peak (464 nm) at 70 °C (Fig. 1B). This absorption is attributed to Cu(II) d–d bands and/or ligand to copper charge transfer transitions (LMCT).32 This led us to speculate whether this phenomenon was caused by structural changes of CuIILH2 caused by temperature changes.
The ligand of CuIILH2 is a closed shell and subsequently EPR-silent (radical silent). Surprisingly, when the reaction temperature was increased to 70 °C, the in situ generated CuIILH2 in an argon atmosphere showed a typical S = 1/2 organic radical signal (g = 2.0043) with hyperfine coupling to two N donors (I = 1) and four H atoms (I = 1/2), which was not observed at room temperature (Fig. 1C). The EPR spectra of PINO may be covered due to the peak location (g = 2.0073) and total width (12 G).
Interestingly, the hyperfine splitting of the two N atoms (AN(1) = 7.8 G, AN(2) = 3.5 G) were unequal and nearly twofold, which means the two N atoms of the ligand have different effects on the spin center.35 For organometallic radical complexes, when heteroatoms and spin centers are in different spatial positions, the values of hyperfine splitting are different (Table S2†).36 Simultaneously, the small hyperfine splitting of the N atom indicates that the EPR signal is caused by the ligand-based delocalization of the π-electron.37 Similar results were observed at 70 °C using directly synthesized CuIILH2 (Fig. S15†). These results suggest that CuIILH2 was distorted and led to a ligand-based radical at 70 °C.
The Cu K-edge XANES spectra of in situ generated CuIILH2 show that the oxidation states of copper decreased (Fig. S16†).38 Furthermore, the variable-temperature EPR experiment of in situ generated CuIILH2 was conducted. Fig. 2 shows that the signal of Cu(II) is gradually enhanced with the decrease in temperature. The results indicated that some Cu(I) (3d10, EPR silent) species were present in the solution at 70 °C, but changed to Cu(II) with the decrease in temperature.11d,39 Based on the above experimental evidence, this phenomenon can be explained by the intramolecular redox reaction of CuIILH2 at 70 °C, which results in the ligand-based unpaired π-electron and Cu(I).39c For convenience, we express CuIILH2 at 70 °C as a CuI-radical species, CuILH2˙.
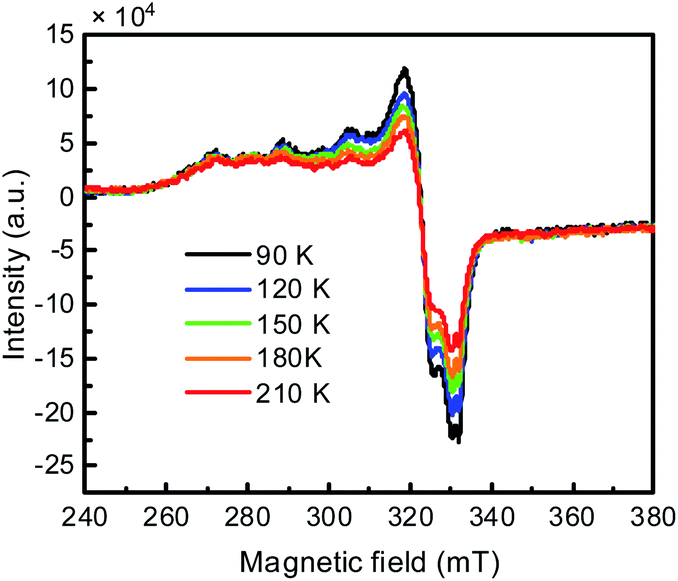 | ||
| Fig. 2 Variable temperature X-band EPR spectra of in situ generated CuIILH2. CuIIL (0.1 mM) and NHPI (0.2 mM) reacted in an argon atmosphere at 70 °C for 2 hours, and then were immediately placed in a 210 K environment for the EPR test. | ||
Subsequently, the cause of an intramolecular redox reaction in CuIILH2 was explored. For electron transfer to occur between the ligand and the central Cu, the ligand's π orbital needs to overlap with the Cu dx2−y2 orbital.39b However, the optimized molecular structure of CuIILH2 is still planar, and the electronic spectral peaks in the long-wavelength region primarily corresponded to ILCT (Fig. S17 and Table S3†). Obviously, intramolecular electron transfer cannot occur in the CuIILH2 plane structure because the ligand π orbital is perpendicular to the Cu dx2−y2 orbital.
Therefore, the molecular structure of CuIILH2 needs to be distorted to achieve antiferromagnetic coupling between the dx2−y2 orbital of Cu(II) and the pz orbital of the ligand. Due to the change in the N atom hybridization from sp2 to sp3 (CuIIL to CuIILH2), the flexibility of CuIILH2 increased.30 This increases the possibility of changes in the ligand's spatial position. The speculated geometry of distorted CuIILH2 (CuILH2˙) is shown in Fig. S18.† The absorption spectrum obtained by excited state calculations is similar to that of in situ generated CuIILH2 at 343 K (Fig. 1B), which proves that the conjecture is reasonable (Fig. S19†). The long-wavelength bands of CuILH2˙ were attributed to ILCT and ligand-to-metal charge transfer (LMCT) transitions (Fig. S20 and Table S4†).16 In addition, the single-point energy calculations showed that the energy of CuILH2˙ is 7.4 kcal mol−1 higher than that of CuIILH2, which explained the different molecular structures caused by the temperature (Fig. 1E).
After the formation of CuILH2˙, the addition of O2 caused a new EPR signal (g = 2.0043, Fig. 1D). Combined with the hyperfine coupling to a N atom and four H atoms on the ligand, we assigned this EPR spectrum to the CuIILH˙ radical (Fig. 1E). Furthermore, the spin density plot of CuIILH˙ also showed that electrons were mainly concentrated in the conjugated system, which is consistent with the EPR data (Fig. 1E). Simultaneously, the EPR spectra of the HOO˙ radical (AN = 14.3, AH(β) = 11.7, g = 2.0061) was captured using DMPO (dimethyl pyridine N-oxide) as a trapping agent (Fig. S21†).40 This indicates that an electron and a proton were transferred from CuILH2˙ to O2. In contrast, O2 cannot be activated at room temperature by CuIILH2.
The reaction between CuIIL and NHPI involves the transfer of two electrons and two protons, and a stepwise thermochemical analysis was conducted. In the process, an electron and a proton of NHPI are transferred to CuIIL to form CuIILH˙; the H˙ transfer in both stepwise electron transfer-proton transfer (ET-PT) (ΔG° = 24.0 kcal mol−1) and PT-ET (ΔG° = 65.6 kcal mol−1) processes involved high-energy intermediates (Scheme S1A†). In contrast, the PCET pathway (ΔG° = 5.6 kcal mol−1, per Hess's Law) has obvious thermodynamic advantages.41 Similarly, the PCET pathway of the process, where an electron and a proton are transferred from NHPI to CuIILH˙ to form CuIILH2 (ΔG° = 23.2 kcal mol−1), is energetically favorable (Scheme S1B†). Furthermore, the total energy (ΔG° = 28.8 kcal mol−1) of the transfer of two electrons and protons from NHPI to CuIIL is relatively low, which indicates that PCET is a possible reaction pathway.
The radicals involved in oxidation were confirmed by the complete inhibition of oxidation by the addition of 10 mol% BHT (free radical scavenger) (Table 1, entry 10); therefore, the identification of reactive oxygen species was carried out by spin trapping. During isobutane oxidation, the EPR spectrum of the tBu-alkoxy radical captured by DMPO (AN = 14.5, AH(β) = 10.5, g = 2.0062) was detected.42 However, this signal was broadened, which resulted in other radical signals being covered (Fig. S22†). However, the adducts of DMPO and the tBu-alkoxy radical (m/z = 186.1494) and DMPO and ˙OH radicals (m/z = 137.0857) are identified by mass spectrometry (Fig. 3A, Fig. S23 and S24†). This indicates the presence of ˙OH radicals in the reaction system.
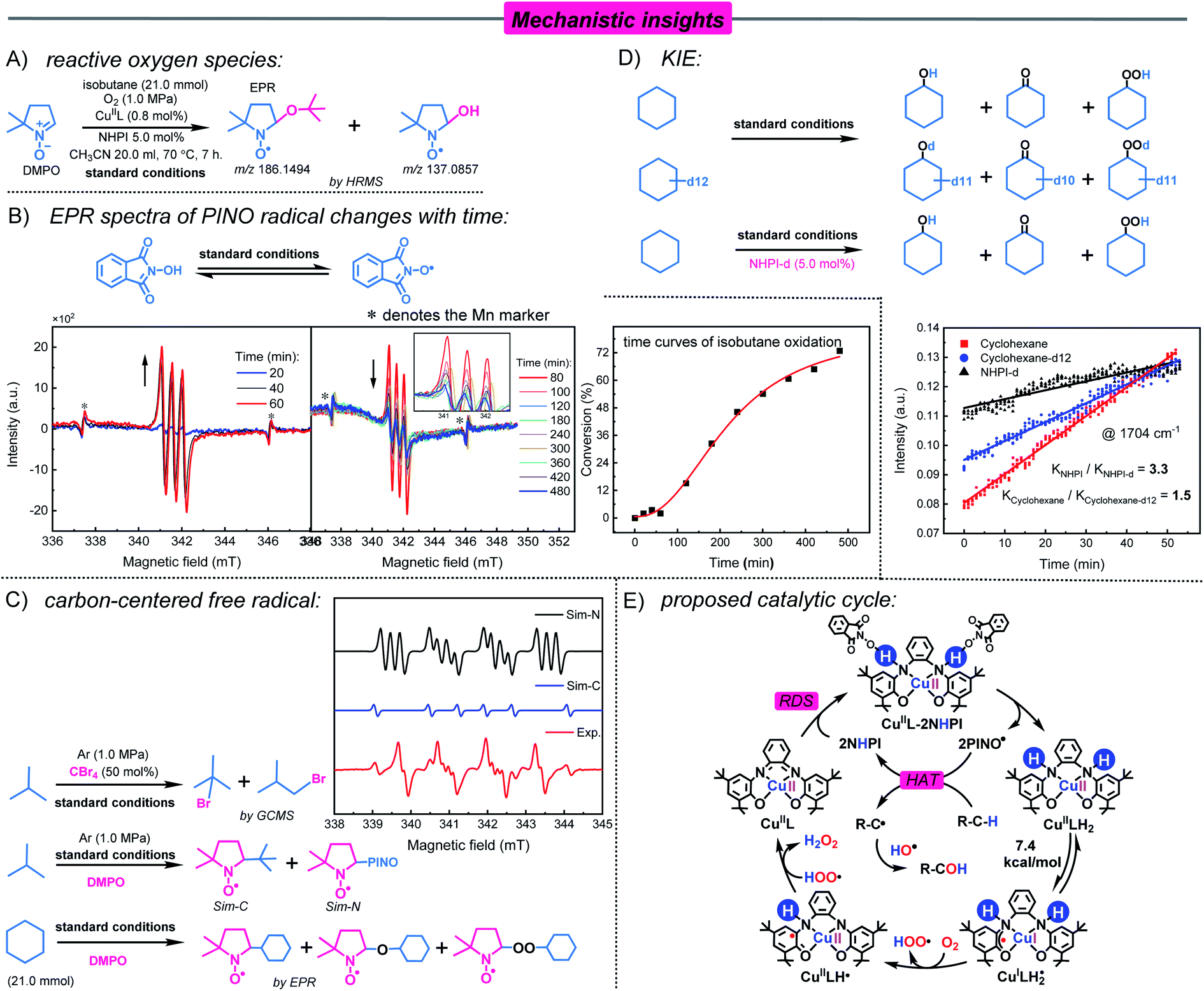 | ||
| Fig. 3 Mechanistic studies for CuIIL–NHPI catalyzed aerobic oxidation. (A) Reactive oxygen species. (B) EPR spectra of PINO radical changes with time and the time curves of the isobutane oxidation reaction. Reaction condition: isobutane (21.0 mmol), CuIIL (0.8 mol%), NHPI (4.0 mol%), solvent (CH3CN, 20.0 mL) and O2 (1.0 MPa) 70 °C. (C) Carbon-centered free radical. Experimental and simulated EPR spectra of the mixture of isobutane (21.0 mmol), CuIIL (0.8 mol%), and NHPI (4.0 mol%) in CH3CN (20.0 mL) under argon (1.0 MPa) at 70 °C. DMPO was used as a spin trapping agent. Simulation parameters (Sim-N, AN1 = 14.1, AN2 = 2.4, AH(β) = 12.6, g = 2.0050; Sim-C, AN = 14.3, AH(β) = 21.8, g = 2.0048). (D) KIE experiment. In situ infrared spectra showing the variations in the peak at 1704 cm−1. Reaction conditions: cyclohexane/cyclohexane-d12 (21.0 mmol), CuIIL (0.8 mol%), NHPI/NHPI-d (4.0 mol%), and O2 (1.0 MPa) in CH2Cl2 (20.0 mL) at 100 °C. (E) Proposed catalytic cycle. | ||
H2O2 was also detected in the oxidation of isobutane by the probe reaction (Fig. S25†). In the contrast experiment, CuIIL efficiently decomposed H2O2 to produce ˙OH radicals (Fig. S26†). To further confirm the existence of ˙OH radicals in the reaction system, Fig. S27A† shows the EPR spectrum of the reaction solution using BMPO (5-tert-butoxycarbonyl 5-methyl-1-pyrroline N-oxide)43 as a capture agent, which is consistent with the ˙OH radical generated by the Fenton reaction captured by BMPO (Fig. S27B†). In addition, ˙OH radical can be quenched by ethanol.44 The reaction solution of oxidation of isobutane was quenched with ethanol, and the ˙C2H4OH radicals were captured by DMPO (Fig. S28A and S28B†). This phenomenon was also consistent with that of the Fenton reaction (Fig. S28C†). These results indicated that an ˙OH radical is the oxygen species in the reaction process.
Nitroxyl radicals are widely used in the aerobic oxidation of C–H bonds, but they have some limitations such as a long initiation period, a short existence time and self-decomposition.20d In the CuIIL–NHPI catalytic system, time-dependent experiments were conducted to investigate the PINO radical by EPR. The PINO radicals are rapidly produced and accumulated to maximum concentrations within 60 min, which are attributed to the bionic catalyst CuIIL superior hydrogen extraction ability (Fig. 3B). Furthermore, the concentration of the PINO radical decreased slowly, lasting up to 480 minutes (Fig. 3B). This indicated the existence of persistent PINO radicals in the CuIIL−NHPI system, which is desirable for efficient reactions.23b,45 Combined with the time curves of isobutane oxidation, the initiation period and rapid reaction phase were synchronized with a change in the PINO radical (Fig. 3B). This further illustrates the importance of the PINO radical. In addition, when isobutane was replaced with cyclohexane-d12, the recovered NHPI contained deuterium products NHPI-d (Fig. S29†). This phenomenon suggests that PINO radicals activate substrates by extracting a hydrogen atom from the C–H bond of the substrate.20e
To obtain direct evidence about the carbon-centered radical, radical capture experiments were performed in an Ar atmosphere. As shown in Fig. 3C, the spin-trap experiments with DMPO revealed the presence of a carbon-centered tert-butyl radical (Sim-C, AN = 14.3, AH(β) = 21.8, g = 2.0048) and a nitrogen-centered radical (Sim-N, AN1 = 14.1, AN2 = 2.4, AH(β) = 12.6, g = 2.0050).42 To our knowledge, this is also the first report of the capture of a tert-butyl radical (C4) during homogeneous catalytic oxidation. However, this signal was unstable due to its high activity. When isobutane was replaced by cyclohexane, we captured ˙C6H11, C6H11O˙, and C6H11OO˙ radicals under an O2 atmosphere (Fig. S30†). In addition, 1-bromo-2-methylpropane and 2-bromo-2-methylpropane were identified by adding 50 mol% CBr4 (Fig. 3C). This indicates that in the CuIIL−NHPI system, C–H bond activation mainly occurred via the formation of a carbon-centered radical.
According to previous studies, the C–H activation approach via the HAT process was likely to be involved in the rate-determining step (RDS).46 The kinetic isotope effect (KIE) experiment was carried out by in situ infrared spectroscopy. Cyclohexane oxidation was used as a model reaction. The reaction kinetics was studied by monitoring the absorption peak of the oxidation product cyclohexanone at 1704 cm−1 (CO bond) over time (Fig. 3D and S31†). A KIE value of 1.5 (KH/KD) was obtained for the oxidation of cyclohexane and cyclohexane-d12 by the CuIIL−NHPI system. Thus, the C–H activation step should not be the rate-determining step.47 However, when NHPI-d was used for oxidation, the value of KIE was 3.3, which indicates that the activation of NHPI is the rate-determining step.48
Based on the experimental results and thermochemical calculations, a possible reaction pathway was outlined (Fig. 3E). First, CuIIL reacts with NHPI (electron and proton donor) through two successive PCET processes to form CuIILH2. This process may be the rate-determining step of the reaction through the KIE experiment. Subsequently, CuIILH2 is converted to a CuI-radical species CuILH2˙ through an intramolecular redox reaction at 70 °C. CuILH2˙ is a highly reactive radical intermediate that reacts with O2 to produce CuIILH˙ and HOO˙ radicals. Next, the catalytic cycle returns to the starting point, with the formation of H2O2 by the transfer of electrons and proton from CuIILH˙ to HOO˙. H2O2 is rapidly decomposed into reactive oxygen species ˙OH under the reaction conditions. Activation of O2 by the CuIIL–NHPI system is similar to the oxidation of alcohol by galactose oxidase. More importantly, this process is also accompanied by the production of a persistent PINO radical for substrate C–H bond activation by HAT. Subsequently, the carbon-centered radical reacts with reactive oxygen species in the reaction system to generate oxidation products.
Conclusions
In summary, we developed the protocol for C–sp(3)–H efficient catalytic aerobic oxidation under mild conditions based on a GOase model compound CuIIL and NHPI. In particular, the CuIIL–NHPI system showed excellent performance for the oxidation of light alkanes (C2–C4). CuIIL can activate NHPI efficiently to generate PINO radicals and CuIILH2 through the PCET pathway. Based on the EPR tests and theoretical calculations, the intramolecular redox process of CuIILH2 has been proved. Throughout the catalytic cycle, O2 is reduced to its active form and the C–sp(3)–H bond of the substrate is converted to a highly active carbon-centered radical by PINO through the HAT pathway. Kinetic experiments showed the activation of NHPI was the rate-determining step. Meanwhile, the generation of PINO radicals was demonstrated by time-resolved EPR tests. It indicated the persistent radicals played an important role in oxidation.Data availability
Data for this paper, including reaction screening and optimization experiments, detailed experimental procedures, spectral data, and computational study details, are provided in the ESI.†Author contributions
X. L. and X. Z. developed the concept, designed these experiments, analyzed the experimental data and wrote the paper. J. S. analyzed the EPR experimental data. H. Y., J. H., C. X., Y. H., Q. H., D. X., and C. X. contributed to catalyst synthesis, catalytic experiments and analyzed the experimental data. X. Z. and H. J. directed the project. All authors discussed the results and provided comments during the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2020YFA0210900), the National Natural Science Foundation of China (No. 21938001 and 21878344), the Guangdong Provincial Key R&D Programme (2019B110206002), and the Local Innovative and Research Teams Project of the Guangdong Pearl River Talents Program (2017BT01C102).Notes and references
- For selected and recent reviews on oxidation reactions, see: (a) Y. Liang, J. Wei, X. Qiu and N. Jiao, Chem. Rev., 2018, 118, 4912–4945 CrossRef CAS PubMed; (b) C. Tang, X. Qiu, Z. Cheng and N. Jiao, Chem. Soc. Rev., 2021, 50, 8067–8101 RSC; (c) E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Glaeser, W. D. Einicke, G. A. Sprenger, U. Beifuss, E. Klemm, C. Liebner, H. Hieronymus, S. F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82–112 CrossRef CAS.
- For selected reviews and studies on C−H functionalization and radical C−H activation, see: (a) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed; (b) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433–9520 CrossRef CAS PubMed; (c) Y. Liu, H. Yi and A. Lei, Chin. J. Chem., 2018, 36, 692–697 CrossRef CAS; (d) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (e) Y. F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653 CrossRef CAS PubMed.
- J. T. Grant, J. M. Venegas, W. P. McDermott and I. Hermans, Chem. Rev., 2018, 118, 2769–2815 CrossRef CAS PubMed.
- For recent reviews on homogeneous catalytic oxidation, see: (a) Z. Zheng, X. Ma, X. Cheng, K. Zhao, K. Gutman, T. Li and L. Zhang, Chem. Rev., 2021, 121, 8979–9038 CrossRef CAS PubMed; (b) L. Marais, H. C. M. Vosloo and A. J. Swarts, Coord. Chem. Rev., 2021, 440, 213958 CrossRef CAS.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC.
- S. R. Docherty, L. Rochlitz, P. A. Payard and C. Coperet, Chem. Soc. Rev., 2021, 50, 5806–5822 RSC.
- For the leading references, see: (a) J. Sheng, B. Yan, W. D. Lu, B. Qiu, X. Q. Gao, D. Wang and A. H. Lu, Chem. Soc. Rev., 2021, 50, 1438–1468 RSC; (b) Y. Wang, P. Hu, J. Yang, Y. A. Zhu and D. Chen, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- Y. Kawamata, M. Yan, Z. Liu, D. H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448–7451 CrossRef CAS PubMed.
- M. S. A. S. Shah, C. Oh, H. Park, Y. J. Hwang, M. Ma and J. H. Park, Adv. Sci., 2020, 7, 2001946 CrossRef PubMed.
- For reviews and studies on galactose oxidase, see: (a) J. Li, I. Davis, W. P. Griffith and A. Liu, J. Am. Chem. Soc., 2020, 142, 18753–18757 CrossRef CAS PubMed; (b) E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed; (c) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901–16910 CrossRef CAS PubMed; (d) J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- For selected reviews on the structure of galactose oxidase, see: (a) W. R. Birmingham and N. J. Turner, ACS Catal., 2018, 8, 4025–4032 CrossRef CAS; (b) J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2363 CrossRef CAS PubMed.
- For the proposed mechanism for the oxidation of alcohols to aldehydes by GOase, see: (a) P. Chaudhuri, M. Hess, T. Weyhermuller and K. Wieghardt, Angew. Chem., Int. Ed., 1999, 38, 1095–1098 CrossRef CAS; (b) H. Oshita and Y. Shimazaki, Chem.–Eur. J., 2020, 26, 8324–8340 CrossRef CAS PubMed.
- W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS.
- N. Kitajima, K. Whang, Y. Moro-oka, A. Uchida and Y. Sasada, J. Chem. Soc., Chem. Commun., 1986, 1504–1505 RSC.
- (a) Y. Wang, J. L. Dubois, B. Hedman, K. O. Hodgson and T. D. P. Stack, Science, 1998, 279, 537–540 CrossRef CAS PubMed; (b) I. L. Fedushkin, O. V. Maslova, A. G. Morozov, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2012, 51, 10584–10587 CrossRef CAS PubMed; (c) T. Glaser, M. Heidemeler, R. Frohlich, P. Hildebrandt, E. Bothe and E. Bill, Inorg. Chem., 2005, 44, 5467–5482 CrossRef CAS PubMed.
- E. Saint-Aman, S. Menage, J. L. Pierre, E. Defrancq and G. Gellon, New J. Chem., 1998, 22, 393–394 RSC.
- P. Chaudhuri, M. Hess, J. Muller, K. Hildenbrand, E. Bill, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS.
- (a) H. Chen, L. Wang, S. Xu, X. Liu, Q. He, L. Song and H. Ji, ACS Catal., 2021, 11, 6810–6815 CrossRef CAS; (b) P. Gandeepan, T. Mueller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- For selected examples with N-oxyl radicals as catalysts, see: (a) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (b) S. Tsujimoto, T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 2001, 2352–2353 RSC; (c) Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed; (d) T. Yoshii, S. Tsuzuki, S. Sakurai, R. Sakamoto, J. Jiang, M. Hatanaka, A. Matsumoto and K. Maruoka, Chem. Sci., 2020, 11, 5772–5778 RSC; (e) M. Petroselli, L. Melone, M. Cametti and C. Punta, Chem.–Eur. J., 2017, 23, 10616–10625 CrossRef CAS PubMed.
- S. Sakaguchi, S. Kato, T. Iwahama and Y. Ishii, Bull. Chem. Soc. Jpn., 1998, 71, 1237–1240 CrossRef CAS.
- Y. Yan, P. Feng, Q. Z. Zheng, Y. F. Liang, J. F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827–5831 CrossRef CAS PubMed.
- (a) E. Baciocchi, M. F. Gerini and O. Lanzalunga, J. Org. Chem., 2004, 69, 8963–8966 CrossRef CAS PubMed; (b) N. Koshino, B. Saha and J. H. Espenson, J. Org. Chem., 2003, 68, 9364–9370 CrossRef CAS PubMed.
- Y. Mo and K. F. Jensen, Chem.–Eur. J., 2018, 24, 10260–10265 CrossRef CAS PubMed.
- C. Zhang, Z. Huang, J. Lu, N. Luo and F. Wang, J. Am. Chem. Soc., 2018, 140, 2032–2035 CrossRef CAS PubMed.
- J. R. Wang, L. Liu, Y. F. Wang, Y. Zhang, W. Deng and Q. X. Guo, Tetrahedron Lett., 2005, 46, 4647–4651 CrossRef CAS.
- E. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- (a) Y. Liu, S. G. Resch, I. Klawitter, G. E. Cutsail, S. Demeshko, S. Dechert, F. E. Kuehn, S. DeBeer and F. Meyer, Angew. Chem., Int. Ed., 2020, 59, 5696–5705 CrossRef CAS PubMed; (b) B. M. Armstrong, R. I. Sayler, B. H. Shupe, T. A. Stich, R. D. Britt and A. K. Franz, ACS Catal., 2019, 9, 1224–1230 CrossRef CAS.
- S. Ghorai, A. Sarmah, R. K. Roy, A. Tiwari and C. Mukherjee, Inorg. Chem., 2016, 55, 1370–1380 CrossRef CAS PubMed.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2000, 200, 991–1008 CrossRef.
- P. Verma, J. Weir, L. Mirica and T. D. P. Stack, Inorg. Chem., 2011, 50, 9816–9825 CrossRef CAS PubMed.
- A. Rana, Y. −M. Lee, X. Li, R. Cao, S. Fukuzumi and W. Nam, ACS Catal., 2021, 11, 3073–3083 CrossRef CAS.
- L. Wang, Y. Zhang, R. Du, H. Yuan, Y. Wang, J. Yao and H. Li, Chemcatchem, 2019, 11, 2260–2264 CrossRef CAS.
- (a) P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS; (b) C. L. Wagner, G. Herrera, Q. Lin, C. T. Hu and T. Diao, J. Am. Chem. Soc., 2021, 143, 5295–5300 CrossRef CAS PubMed.
- S. Roy, B. Dittrich, T. Mondal, D. Koley, A. C. Stueckl, B. Schwederski, W. Kaim, M. John, S. K. Vasa, R. Linser and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 6180–6183 CrossRef CAS PubMed.
- (a) K. C. Mondal, H. W. Roesky, A. C. Stuckl, F. Ehret, W. Kaim, B. Dittrich, B. Maity and D. Koley, Angew. Chem., Int. Ed., 2013, 52, 11804–11807 CrossRef CAS PubMed; (b) B. Kirste, Magn. Reson. Chem., 2016, 54, 835–841 CrossRef PubMed.
- E. Borfecchia, P. Beato, S. Svelle, U. Olsbye, C. Lamberti and S. Bordiga, Chem. Soc. Rev., 2018, 47, 8097–8133 RSC.
- (a) K. Rajabimoghadam, Y. Darwish, U. Bashir, D. Pitman, S. Eichelberger, M. A. Siegler, M. Swart and I. Garcia−Bosch, J. Am. Chem. Soc., 2018, 140, 16625–16634 CrossRef CAS PubMed; (b) W. Kaim, Dalton Trans., 2003, 761–768 RSC; (c) J. Rall, M. Wanner, M. Albrecht, F. M. Hornung and W. Kaim, Chem.–Eur. J., 1999, 5, 2802–2809 CrossRef CAS.
- J. M. Fontmorin, R. C. B. Castillo, W. Z. Tang and M. Sillanpaa, Water Res., 2016, 99, 24–32 CrossRef CAS PubMed.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarstrom, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- S. Wu, Y. He, C. Wang, C. Zhu, J. Shi, Z. Chen, Y. Wan, F. Hao, W. Xiong, P. Liu and H. Luo, ACS Appl. Mater., 2020, 12, 26733–26745 CrossRef CAS PubMed.
- J. Chang, R. D. Taylor, R. A. Davidson, A. Sharmah and T. Guo, J. Phys. Chem. A, 2016, 120, 2815–2823 CrossRef CAS PubMed.
- (a) R. C. Kukreja, E. Okabe, G. M. Schrier and M. L. Hess, Arch. Biochem. Biophys., 1988, 261, 447–457 CrossRef CAS PubMed; (b) M. B. Yim, P. B. Chock and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5006–5010 CrossRef CAS PubMed.
- (a) J. Lalevee, N. Blanchard, M. El−Roz, X. Allonas and J. P. Fouassier, Macromolecules, 2008, 41, 2347–2352 CrossRef CAS; (b) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS.
- Y. Liu, B. Shi, Z. Liu, R. Gao, C. Huang, H. Alhumade, S. Wang, X. Qi and A. Lei, J. Am. Chem. Soc., 2021, 143, 20863–20872 CrossRef CAS PubMed.
- (a) C. Y. Huang, J. Li, W. Liu and C. J. Li, Chem. Sci., 2019, 10, 5018–5024 RSC; (b) A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02606f |
| This journal is © The Royal Society of Chemistry 2022 |