
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bond-induced quantum interference in single-molecule junctions of regioisomers†
Lingbing
Ge‡
a,
Songjun
Hou‡
b,
Yaorong
Chen‡
c,
Qingqing
Wu
b,
Lanxin
Long
a,
Xingzhou
Yang
a,
Yu
Ji
a,
Luchun
Lin
c,
Guodong
Xue
a,
Junyang
Liu
 c,
Xiaodong
Liu
*a,
Colin J.
Lambert
*b,
Wenjing
Hong
*c and
Yonghao
Zheng
*a
c,
Xiaodong
Liu
*a,
Colin J.
Lambert
*b,
Wenjing
Hong
*c and
Yonghao
Zheng
*a
aSchool of Optoelectronic Science and Engineering, University of Electronic Science and Technology of China (UESTC), Chengdu 610054, People's Republic of China. E-mail: zhengyonghao@uestc.edu.cn; xdliu@uestc.edu.cn
bDepartment of Physics, Lancaster University, Lancaster LA1 4YB, UK. E-mail: c.lambert@lancaster.ac.uk
cState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, People's Republic of China. E-mail: whong@xmu.edu.cn
First published on 2nd August 2022
Abstract
Solvents can play a significant role in tuning the electrical conductance of single-molecule junctions. In this respect, protic solvents offer the potential to form hydrogen bonds with molecular backbones and induce electrostatic gating via their dipole moments. Here we demonstrate that the effect of hydrogen bond formation on conductance depends on whether transport through the junction is controlled by destructive quantum interference (DQI) or constructive quantum interference (CQI). Furthermore, we show that a protic solvent can be used to switch the conductance of single-molecule junctions between the two forms of quantum interference. To explore this possibility, two regioisomers (BIT-Zwitterion and BIT-Neutral) were synthesized and their single-molecule conductances in aprotic and protic solvents were investigated using a scanning-tunneling-microscope-based break junction technique, combined with density functional theory and quantum transport theory. We find that the protic solvent twists the geometry of BIT-Zwitterion by introducing intermolecular hydrogen bonds between the solvent and target molecule. Moreover, it increases the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the molecule by imposing different electrostatic gating on the delocalized HOMO and localized LUMO, leading to a lower conductance compared to that in aprotic solvent. In contrast, the conductance of BIT-Neutral increases due to a transformation from DQI to CQI originating from a change from a planar to a folded conformation in the protic solvent. In addition, the stacking between the two folded moieties produces an extra through-space transport path, which further contributes to conductance. This study demonstrates that combinations of protic solvents and regioisomers present a versatile route to controlling quantum interference and therefore single-molecule conductance, by enabling control of hydrogen bond formation, electrostatic gating and through-space transport.
Introduction
The field of single-molecule electronics1 focuses on understanding and controlling charge transport through single molecules that could act as functional components.2–4 To date, significant progress has been made in clarifying the correlations between molecular structure and charge transport,5,6 asymmetric charge transport phenomena under applied bias,7 and the dependence of charge-transport properties on HOMO–LUMO gaps.8 These studies provide important impetus for the development of functional molecular devices, such as rectifiers,9,10 transistors11 and switches.4,12,13 It is known that weak non-covalent intramolecular hydrogen bonds can be broken in protic solvents due to the formation of additional intermolecular interactions14,15 and therefore it is natural to ask if such solvent could be used to tune charge transport through a single-molecule junction. From a fundamental viewpoint, it is also of interest to determine if the effect of hydrogen bonds depends on whether transport through the junction is controlled by destructive quantum interference (DQI) or constructive quantum interference (CQI) and if it is possible to use solvents to switch between the two interference patterns.To investigate this possibility, we functionalized [2,2′-Bi-1H-indene]-3,3′-dihydroxy-1,1-dione (BIT-OH2)16 with 4-(Methylthio)aniline under different conditions to form two regioisomers (zwitterionic configuration BIT-Zwitterion17 and neutral configuration BIT-Neutral). These were chosen, because our theoretical calculation predicts that in the absence of a solvent, transport through the BIT-Zwitterion is controlled by CQI, whereas for the BIT-Neutral, it is controlled by DQI (Scheme 1). Although both molecules have similar types of hydrogen bonds, BIT-Zwitterion possesses stronger intramolecular O–H bonds compared to BIT-Neutral. Furthermore, the two isomers have different charge distributions and distinct electrostatic potentials. Our theoretical calculations show that BIT-Zwitterion possesses a much larger dipole moment and a more localized charge distribution than BIT-Neutral. Moreover, the intramolecular hydrogen bonds in both molecules can be broken in protic solvents, as evidenced by UV-vis-NIR absorption and single crystal structures. Hence, we used a scanning-tunnelling-microscope-based break junction (STM-BJ) technique to measure the conductance of BIT-Zwitterion and BIT-Neutral under different solvent environments, in which the formation of intermolecular hydrogen bonds and breaking of intramolecular hydrogen bonds can be controlled without changing their chemical structures (Scheme 1). The different hydrogen bonds in the two regioisomers lead to significantly different intermolecular interactions between dissolved molecules and solvent. In the protic solvent, BIT-Zwitterion shows a lower conductance compared with that in the aprotic solvent, while in the case of BIT-Neutral, the conductance in the protic solvent is significantly higher than that in an aprotic solvent. More specifically, the protic solvent causes the conductance decrease of BIT-Zwitterion compared to that in an aprotic solvent, because it forms intermolecular hydrogen bonds which induces a twist in the BIT-Zwitterion. Furthermore, the dipole moments of these hydrogen bonds impose different electrostatic gating on the delocalized HOMO and localized LUMO resulting in increased HOMO–LUMO gap of BIT-Zwitterion, which further contributes to a lower conductance. In contrast, the protic solvent causes the conductance increase of BIT-Neutral compared to that in an aprotic solvent, because it induces a transformation from DQI to CQI due to a change from a planar to a folded conformation in the protic solvent. Furthermore, an extra through-space transport path is created between the two folded moieties, leading to an additional increase in the conductance of BIT-Neutral. These distinct behaviors of BIT-Zwitterion and BIT-Neutral demonstrate that their response to a protic solvent indeed reflects the control of their transport properties by quantum interference.
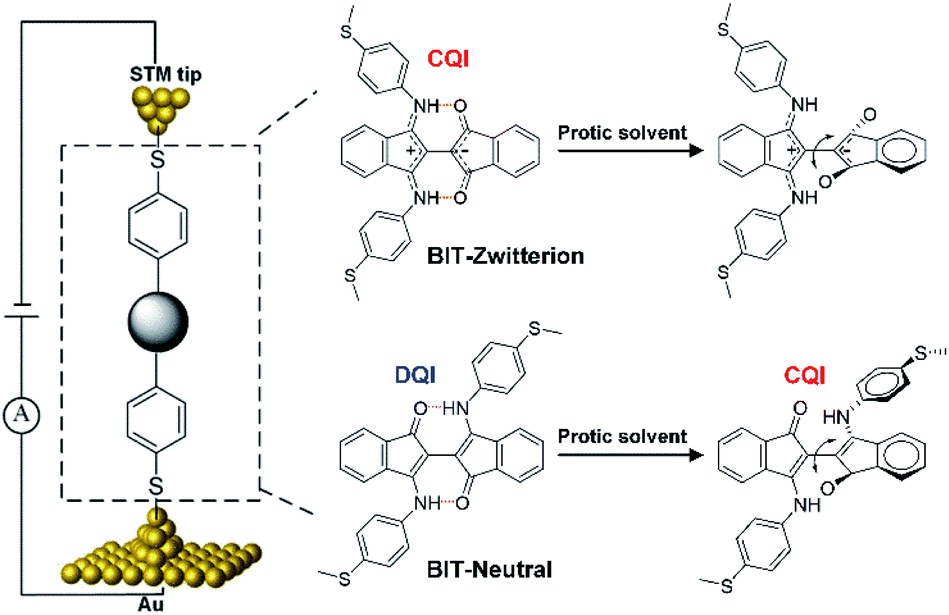 | ||
| Scheme 1 Schematic of STM-BJ setup and the two BIT isomers. | ||
Results and discussion
Material syntheses and characterizations
The target molecules were synthesized according to the synthetic routes illustrated in Scheme S1,† and as described in detail in Section S1 of ESI.† Both compounds can be obtained in reasonable yields and were characterized by 1H and 13C nuclear magnetic resonance (NMR) spectroscopies (see Fig. S1 and S2†). These two molecules exhibit intramolecular hydrogen bonds and thus have planar cores in aprotic solvents. Moreover, we can easily control the formation and destruction of intramolecular hydrogen bonds by adding the protic solvents, as indicated by UV-vis-NIR absorption spectroscopies and single-crystal X-ray diffraction analyses.The optical properties of BIT-Zwitterion and BIT-Neutral in dichloromethane (DCM) and ethanol (EtOH) were investigated by UV-vis-NIR spectroscopy at room temperature, and their absorption spectra are shown in Fig. S3c.† In aprotic solvents like DCM and 1,2,4-trichlorobenzene (TCB), BIT-Neutral displays two intense absorption bands in the UV region with the absorption peaks (λmax) at 286 and 367 nm, along with weak absorption in the visible region (Table S1†). The less pronounced absorption from 448 to 740 nm may be attributed to intramolecular charge transfer interactions. In contrast, BIT-Zwitterion shows a broader absorption with a similar profile, and the absorption is extended into the NIR region with a λmax at 717 nm compared to the BIT-Neutral (570 nm) due to the stronger intramolecular charge transfer interactions of the zwitterionic state. Interestingly, both isomers dissolved in protic solvents present obvious changes in the absorption spectra. For BIT-Zwitterion, the absorption intensity in the NIR region deceases after adding the protic solvent, which suggests that in protic solvents, such as ethanol and mixed solvent of ethanol and propylene carbonate (PC), BIT-Zwitterion prefers a twisted conformation (Fig. S3a†), which agrees with our previous studies.17 Notably, similar solvent-dependent changes are also observed for BIT-Neutral (Fig. S3b†), which can be ascribed to the competition between intramolecular and intermolecular hydrogen bonds resulting in the destruction of the planar molecular structure, in agreement with the single crystal results (Fig. 1c and S5†). As shown in Fig. 1b, BIT-Zwitterion and BIT-Neutral were dissolved in the solvents used for single-molecule testing and characterized by UV-vis-NIR absorption spectroscopy (Fig. 1a), which suggests that both BIT-Zwitterion and BIT-Neutral prefer twisted configurations. Furthermore, cyclic voltammetry (CV) measurements were performed to investigate the electrochemical properties of these two regioisomers in dry DCM (see Table 1 and Fig. S4†). Both BIT-Zwitterion and BIT-Neutral display two reversible one-electron oxidation waves. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) values of BIT-Zwitterion are calculated to be −4.82 and −3.52 eV, respectively, and those for BIT-Neutral are −4.89 and −3.19 eV, respectively.
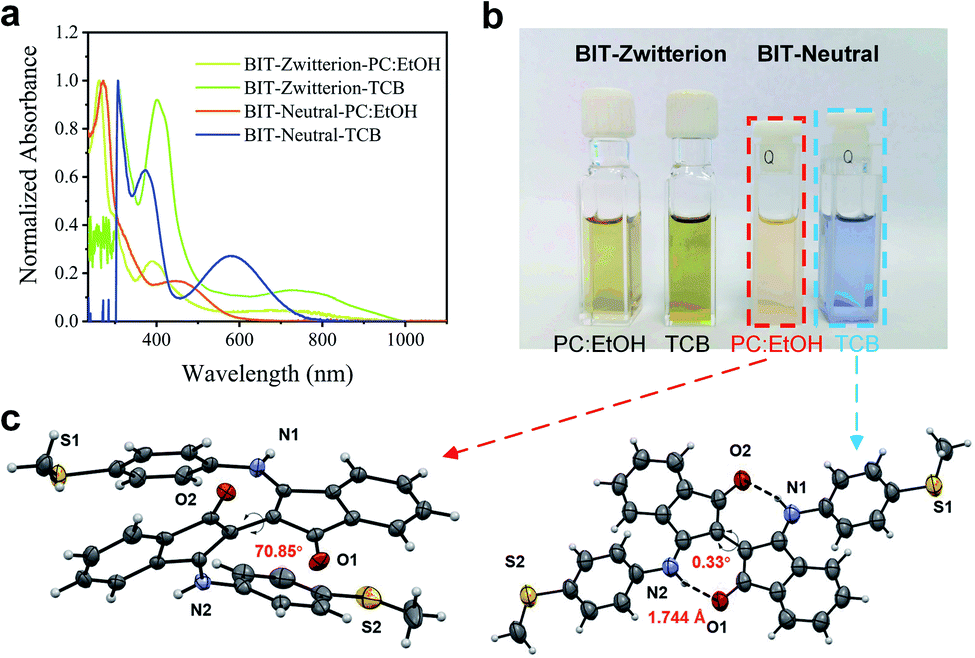 | ||
| Fig. 1 The optical and crystallographic properties of BIT-Zwitterion and BIT-Neutral. (a) Normalized UV-vis-NIR absorption spectra and (b) photographs of BIT-Zwitterion and BIT-Neutral in PC:EtOH and TCB solutions. (c) The single crystal structures of BIT-Neutral with twisted and coplanar configurations. | ||
Single-molecule conductance measurements
The electrical properties of single-molecule junctions were measured by the STM-BJ technique. The solutions of target molecules (0.1 mM) were separately prepared in TCB or a mixed solvent of PC and ethanol (PC:EtOH, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v) to investigate their charge transport properties when placed in a molecular junction. To avoid the noise caused by the polarity of the solvent, the gold tips were etched and encapsulated with Apiezon wax. As shown in Fig. 2c and d, both the two-dimensional (2D) conductance versus relative displacement histograms of BIT-Zwitterion under different solvents exhibit a clear peak at G0 (quantum conductance, G0 = 2e2/h), associated with the gold contact.18 The evident conductance intensity clouds of the molecules indicate that the molecular junctions are well established during the break–junction cycles. Compared with the experiment under TCB solvent (Fig. 2c), PC:EtOH shows two conductance intensity clouds around 10−2G0, which is considered to be the conductance of mercaptoaminobenzene (Fig. S8†), and around 10−4G0, which presents a more obvious low conductance distribution (Fig. 2d). Fig. 2a compares the typical individual conductance traces from BIT-Zwitterion in TCB and PC:EtOH solutions. These traces show that the conductance of BIT-Zwitterion in PC:EtOH is lower than that in TCB. Fig. 2b presents 1D conductance histograms generated from over 2000 independent traces. Through Gaussian fitting, Fig. S9a and b† reveal that the conductance of BIT-Zwitterion in TCB is located at 10−3.67G0. In contrast, when the solvent is changed to the mixture of PC and EtOH, BIT-Zwitterion yields a lower conductance peak at 10−3.96G0. From the 2D conductance versus relative displacement histograms of BIT-Zwitterion in TCB and PC:EtOH (inset of Fig. 2c and d), the peaks of the length displacement distributions of junctions are located at about 0.9 nm. With 0.5 nm distance caused by the gold–gold snap-back effect, the molecular junctions are determined to be about 1.4 nm, which is consistent with the length of molecules, indicating that the characteristics of conductance feature mainly come from the fully-extended molecule.19 In other words, the formation or elimination of hydrogen bonds has an obvious effect on conductance, which demonstrates that the solvent gating effect is significant for BIT-Zwitterion in single-molecule junctions.
:2, v/v) to investigate their charge transport properties when placed in a molecular junction. To avoid the noise caused by the polarity of the solvent, the gold tips were etched and encapsulated with Apiezon wax. As shown in Fig. 2c and d, both the two-dimensional (2D) conductance versus relative displacement histograms of BIT-Zwitterion under different solvents exhibit a clear peak at G0 (quantum conductance, G0 = 2e2/h), associated with the gold contact.18 The evident conductance intensity clouds of the molecules indicate that the molecular junctions are well established during the break–junction cycles. Compared with the experiment under TCB solvent (Fig. 2c), PC:EtOH shows two conductance intensity clouds around 10−2G0, which is considered to be the conductance of mercaptoaminobenzene (Fig. S8†), and around 10−4G0, which presents a more obvious low conductance distribution (Fig. 2d). Fig. 2a compares the typical individual conductance traces from BIT-Zwitterion in TCB and PC:EtOH solutions. These traces show that the conductance of BIT-Zwitterion in PC:EtOH is lower than that in TCB. Fig. 2b presents 1D conductance histograms generated from over 2000 independent traces. Through Gaussian fitting, Fig. S9a and b† reveal that the conductance of BIT-Zwitterion in TCB is located at 10−3.67G0. In contrast, when the solvent is changed to the mixture of PC and EtOH, BIT-Zwitterion yields a lower conductance peak at 10−3.96G0. From the 2D conductance versus relative displacement histograms of BIT-Zwitterion in TCB and PC:EtOH (inset of Fig. 2c and d), the peaks of the length displacement distributions of junctions are located at about 0.9 nm. With 0.5 nm distance caused by the gold–gold snap-back effect, the molecular junctions are determined to be about 1.4 nm, which is consistent with the length of molecules, indicating that the characteristics of conductance feature mainly come from the fully-extended molecule.19 In other words, the formation or elimination of hydrogen bonds has an obvious effect on conductance, which demonstrates that the solvent gating effect is significant for BIT-Zwitterion in single-molecule junctions.
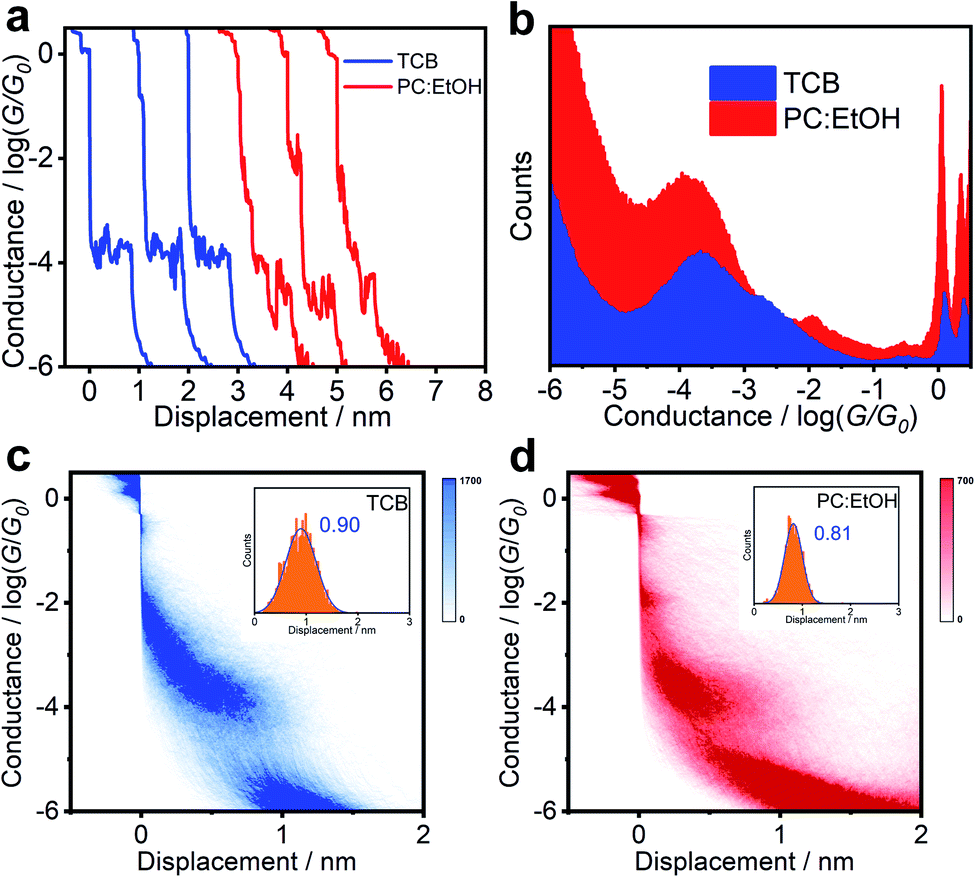 | ||
| Fig. 2 Results of BIT-Zwitterion without any data selection. (a) Conductance-displacement traces, and (b) 1D conductance histograms of BIT-Zwitterion in TCB and PC:EtOH solutions. (c and d) 2D conductance-displacement histograms, and relative displacement distributions (inset) of BIT-Zwitterion in (c) TCB and (d) PC:EtOH solutions. | ||
The response of BIT-Neutral to the different solvents is in marked contrast with the above behaviour. As shown in Fig. 3b and S9c and d,† after changing the aprotic solvent (TCB) with protic solvent (PC:EtOH), the statistical electrical signal analysis of BIT-Neutral molecular junction shows that the conductance increases from 10−4.39G0 to 10−3.96G0, which is inconsistent with the conductance change compared to BIT-Zwitterion. Fig. 3c and d display the 2D conductance versus relative displacement histograms of BIT-Neutral constructed from over 2000 single-molecule conductance-displacement traces (Fig. 3a). The BIT-Neutral molecule becomes difficult to be connected to the metallic electrodes in a polar solvent environment, which arises from the twisted core between two terminal groups, compared to zwitterionic configuration with well planarity. Therefore, we chose the data with distinct plateau which means good connection to gold electrodes. Moreover, the length of BIT-Neutral junction under the polar solvent condition is reduced to be ∼0.5 nm (inset of Fig. 3d), while that of BIT-Zwitterion remains unchanged. The difference in the junction length suggests the destruction of molecular planarity due to the cleavage of the intramolecular hydrogen bond, which has a greater impact on BIT-Neutral (confirmed by single ctrystals from 1.71 to 1.04 nm). More interestingly, from the results of single crystal structures, the twisted form of BIT-Neutral appears to be folded, so the conductance variation originates from the length changes and the existence of π–π stacking to compensate for the conductance (Fig. 1c). These results demonstrate that the solvent gating effect is not significant for BIT-Neutral in single-molecule junctions.
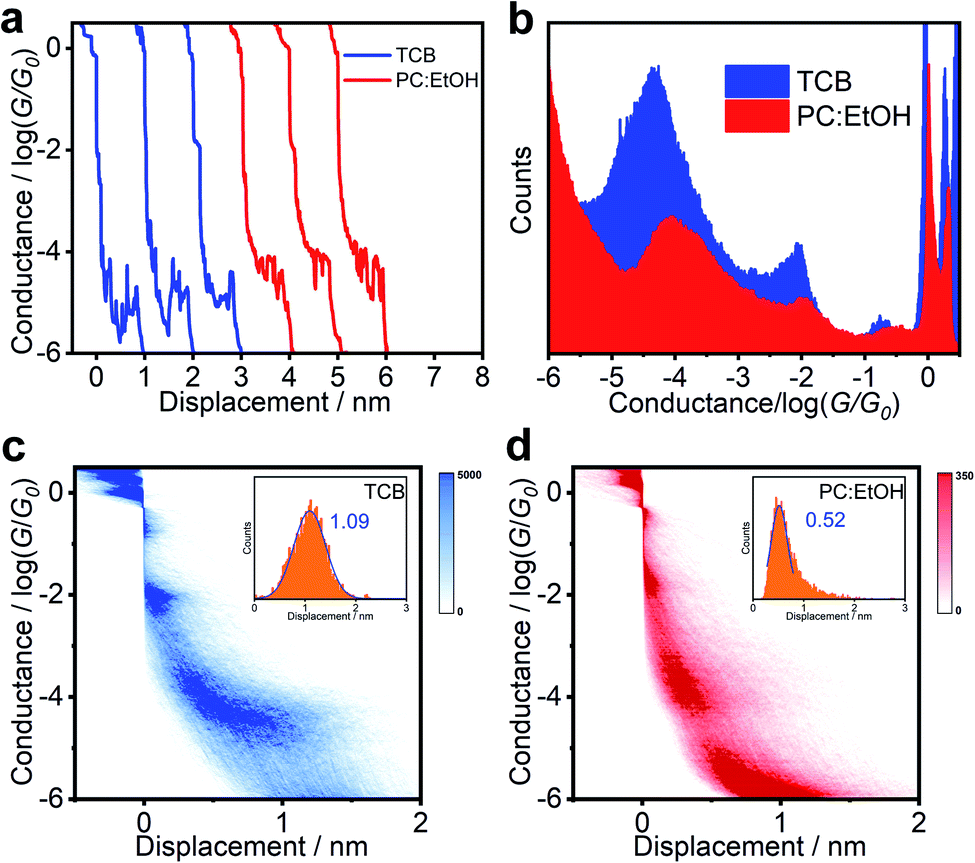 | ||
| Fig. 3 Results of BIT-Neutral without any data selection. (a) Conductance-displacement traces, and (b) 1D conductance histograms of BIT-Neutral in TCB and PC:EtOH solutions. (c and d) 2D conductance-displacement histograms, and relative displacement distributions (inset) of BIT-Neutral in (c) TCB and (d) PC:EtOH solutions. | ||
DFT calculations
To clarify the mechanisms for the effect of the protic solvent on regioisomers, the electronic structures and transmission functions T(E) of the isomers are calculated by combining density functional theory (DFT)20 with quantum transport theory21 (see Methods for further details in Section S7 of ESI†). The cores of both BIT molecules possess planar groups formed from fused 5- and 6-membered rings. First, the BIT-Zwitterion molecule is fully relaxed in the gas phase, where it is found that the pendant groups are coplanar forming a well conjugated structure. Then the influence of protic solvent (ethanol) on BIT-Zwitterion is investigated by employing a SMD solvation model.22,23 After relaxation, this results in a twisted geometry, with an angle of ∼30° between the planes of the planar groups. Since the HOMO–LUMO (HL) gap creates an effective energy barrier to electron transport through a molecular junction, we studied the change in the HL gap due to this twist, both with and without protic solvent molecules. As shown in Fig. 4a, the HL gap of gas phase BIT-Zwitterion is 0.88 eV and reduces to 0.48 eV when the planar groups are artificially twisted to an angle of 30°, in the absence of the solvent. Fig. S10† shows that this is a reflection of the fact that as the angle increases, the coupling between these two pendant moieties decreases and the HL gap gradually shrinks. When BIT-Zwitterion is surrounded by solvent molecules, the HL gap is further impacted by the gating effect of the surrounded protic solvent.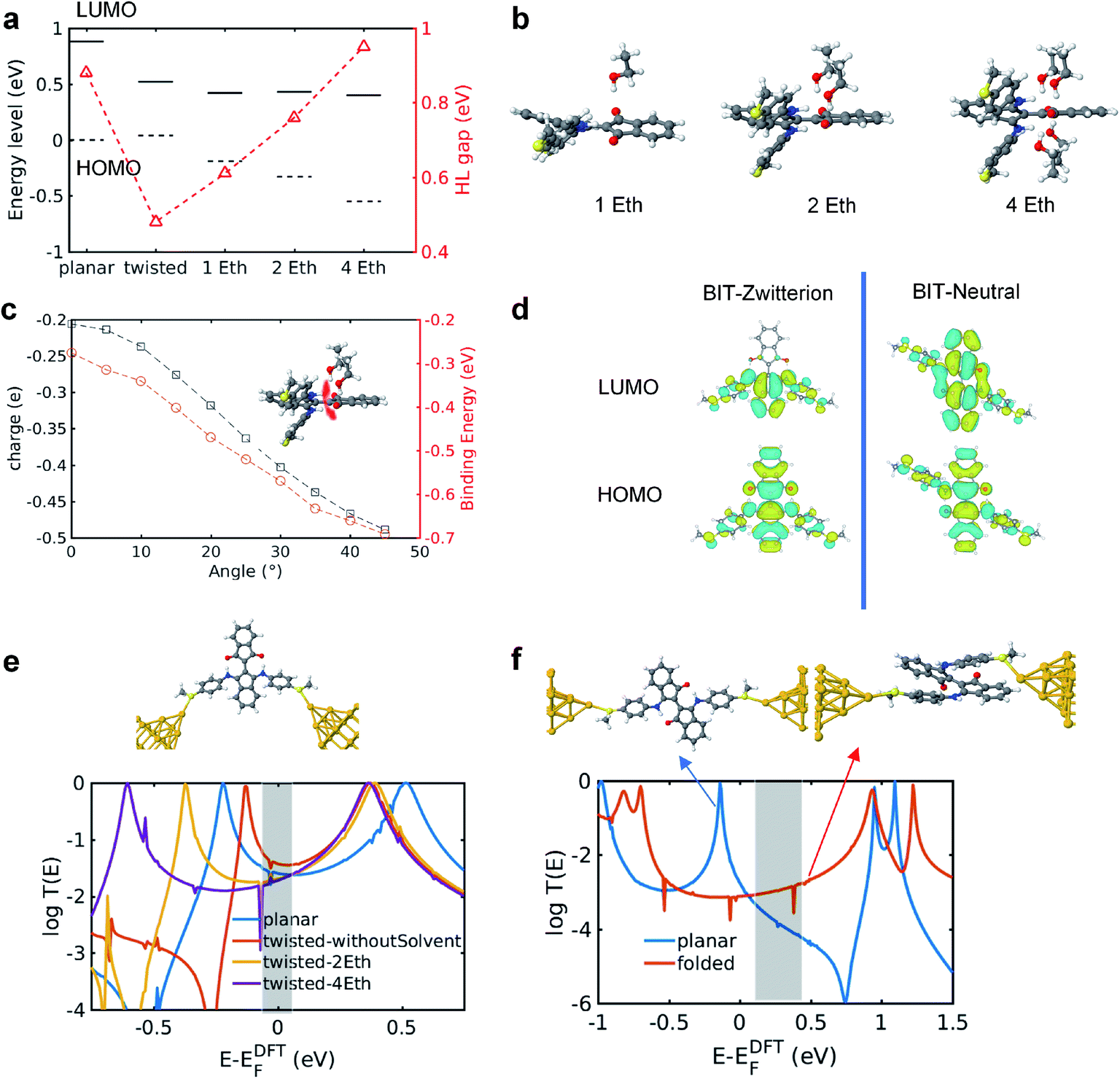 | ||
| Fig. 4 Electronic properties of BIT-Zwitterion and BIT- Neutral. (a) The dependence of HOMO and LUMO energy levels on twist angle and on the presence of 1, 2 and 4 ethanol molecules. The evolution of the HL gap is indicated by red triangles. (b) The relaxed conformations with one, two and four ethanol molecules binding to the pendant group. (c) The charge located on the pendant group and the binding energy between BIT-Zwitterion and two ethanol molecules as a function of rotation angle. (d) The HOMO and LUMO for BIT-Zwitterion and BIT-Neutral. (e) The transmission functions for planar and twisted BIT-Zwitterion without solvent, and in the presence of two ethanol and four ethanol molecules as shown in (b). ‘planar’ BIT-Zwitterion indicates results obtained in aprotic solvent. (f) The transmission functions for planar and folded BIT-Neutral. The range of possible Fermi energies yielding agreement with experimental trends is indicated by the shaded grey area. ‘planar’ BIT-Neutral indicates results obtained in aprotic solvent and ‘folded’ BIT-Neutral indicates results obtained in protic solvent. | ||
To distinguish this from the effect of twisting, it is convenient to start from an artificially twisted BIT-Zwitterion, and then compute the change in the HL gap, when a succession of 1, 2 and 4 ethanol molecules are placed close to the pendant group. The proton of the hydroxyl group initially points to the oxygen of the pendant group (Fig. 4b). This leads to the formation of hydrogen bonds after the geometrical relaxation. As shown in Fig. 4c, the binding energy between BIT-Zwitterion and the two ethanol molecules increases from −0.28 eV to −0.69 eV as the twist angle increases from 0° to 30°. This indicates a robust gating effect from the protic solvent originating from stronger coulomb interaction due to the increasing negative charge located on pendant group (Fig. 4c). The proton of protic solvent ethanol imposes a positive electrostatic potential on the molecular orbitals, whose effect on a given energy level depends on the spatial distribution of the corresponding molecular orbital. In the case of BIT-Zwitterion, the HOMO is delocalized nearly evenly on both the backbone and the pendant group, while the LUMO is localized mainly on the backbone (Fig. 4d). For BIT-Zwitterion, since the proton is close to pendant group in Fig. 4d, the HOMO shifts downwards in energy, while the LUMO energy is almost unchanged. This gating effect causes the HL gap to increase from 0.48 eV to 0.6 eV and 0.76 eV for one and two ethanol respectively and further to 0.95 eV in the presence of four ethanols; the latter is higher than the HL gap of planar BIT-Zwitterion (Fig. 4a). This behavior is rationalized with the framework of perturbation theory.15,24 The BIT-Zwitterion contains two parts (a pendant group and the backbone) which could be regarded as a two-level system (Fig. S11†). Based on the DFT-calculated orbitals, the HOMO and LUMO amplitudes on each moiety are of the form 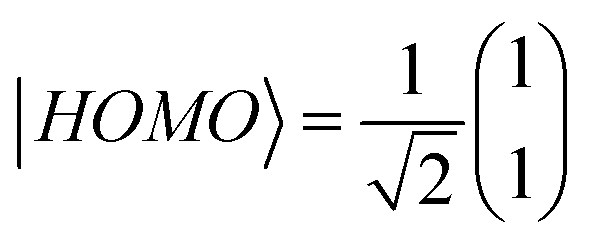 and
and 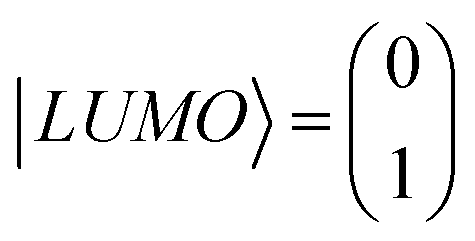 respectively. A positive-gating perturbation acting on the upper level is described by a Hamiltonian
respectively. A positive-gating perturbation acting on the upper level is described by a Hamiltonian 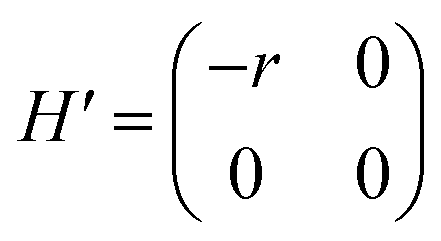 , where r is a small positive value. Then the energy level shifts are given by
, where r is a small positive value. Then the energy level shifts are given by 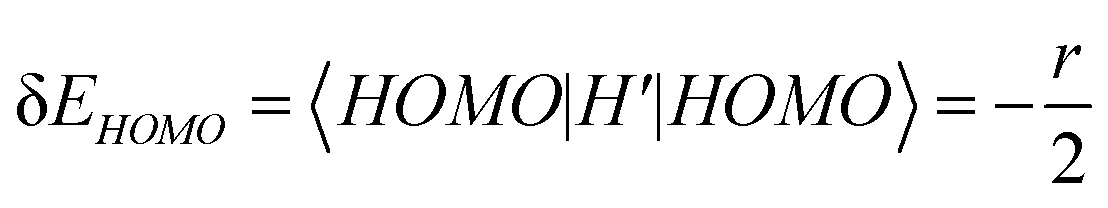 , and
, and 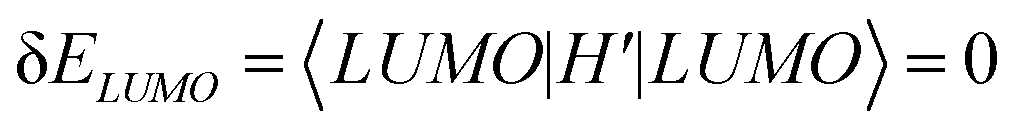 . Consequently, we predict an increase in the HL gap of
. Consequently, we predict an increase in the HL gap of 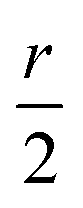 . The second type of hydrogen bond is also considered in this two-level model and DFT calculation where the oxygen of the hydroxyl points towards the nitrogen of the backbone. Similar increase in the HL gap is observed (see Section S8 of ESI and Table S2† where the second type and the mixture of two types of hydrogen bonds are both calculated). In contrast, in the case of BIT-Neutral, both the HOMO and LUMO are distributed evenly across the whole molecule resulting in almost no change in the HL gap in the presence of protic solvent (Fig. 4d).
. The second type of hydrogen bond is also considered in this two-level model and DFT calculation where the oxygen of the hydroxyl points towards the nitrogen of the backbone. Similar increase in the HL gap is observed (see Section S8 of ESI and Table S2† where the second type and the mixture of two types of hydrogen bonds are both calculated). In contrast, in the case of BIT-Neutral, both the HOMO and LUMO are distributed evenly across the whole molecule resulting in almost no change in the HL gap in the presence of protic solvent (Fig. 4d).
The transmission functions of BIT-Zwitterion and BIT-Neutral and the corresponding junctions are shown in Fig. 4e and f, with the latter showing a clear DQI transmission dip at an energy of 0.75 eV (relative to the DFT-predicted Fermi energy). The two resonances of each curve correspond to the HOMO and LUMO of the gas-phase molecule. For the twisted BIT-Zwitterion without protic solvent, the decreased HL gap leads to an increased transmission over a large energy range in the HL gap (red curve in Fig. 4e) compared to the planar geometry (blue curve). However, when the gating effect of the protic solvent is considered, the increased HL gap ends up with a decreased conductance for two ethanol molecules (yellow curve) and decreases further with four ethanol molecules (purple curve) agreeing well with the experiment, where a decrease in conductance is observed after changing the aprotic solvent with protic solvent (Table 1). In the case of planar BIT-Neutral, a transmission dip (blue curve in Fig. 4f) appears in the vicinity of LUMO resonance. This arises in part from DQI between the HOMO and LUMO, as indicated by an orbital product rule, which notes that DQI occurs when the terminal amplitudes of the HOMO and LUMO at opposite ends of the molecule possess the same symmetry.25–30 Indeed, as shown in the right panel of Fig. 4d and the left panel of Fig. S12,† for both the HOMO and LUMO, the amplitude on the left end of the molecule is opposite relative to that on the right. When BIT-Neutral becomes folded in protic solvent, the terminal amplitudes of the HOMO and LUMO now have opposite symmetries (right panel of Fig. S12†) and the DQI dip is removed, as shown by the red curve in Fig. 4f. This CQI increases the conductance, even though the gap between the HL resonances is increased by around 0.5 eV. Transport is also facilitated by the stacking between the two folded moieties, which promotes through-space tunnelling. These features combine to increase the transmission coefficient over a large energy range (indicated by the shaded grey region in Fig. 4f) in the HL gap. Past comparisons between theory and experiments suggest that the Fermi energy lies in the vicinity of the middle of the HOMO–LUMO gap. It should be noted that determining the precise alignment of molecular orbitals with the Fermi energy of gold electrodes is challenging from both experimental and theoretical points of view, due to several factors such as the charge transfer between the electrodes and the molecule, and the screening charges in the electrodes shifting molecular energy levels, which depend on the unknown atomic-scale shapes of the electrodes and the environmental effects. In practice, it seems that the only solution is to compare transmission curves with transport measurements and identify ranges of possible Fermi energies, such as the shaded regions in Fig. 4, which agree with the measured trends. In this sense, DFT is not predictive, but can be useful in post-rationalising experiments. Regarding the interference effects of BIT-Neutral, numerical evidence shows a change in the relative phase in the LUMO at the S attachment points due to twisting of the BIT-Neutral (Fig. S12†). Here we provide further physical explanation as follows.
The two branches are connected to the upper and lower parts of backbone respectively as indicated by the black circle in Fig. 5a and b. Since the backbone part of the HOMO (blue dashed circle in Fig. 5a) is symmetric along the black dashed line shown in Fig. 5a, the phase of the two –S anchors does not change before and after folding, as demonstrated in Fig. 5a. However, since the backbone part of the LUMO (blue dashed circle in Fig. 5b) is asymmetric along the dashed black line shown in Fig. 5b, the phase of the two –S anchors change from opposite to the same, before and after folding, as revealed by Fig. 5b. The key point for the above results is that the spatial distribution of molecular states on the backbone does not follow the geometrical rotation of the upper part of the molecule (see the unchanged colors in blue dashed circles in Fig. 5a and b). More specifically, as shown in Fig. 5a and b, for both the HOMO and LUMO, the phase of backbone persists, which means the colors do not change. However, the branch 1 rotates to the right part from the left geometrically. Therefore, the phase on the –S anchor of the HOMO persists while that of LUMO changes. According to the orbital product rule (see details in ref. 25), the quantum interference (QI) of BIT-Neutral will change from DQI to CQI after folding. In contrast, in the case of BIT-Zwitterion, the two branches are both connected to the lower parts of the backbone shown in Fig. 4d. Therefore, the rotation of the upper part will not affect the phase of the lower part of the two branches, which means the QI pattern will not change before and after the twist.
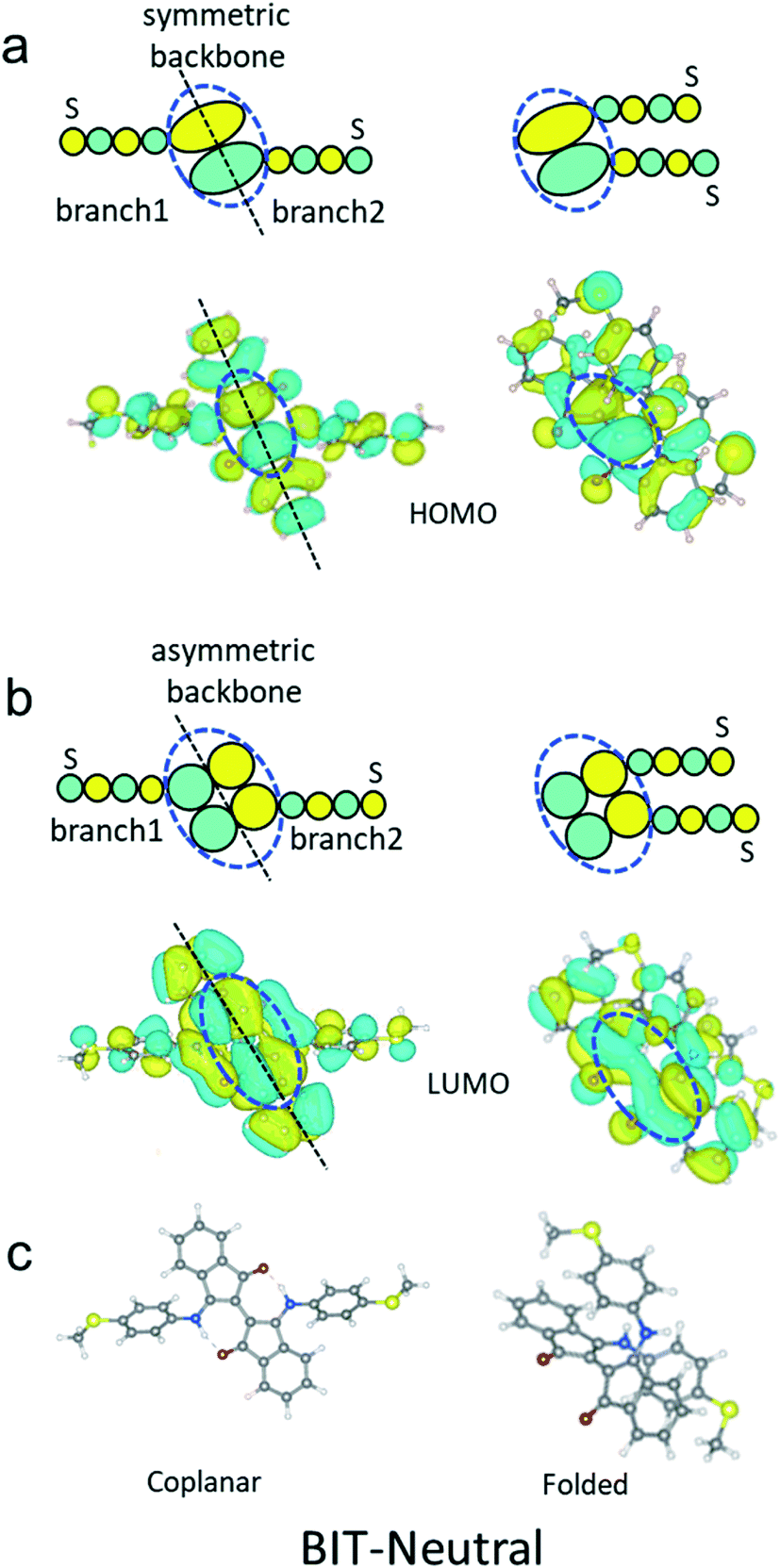 | ||
| Fig. 5 Symmetries of molecular orbitals HOMO and LUMO for coplanar and folded BIT-Neutral. (a) Schematic and DFT-based HOMO before and after folding. (b) Schematic and DFT-based LUMO before and after folding. (c) Structures of coplanar and folded BIT-Neutral. The part in the blue dashed circle is denoted as ‘backbone’, the left linker and anchor –S are marked as ‘branch1’ and the right linker and anchor –S are marked as ‘branch2’. | ||
Conclusions
To conclude, two regioisomers were synthesized and their single-molecule conductance properties were characterized. Both molecules possess hydrogen bonds, but the effects of hydrogen bonds are completely different, because in the absence of a solvent, transport through BIT-Zwitterion is controlled by CQI, whereas in BIT-Neutral, it is controlled by DQI. We find that the protic solvent twists the geometry of BIT-Zwitterion by introducing an intermolecular hydrogen bond between the solvent and target molecule, leading to a decrease in the HL gap. On the other hand, it increases the HL gap of the molecule by imposing a different electrostatic gating on the delocalized HOMO and localized LUMO. These competing effects lead to a lower conductance compared to that in aprotic solvent. In contrast, the conductance of BIT-Neutral increases due to a transformation from DQI to CQI originating from a change from a planar to a folded conformation in the protic solvent. In addition, the stacking between the two folded moieties produces an extra through-space transport path, which further contributes to conductance. This study demonstrates that combinations of protic solvents and regioisomers present a versatile route to controlling single-molecule conductance and quantum interference, by enabling control of hydrogen bond formation, electrostatic gating and through-space transport.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI materials.†Author contributions
Y. Z., C. L., W. H. and X. L. conceived the idea, designed the experiments and co-supervised the project. L. G. carried out material synthesis, characterization. L. G., S. H. and Q. W. wrote the original draft. S. H., Q. W. and G. X. performed the calculations. J. L., Y. C. and L. L. carried out STM-BJ experiments. L. L. and X. Y. participated in discussions and data analysis. Y. J. performed the UV-vis-NIR absorption and CV measurements. All authors contributed to the discussions and gave comments on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by National Natural Science Foundation of China (61805034 and 21722305) and the UK EPSRC, through grant nos. EP/M014452/1, EP/P027156/1 and EP/N03337X/1.Notes and references
- N. Xin, J. Guan, C. Zhou, X. Chen, C. Gu, Y. Li, M. A. Ratner, A. Nitzan, J. F. Stoddart and X. Guo, Nat. Rev. Phys., 2019, 1, 211–230 Search PubMed.
- I. Diez-Perez, J. Hihath, T. Hines, Z. S. Wang, G. Zhou, K. Mullen and N. Tao, Nat. Nanotechnol., 2011, 6, 226–231 CAS.
- T. Hines, I. Diez-Perez, H. Nakamura, T. Shimazaki, Y. Asai and N. Tao, J. Am. Chem. Soc., 2013, 135, 3319–3322 CAS.
- M. C. Walkey, C. R. Peiris, S. Ciampi, A. C. Aragonès, R. B. Dominguez-Espindola, D. Jago, T. Pulbrook, B. W. Skelton, A. N. Sobolev, I. Diez Perez, M. J. Piggott, G. A. Koutsantonis and N. Darwish, ACS Appl. Mater. Interfaces, 2019, 11, 36886–36894 CAS.
- S. Pan, Q. Fu, T. Huang, A. Zhao, B. Wang, Y. Luo, J. Yang and J. Hou, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15259 CAS.
- L. Venkataraman, J. E. Klare, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nature, 2006, 442, 904–907 CAS.
- H. Ren, G. Zhang, N. Lin, L. Deng, Y. Luo and F. Huang, Phys. Chem. Chem. Phys., 2016, 18, 26586–26594 CAS.
- J. Liu, X. Zhao, Q. Al-Galiby, X. Huang, J. Zheng, R. Li, C. Huang, Y. Yang, J. Shi, D. Z. Manrique, C. J. Lambert, M. R. Bryce and W. Hong, Angew. Chem., Int. Ed., 2017, 56, 13061–13065 CAS.
- J. Hihath, C. Bruot, H. Nakamura, Y. Asai, I. Díez-Pérez, Y. Lee, L. Yu and N. Tao, ACS Nano, 2011, 5, 8331 CAS.
- D. Q. Andrews, G. C. Solomon, R. P. V. Duyne and M. A. Ratner, J. Am. Chem. Soc., 2008, 130, 17309–17319 CAS.
- B. Limburg, J. O. Thomas, J. K. Sowa, K. Willick, J. Baugh, E. M. Gauger, G. A. D. Briggs, J. A. Mol and H. L. Anderson, Nanoscale, 2019, 11, 14820–14827 CAS.
- F. Schwarz, G. Kastlunger, F. Lissel, C. Egler-Lucas, S. N. Semenov, K. Venkatesan, H. Berke, R. Stadler and E. Lortscher, Nat. Nanotechnol., 2016, 11, 170–176 CAS.
- N. Xin, J. Wang, C. Jia, Z. Liu, X. Zhang, C. Yu, M. Li, S. Wang, Y. Gong, H. Sun, G. Zhang, Z. Liu, G. Zhang, J. Liao, D. Zhang and X. Guo, Nano Lett., 2017, 17, 856–861 CAS.
- D. C. Milan, O. A. Al-Owaedi, M.-C. Oerthel, S. Marqués-González, R. J. Brooke, M. R. Bryce, P. Cea, J. Ferrer, S. J. Higgins, C. J. Lambert, P. J. Low, D. Z. Manrique, S. Martin, R. J. Nichols, W. Schwarzacher and V. M. García-Suárez, J. Phys. Chem. C, 2016, 120, 15666–15674 CAS.
- Z. Tang, S. Hou, Q. Wu, Z. Tan, J. Zheng, R. Li, J. Liu, Y. Yang, H. Sadeghi, J. Shi, I. Grace, C. J. Lambert and W. Hong, Sci. Bull., 2020, 65, 944–950 CAS.
- L. Dou, Y. Zheng, X. Shen, G. Wu, K. Fields, W. C. Hsu, H. Zhou, Y. Yang and F. Wudl, Science, 2014, 343, 272–277 CAS.
- Y. Zheng, M. S. Miao, Y. Zhang, T. Q. Nguyen and F. Wudl, J. Am. Chem. Soc., 2014, 136, 11614–11617 CAS.
- L. Lin, C. Tang, G. Dong, Z. Chen, Z. Pan, J. Liu, Y. Yang, J. Shi, R. Ji and W. Hong, J. Phy. Chem. C, 2021, 125, 3623–3630 CAS.
- J. Liu, X. Zhao, J. Zheng, X. Huang, Y. Tang, F. Wang, R. Li, J. Pi, C. Huang, L. Wang, Y. Yang, J. Shi, B.-W. Mao, Z. Q. Tian, M. R. Bryce and W. Hong, Chem, 2019, 5, 390–401 CAS.
- J. e. M. Soler, E. Artacho, J. D. Gale, A. Garćıa, J. Junquera, P. Ordejon and D. Śanchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CAS.
- J. Ferrer, C. J. Lambert, V. M. García-Suárez, D. Z. Manrique, D. Visontai, L. Oroszlany, R. Rodríguez-Ferradás, I. Grace, S. W. D. Bailey, K. Gillemot, H. Sadeghi and L. A. Algharagholy, New J. Phys., 2014, 16, 093029 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03; Gaussian Inc.: Wallingford, Ct. 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CAS.
- P. Puczkarski, Q. Wu, H. Sadeghi, S. Hou, A. Karimi, Y. Sheng, J. H. Warner, C. J. Lambert, G. A. D. Briggs and J. A. Mol, ACS Nano, 2018, 12, 9451–9460 CAS.
- C. J. Lambert and S. X. Liu, Chem.– Eur. J., 2018, 24, 4193–4201 CAS.
- C. J. Lambert, Chem. Soc. Rev., 2015, 44, 875–888 CAS.
- C. J. Lambert, Quantum Transport in Nanostructures and Molecules, IOP Publishing, 2021 Search PubMed.
- K. Yoshizawa, Acc. Chem. Res., 2012, 45, 1612–1621 CAS.
- H. Zheng, S. Hou, C. Xin, Q. Wu, F. Jiang, Z. Tan, X. Zhou, L. Lin, W. He, Q. Li, J. Zheng, L. Zhang, J. Liu, Y. Yang, J. Shi, X. Zhang, Y. Zhao, Y. Li, C. Lambert and W. Hong, Nat. Commun., 2019, 10, 5458 Search PubMed.
- R. Almughathawi, S. Hou, Q. Wu, Z. Liu, W. Hong and C. Lambert, ACS Sens., 2021, 6, 470–476 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1963994 and 1991442. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03229e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |