
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic enantioselective synthesis of fluoromethylated stereocenters by asymmetric hydrogenation†
Jianping
Yang‡
a,
Sudipta
Ponra‡
a,
Xingzhen
Li
b,
Bram B. C.
Peters
 a,
Luca
Massaro
a,
Taigang
Zhou
*b and
Pher G.
Andersson
*ac
a,
Luca
Massaro
a,
Taigang
Zhou
*b and
Pher G.
Andersson
*ac
aDepartment of Organic Chemistry, Stockholm University, Arrhenius Laboratory, 106 91, Stockholm, Sweden. E-mail: pher.andersson@su.se
bCollege of Chemistry and Chemical Engineering, Southwest Petroleum University, Chengdu, Sichuan 610500, China. E-mail: tgzhou@swpu.edu.cn
cSchool of Chemistry and Physics, University of Kwazulu-Natal, Private Bag X54001, Durban, 4000, South Africa
First published on 29th June 2022
Abstract
Fluoromethyl groups possess specific steric and electronic properties and serve as a bioisostere of alcohol, thiol, nitro, and other functional groups, which are important in an assortment of molecular recognition processes. Herein we report a catalytic method for the asymmetric synthesis of a variety of enantioenriched products bearing fluoromethylated stereocenters with excellent yields and enantioselectivities. Various N,P-ligands were designed and applied in the hydrogenation of fluoromethylated olefins and vinyl fluorides.
Introduction
Organofluorine compounds, on the basis of their special chemical and biological properties, are widely used in pharmaceuticals, agrochemistry, and materials science.1 In pharmaceuticals, the incorporation of a fluorine atom or fluorinated group into a biologically active compound usually modifies the biological and physicochemical properties by improving potency, lipophilicity, metabolic stability, binding affinity, and bioavailability.2 As a result, fluoromethylated analogues have become a potential class of drug candidates in isostere-based drug design.3 In terms of bioisosterism, monofluoromethyl (CH2F) and difluoromethyl (CHF2) groups are inert, isosteric and isopolar to an OH or SH group in biologically active compounds and pharmaceuticals.4 The trifluoromethyl (CF3) group could be considered to be bioisosteric with an ethyl group or a potential nitro bioisostere based on its topological, steric, and electronic effect.3c,5 In addition, mono/difluoromethylated analogues can also serve as a hydrogen donor in binding enzyme active sites. As a result, a variety of structurally diverse CH2F, CHF2, and CF3 containing drugs have been developed (Fig. 1).1f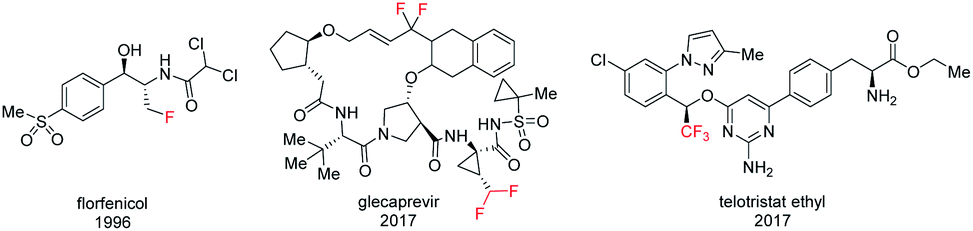 | ||
| Fig. 1 Fluoromethylated drugs. | ||
Hence, in modern organic chemistry and in drug discovery the development of versatile fluoromethylated molecules in an efficient fashion (especially in enantioenriched version) are very active research areas. Although distinct approaches6 are available for the asymmetric construction of the C(sp3)–CF3 function, little attention has been devoted towards asymmetric construction of the C(sp3)–CH2F and C(sp3)–CHF2 functions. The most common used strategies for the construction of the C(sp3)–CH2F stereogenic center are monofluoromethylation using 1-fluorobis (phenylsulfonyl)methane (FBSM), fluoro-(phenylsulfonyl)methane (FSM), 2-fluoro-1,3-benzodithiole-1,1,3,3-tetraoxide (FBDT), or α-fluoro-α-nitro(phenylsulfonyl)-methane as the fluoromethide equivalent (Scheme 1A).7 Other strategies consist of diasteroselective monofluoromethylation of chiral N-(tert-butylsulfinyl) aldimines/ketimines using fluoromethyl phenyl sulfone.8 Enantioenriched difluoromethylated compounds are synthesized by reacting nucleophiles or electrophiles with difluoromethylation reagents, for example, PhSO2CF2H, TMSCF2SPh, Me3SiCF2H, Me3SiCF2SO2Ph, HCF2SO2Cl, etc., or asymmetric addition of CF2H containing prochiral compounds such as imines, olefins, and carbonyl groups (Scheme 1B).9 However, the existing methods often require complex reaction conditions. Reduction of fluoromethylalkenes, on the other hand, remains unexplored but could be a broadly effective strategy for the construction of enantioenriched stereogenic centers bearing either CH2F, CHF2 or CF3 group by using a single general strategy.6r,9a,10
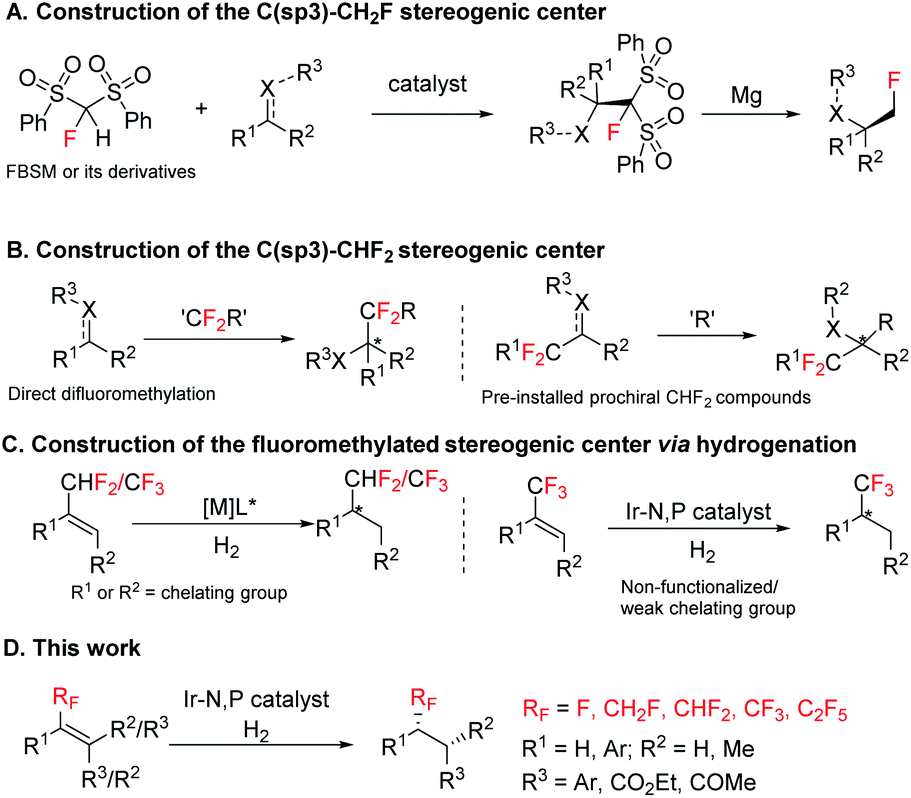 | ||
| Scheme 1 Strategies for the construction of fluoromethylated stereocenters. (A) Construction of the C(sp3)–CH2F stereogenic center; (B) construction of the C(sp3)–CHF2 stereogenic center; (C) construction of the fluoromethylated stereogenic center via hydrogenation; (D) This work. | ||
In asymmetric catalysis, enantioselective hydrogenation of alkenes using an appropriate transition metal catalyst and chiral ligand is one of the most fundamental and atom-economical processes. Rh and Ru catalysts are widely used for asymmetric hydrogenation of olefins having strong coordinating functional group such as amides or carboxylic acids in close proximity to the double bond.11 For olefins having weak coordinating groups or non-coordinating groups, Ir complexes are the most effective catalyst.12 Several RuII,13 RhI,14 and PdII (ref. 15) complexes were found effective for hydrogenation of some specific CF3 substituted olefins with a coordinating group near the substrate double bond (Scheme 1C, left). Fortunately, Ir complexes complement the substrate limitations of Rh/Ru catalyzed enantioselective hydrogenation and are efficient catalysts for enantioselective hydrogenation of CF3 substituted unfunctionalized olefins or CF3 substituted olefins with the weak chelating group.16
Herein, we report a direct catalytic, and highly enantioselective hydrogenation of fluoromethylated olefins for the efficient synthesis of a wide array of chiral building blocks containing fluoromethyl groups.
Results and discussion
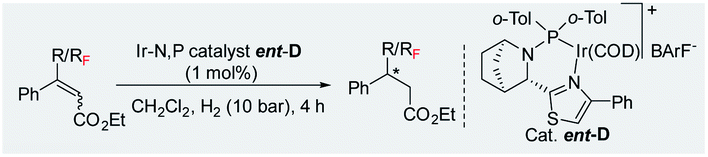
Difluoromethylated olefins were first chosen as the fluoromethylated olefin substrate for our study. We used (E)-ethyl 4,4-difluoro-3-phenylbut-2-enoate 1a as the model substrate and an iridium complex with a bicyclic backbone ligand as the catalyst for this asymmetric hydrogenation (Table 1). Hydrogenation of 1a using azabicyclo iridium oxazoline phosphine complex A (1 mol% catalyst, CH2Cl2, 10 bar H2) gave excellent conversion in 4 h but poor enantioselectivity (95% conversion, 21% ee) of the desired product 2a (entry 1). However, the thiazole N,P–iridium complex B dramatically increased the enantioselectivity (91% ee) with very good conversion (91%, entry 2). Based on our previous knowledge of iridium–N,P catalyzed asymmetric hydrogenation,16b we investigated the effect of varying the substituents on phosphine. Replacing the aliphatic iPr group with aromatic group (Ph) resulted in a slight change of enantioselectivity to 92% ee with 72% conversion (entry 3). However, replacing the phenyl group with ortho-tolyl group on the bicyclic thiazole iridium–N,P catalyst (catalyst D) resulted in complete conversion (99%) to the desired product 2a with the same level of enantioselectivity (92% ee, entry 4).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | H2 (bar) | Solvent | Time (h) | Conversion (%) | ee (%) |
| a Reaction conditions: 0.05 mmol of 1a, 0.5 mL solvent. The conversion was determined by 1H-NMR. Enantiomeric excess was determined by GCMS using a chiral stationary phase. | ||||||
| 1 | A (1.0) | 10 | CH2Cl3 | 4 | 95 | 21 |
| 2 | B (1.0) | 10 | CH2Cl2 | 4 | 91 | 91 |
| 3 | C (1.0) | 10 | CH2Cl2 | 4 | 72 | 92 |
| 4 | D (1.0) | 10 | CH2Cl2 | 4 | 99 | 92 |
| 5 | D (0.5) | 5 | CH2Cl2 | 4 | 99 | 92 |
| 6 | D (0.5) | 5 | Toluene | 4 | 99 | 93 |
| 7 | D (0.5) | 5 | PhCF3 | 4 | 99 | 94 |
| 8 | E (0.5) | 5 | PhCF3 | 4 | 99 | 94 |
| 9 | F (0.5) | 5 | PhCF3 | 4 | 99 | 95 |
| 10 | G (0.5) | 5 | PhCF 3 | 4 | 99 | 96 |
| 11 | H(0.5) | 5 | PhCF3 | 4 | 17 | 90 |
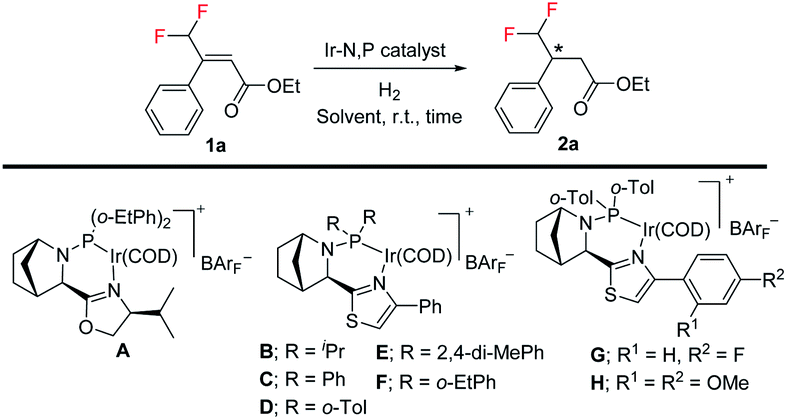
Further optimization of the reaction conditions with catalyst D was carried out by lowering the catalyst loading from 1.0 mol% to 0.5 mol% as well as the H2 pressure from 10 bar to 5 bar, respectively (Table 1, entry 5, for details, see ESI†). Using PhCF3 as solvent (entry 7) provided slightly better enantioselectivity (94% ee). To further increase enantioselectivity, we prepared a few new catalysts by varying the electronic density and steric hindrance on phosphorus. Catalyst with 2,4-di-MePh substituent (catalyst E, entry 8) gave the same result as complex D. Changing the ortho-tolyl group to an o-Etphenyl group afforded new thiazole N,P–iridium complex F with slightly improved enantioselectivity (95% ee, entry 9). Gratifyingly, adding a small electron-withdrawing (F) substituent on the aromatic ring of thiazole moiety (catalyst G) provides the best result in terms of enantioselectivity (96% ee) and conversion (99%, entry 10). On the other hand, the electron-donating (OMe) substituent on aromatic ring of thiazole moiety (catalyst H) led to much lower conversion (17%) and slightly lower enantioselectivity of 90% ee (entry 11). Thus, among these effectively designed new catalyst, a phenyl ring with F atom at para position on thiazole moiety and ortho-tolyl group on phosphorus (catalyst G, 0.5 mol%) in PhCF3 under 5 bar H2 pressure for 4 h provided the superior result in enantioselectivity (96% ee) with excellent 99% conversion (entry 10).
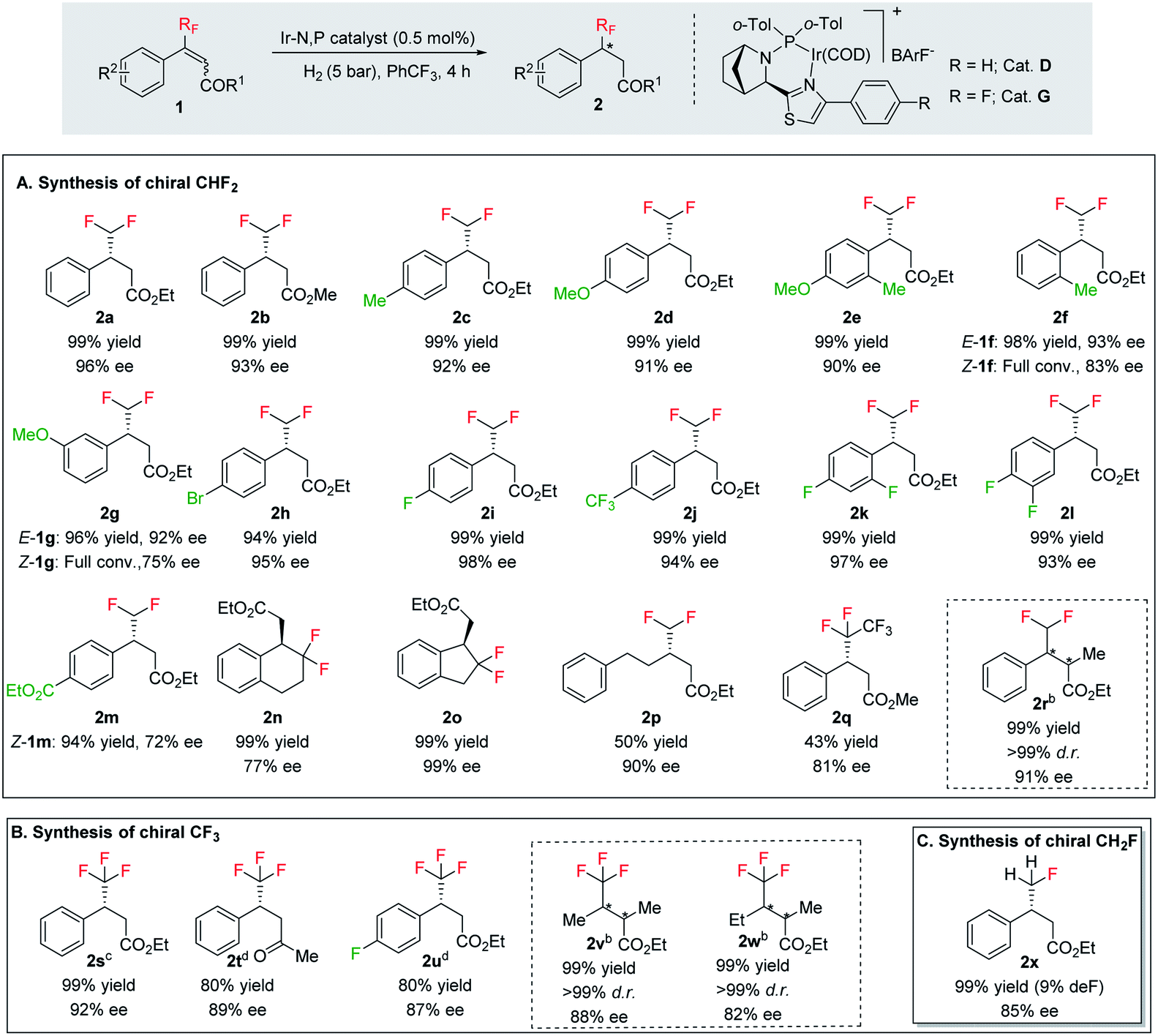
With the optimized reaction conditions established, we evaluated the hydrogenation of various (E)-fluorinated olefins 1 having different substituents (Table 2). A variety of difluoromethylated olefins (E)-1a–1l having different ester groups and with either electron-donating or electron-withdrawing substituents on the phenyl rings were successfully hydrogenated to deliver the desired products 2a–2l in excellent yield (94–99%) and enantioselectivities (90–98% ee). When evaluating the Z-isomer (Z-1f, Z-1g and Z-1m), lower enantioselectivities but the same major enantiomers were observed (83% ee, 75% ee and 72% ee, respectively). Interestingly, substrates with electron-withdrawing substituents seem advantageous for higher enantioselectivity. Carbocyclic CHF2 olefins (1n–1o) were hydrogenated in excellent yield but with significant variations in enantioselectivities. Benzo-fused cyclohexyl ring substrate 1n gave 77% ee while substrate with five-member ring (1o) provided 99% ee. Aliphatic CHF2 olefins were also tested and they generally resulted in lower reactivity. Nevertheless, we managed to hydrogenate compound 1p with a moderate conversion and good ee (90%). When the H on CHF2 group was replaced by strong electron-withdrawing CF3 group, the olefin was also hydrogenated much sluggishly and provided 2q in only 43% yield with 81% ee. After successfully hydrogenating various trisubstituted (E)-CHF2 olefins, tetrasubstituted CHF2 olefin (1r) was efficiently hydrogenated (2r) in 99% yield, excellent diastereoselectivity (>99% d.r.) and enantioselectivity (91% ee). The effectiveness of this stereoselective hydrogenation process was further investigated by evaluating various CF3 containing olefins to produce chiral CF3 alkanes. Various CF3 containing trisubstituted β,β-unsaturated esters or ketone were successfully hydrogenated under the standard conditions with good yields (80–99%) and enantioselectivities (87–96% ee, 2s–2u). Similarly, the developed protocol was equally efficient for tetra-substituted CF3 containing aliphatic olefins (1v and 1w) which were efficiently hydrogenated in excellent yields (99%) and diastereoselectivities (>99% d.r.) with high enantioselectivity (88% and 82% ee respectively). In addition, CH2F containing olefin (1x) was also hydrogenated in an exceptionally good yield (99%) with good enantioselectivity (84% ee) and slight defluorination (9%). The successful examples in Table 2 emphasizes that this azabicyclo iridium thiazole phosphine catalyst is very general for various fluoromethylated olefins.
| a Reaction conditions: 0.15 mmol of E-substrate, 0.5 mol% catalyst G, 5 bar H2, 1.5 mL PhCF3, 4 h. b 1.0 mol% catalyst ent-D, 100 bar H2. c 0.5 mol% catalyst D, 10 bar H2, 1.5 mL CH2Cl2. d 2.0 mol% catalyst D, 10 bar H2, 1.5 mL CH2Cl2. Yields are isolated hydrogenated product. Enantiomeric excess was determined by SFC or GCMS using chiral stationary phases. |
|---|
|
To further study the effectiveness of this developed method for the catalytic asymmetric synthesis of fluoromethylated stereogenic centers, a different class of olefins (vinyl fluoride), which affords the chiral monofluorinated molecule, was also evaluated. For these vinyl fluorides, catalyst B (1 mol%) was the most suitable catalyst using 20 bar H2 pressure for 24 h (see ESI† for optimization details). Employing the newly optimized reaction conditions, a variety of unfunctionalized naphthalene fused vinyl-fluoride substrates were efficiently hydrogenated in excellent enantioselectivity (90–98% ee, Table 3, 4a–h) although in some cases the conversions are low (3c, 73%; 3d, 40%; 3e, 40%; 3h, 70%). Notably, substrates having the bulky secondary (iPr, Cy) substituent were hydrogenated in high levels of stereoselectivity (4d–e). Both substrates with electron-donating (Me, OMe) or electron-withdrawing (F) substituents were tolerated (3f–h), however; substrates bearing electron-donating substituents were slightly more favorable in terms of reactivity (3f–g). A small amount of de-F byproduct (3–11%) were detected in the hydrogenation, however considering the challenges generally associated with hydrogenation of vinyl-fluoride, this efficient hydrogenation still highlights this catalytic protocol as general for fluorine-containing olefins to synthesize enantioenriched fluoromethylated compounds.
Interestingly, in this work, an enantioconvergent outcome was observed, where the E and Z isomers of fluoromethylated olefins were successfully hydrogenated using catalyst ent-D. Both isomers produced the same enantiomer in favor. The three different types of fluoromethylated olefins, including CH2F, CHF2 and CF3 groups, underwent enantioconvergent hydrogenation (Table 4, entries 1–3). However, removal of fluorine from the substrate (Table 4, entry 4) provided an enantiodivergent hydrogenation outcome (Table 4, entry 4), which suggested fluorine played an important role in the enantio-discrimination step. Our recent work on an efficient convergent hydrogenation using Ir–N,P complexes with a weak chelating group on the double bond suggested that α-prochiral olefins underwent an enantioconvergent hydrogenation while β-prochiral olefins reacted in an enantiodivergent manner.17 In this case, conversely, β-prochiral fluoromethylated olefin react in an enantioconvergent manner. We speculate that this could be due to the chelation effect or the electronic effect of the fluorine atom. Further investigations are still in progress.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Olefin | Isomer | Product | Conversion (%) | ee (%) |
| a Reaction conditions: 0.05 mmol of substrate, 1.0 mol% catalyst ent-D, 0.5 mL PhCF3, 10 bar H2. Enantiomeric excess was determined by SFC or GCMS using chiral stationary phases. | |||||
| 1 |
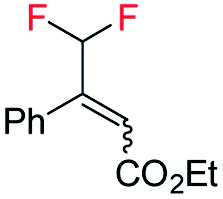
|
E-1a |
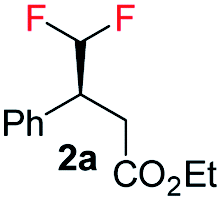
|
99 | 92 (S) |
| Z-1a | 99 | 55 (S) | |||
E/Z (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
99 | 71 (S) | |||
| 2 |
|
E-1s |
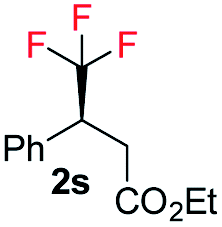
|
99 | 92 (S) |
| Z-1s | 99 | 26 (S) | |||
|
E/Z (1:1) |
99 | 56 (S) | |||
| 3 |
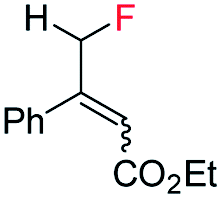
|
E-1x |
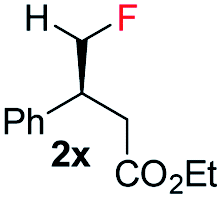
|
99 (9% de-F) | 84 (S) |
| Z-1x | 69 (32% de-F) | 56 (S) | |||
| E/Z (1:1) | 99 (30% de-F) | 76 (S) | |||
| 4 |
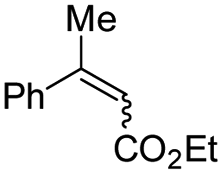
|
E-6 |
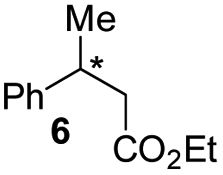
|
99 | 99 (S) |
| E-6 | 99 | 62 (R) | |||
The efficacy of the asymmetric synthesis of fluoromethylated compounds were investigated in gram-scale under standard reaction conditions. Product 2a was obtained in 97% yield with 96% ee (Scheme 2). This synthesized enantioenriched fluromethylated compound was transformed into a variety of many useful chiral fluorinated derivatives, such as alcohol, aldehyde, acid, Weinreb amide, ketone and nitrile (Scheme 2A) with almost perfect retention of enantiopurity. Interestingly, acid 11 provided (S)-3-(dichloromethyl)-2,3-dihydro-1H-inden-1-one 13 under Friedel–Crafts reaction condition. In the presence of AlCl3, difluoromethyl group underwent halogen exchange while preserving enantiomeric purity. Based on these successful transformations, some difluoromethylated natural products were accessed (Scheme 2B). Weinreb amide 14 was further transformed into difluorinated analogue of natural products 15. Synthetically versatile intermediate alcohol was transferred into bromide 17 which could be further transformed into the difluorinated analogue of alpha-curcumene 18.18
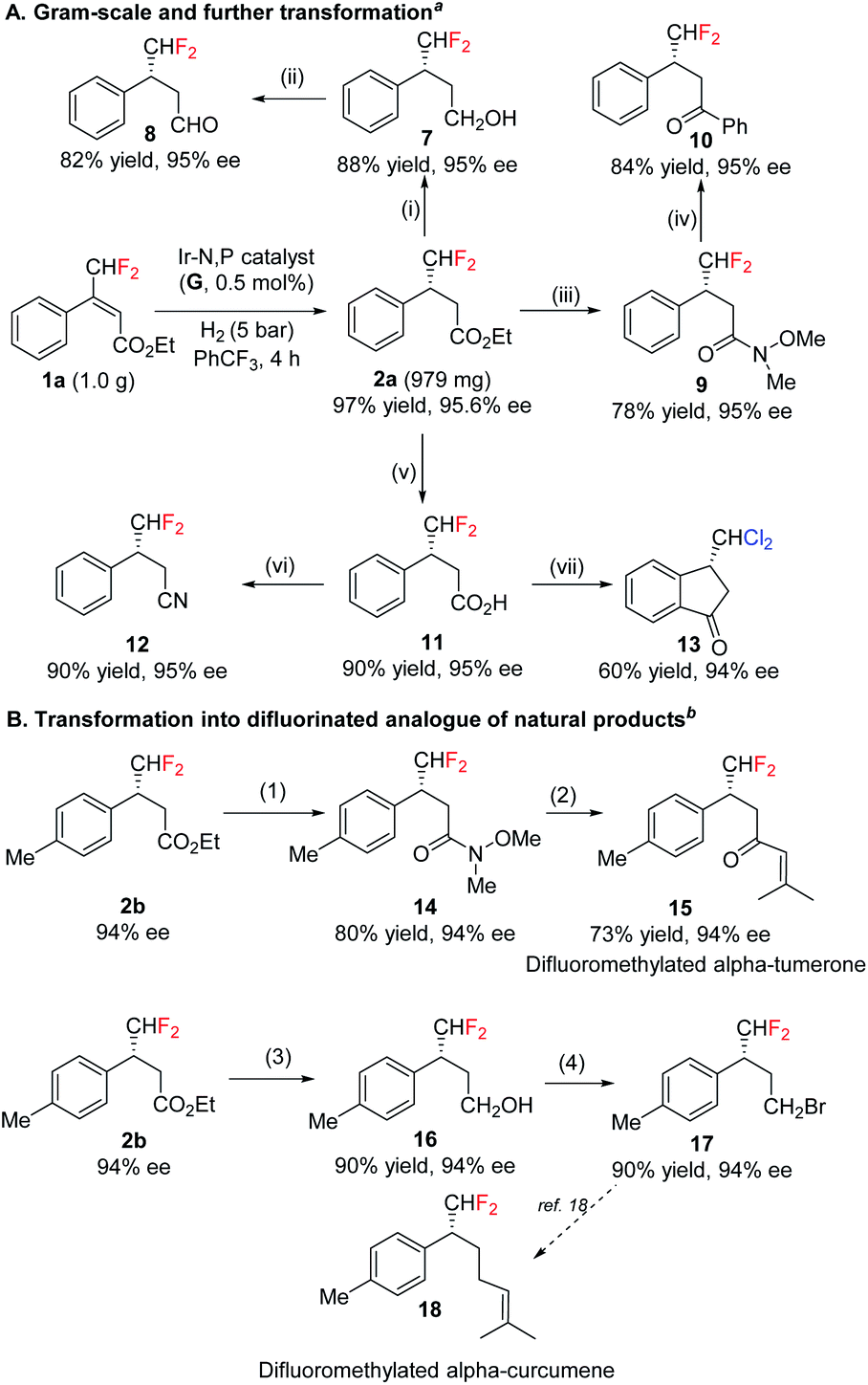 | ||
| Scheme 2 Synthesis of chiral difluoromethylated compounds having different functional group. (A) Gram-scale and further transformation; (B) transformation into difluorinated analogue of natural products. aReaction conditions: (i) LiAlH4, THF, 0 °C; (ii) DMP, DCM, r.t.; (iii) iPrMgBr, NH(OMe)Me·HCl, THF, 0 °C; (iv) PhMgCl, THF, 0 °C; (v) 2 M NaOH (aq.), MeOH, reflux; (vi) NH4HCO3, Boc2O, dioxane, r.t.; NEt3, (COCF3)2O, DCM, 0 °C – r.t.; (vii) cyanuric trichloride, pyridine, AlCl3, DCM. bReaction conditions: (1) iPrMgBr, NH(OMe)Me·HCl, THF, 0 °C; (2) 1-bromo-2-methylprop-1-ene, nBuLi, −78–0 °C; (3) LiAlH4, THF, 0 °C; (4) CBr4, PPh3, r.t. | ||
Conclusions
In summary, we have developed a catalytic, asymmetric methodology to synthesize various products bearing fluoromethylated stereocenters, which are important bioisostere in drug discovery. Different types of fluoromethylated olefins and vinyl fluorides were hydrogenated successfully by effective new catalyst design. In addition, an interesting enantioconvergency was observed, which indicated that fluorine has the potential to control the enantioselectivity due to its special properties.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
P. G. Andersson and T. Zhou supervised the project and conceived experiments. J. Yang and S. Ponra designed the project, optimized the reaction, performed the major of experiments, and prepared the Supporting Information. X. Li, B. B. C. Peters, and L. Massaro prepared some of the starting materials and evaluated some hydrogenation reactions. P. G. Andersson, J. Yang, and S. Ponra wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Swedish Research Council (VR), the Knut and Alice Wallenberg Foundation (KAW 2016:0072 & KAW 2018:0066) and Stiftelsen Olle Engkvist Byggmästare for their financial support. T. Z. is grateful for the financial support from the Sichuan Science and Technology Program (2021YFH0161), National Natural Science Foundation of China (No. 21811530004), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT CH2017-7226). We thank Dr Thishana Singh, School of Chemistry and Physics, University of Kwazulu-Natal, South Africa, for proofreading and editing the manuscript.References
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, John Wiley & Sons, 2013 CrossRef; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, 2009 CrossRef; (c) K. Uneyama, Organofluorine Chemistry, John Wiley & Sons, 2008 Search PubMed; (d) T. Hiyama, Organofluorine Compounds: Chemistry and Applications, Springer Science & Business Media, 2000 CrossRef; (e) P. V. Reddy, Organofluorine Compounds in Biology and Medicine, Newnes, 2015 Search PubMed; (f) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (g) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, Iscience, 2020, 23, 101467 CrossRef CAS PubMed; (h) A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 2081–2086 RSC.
- (a) S. Swallow, in Progress in Medicinal Chemistry, ed. G. Lawton and D. R. Witty, Elsevier, 2015, vol. 54, pp. 65–133 Search PubMed; (b) H. J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed; (c) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS; (d) K. L. Kirk, J. Fluorine Chem., 2006, 127, 1013–1029 CrossRef CAS; (e) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (f) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (h) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (i) J. Han, A. M. Remete, L. S. Dobson, L. Kiss, K. Izawa, H. Moriwaki, V. A. Soloshonok and D. O'Hagan, J. Fluorine Chem., 2020, 239, 109639 CrossRef CAS.
- (a) T. Taguchi and H. Yanai, in Fluorine in Medicinal Chemistry and Chemical Biology, 2009, pp. 257–290 Search PubMed; (b) A. Choudhary and R. T. Raines, ChemBioChem, 2011, 12, 1801–1807 CrossRef CAS PubMed; (c) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) M. Drouin and J.-F. Paquin, Beilstein J. Org. Chem., 2017, 13, 2637–2658 CrossRef CAS PubMed; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- C.-C. Tseng, G. Baillie, G. Donvito, M. A. Mustafa, S. E. Juola, C. Zanato, C. Massarenti, S. Dall'Angelo, W. T. A. Harrison, A. H. Lichtman, R. A. Ross, M. Zanda and I. R. Greig, J. Med. Chem., 2019, 62, 5049–5062 CrossRef CAS PubMed.
- (a) Z. Wang, C. Yang, J. Chen, F. Yang, R. Khan, Y. Yang, X. Qiao, Z. Su and B. Fan, Asian J. Org. Chem., 2021, 10, 1530–1535 CrossRef CAS; (b) T.-Z. Zhu, P.-L. Shao and X. Zhang, Org. Chem. Front., 2021, 8, 3705–3711 RSC; (c) Y. Min, J. Sheng, J. L. Yu, S. X. Ni, G. Ma, H. Gong and X. S. Wang, Angew. Chem., 2021, 133, 10035–10040 CrossRef; (d) A. Varenikov, E. Shapiro and M. Gandelman, Org. Lett., 2020, 22, 9386–9391 CrossRef CAS PubMed; (e) A. Varenikov and M. Gandelman, J. Am. Chem. Soc., 2019, 141, 10994–10999 CrossRef CAS PubMed; (f) A. Varenikov and M. Gandelman, Nat. Commun., 2018, 9, 3566 CrossRef PubMed; (g) M. Brambilla and M. Tredwell, Angew. Chem., Int. Ed., 2017, 56, 11981–11985 CrossRef CAS PubMed; (h) Y. Liang and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 9523–9526 CrossRef CAS PubMed; (i) C. Jiang, L. Wang, H. Zhang, P. Chen, Y.-L. Guo and G. Liu, Chem, 2020, 6, 2407–2419 CrossRef CAS; (j) S. Okusu, K. Hirano, Y. Yasuda, J. Tanaka, E. Tokunaga, H. Fukaya and N. Shibata, Org. Lett., 2016, 18, 5568–5571 CrossRef CAS PubMed; (k) T. Nishimine, K. Fukushi, N. Shibata, H. Taira, E. Tokunaga, A. Yamano, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2014, 53, 517–520 CrossRef CAS PubMed; (l) D.-F. Lu, C.-L. Zhu and H. Xu, Chem. Sci., 2013, 4, 2478–2482 RSC; (m) S. Wu, W. Zeng, Q. Wang and F.-X. Chen, Org. Biomol. Chem., 2012, 10, 9334–9337 RSC; (n) T. Furukawa, T. Nishimine, E. Tokunaga, K. Hasegawa, M. Shiro and N. Shibata, Org. Lett., 2011, 13, 3972–3975 CrossRef CAS PubMed; (o) Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 10769–10772 CrossRef CAS PubMed; (p) H. Kawai, A. Kusuda, S. Nakamura, M. Shiro and N. Shibata, Angew. Chem., Int. Ed., 2009, 48, 6324–6327 CrossRef CAS PubMed; (q) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed; (r) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed.
- (a) T. Furukawa, J. Kawazoe, W. Zhang, T. Nishimine, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Angew. Chem., 2011, 123, 9858–9862 CrossRef; (b) T. Fukuzumi, N. Shibata, M. Sugiura, H. Yasui, S. Nakamura and T. Toru, Angew. Chem., 2006, 118, 5095–5099 CrossRef; (c) K. Matsuzaki, T. Furukawa, E. Tokunaga, T. Matsumoto, M. Shiro and N. Shibata, Org. Lett., 2013, 15, 3282–3285 CrossRef CAS PubMed; (d) T. Furukawa, N. Shibata, S. Mizuta, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2008, 47, 8051–8054 CrossRef CAS PubMed; (e) J.-H. Lin and J.-C. Xiao, Tetrahedron Lett., 2014, 55, 6147–6155 CrossRef CAS; (f) Y. S. Kim, Synlett, 2014, 25, 2816–2817 CrossRef CAS; (g) S. Zhang, Y. Zhang, Y. Ji, H. Li and W. Wang, Chem. Commun., 2009, 4886–4888, 10.1039/B907399J; (h) M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed; (i) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- (a) Y. Li, C. Ni, J. Liu, L. Zhang, J. Zheng, L. Zhu and J. Hu, Org. Lett., 2006, 8, 1693–1696 CrossRef CAS PubMed; (b) J. Liu, L. Zhang and J. Hu, Org. Lett., 2008, 10, 5377–5380 CrossRef CAS PubMed; (c) J. Liu, Y. Li and J. Hu, J. Org. Chem., 2007, 72, 3119–3121 CrossRef CAS PubMed.
- (a) F. Gao, B. Li, Y. Wang, Q. Chen, Y. Li, K. Wang and W. Yan, Org. Chem. Front., 2021, 8, 2799–2819 RSC; (b) C. Ni, F. Wang and J. Hu, Beilstein J. Org. Chem., 2008, 4, 21 CrossRef PubMed; (c) X. Shen, W. Zhang, C. Ni, Y. Gu and J. Hu, J. Am. Chem. Soc., 2012, 134, 16999–17002 CrossRef CAS PubMed; (d) P. Zhang and C. Wolf, Angew. Chem., Int. Ed., 2013, 52, 7869–7873 CrossRef CAS PubMed; (e) C. Batisse, A. Panossian, G. Hanquet and F. R. Leroux, Chem. Commun., 2018, 54, 10423–10426 RSC; (f) K. Aikawa, S. Yoshida, D. Kondo, Y. Asai and K. Mikami, Org. Lett., 2015, 17, 5108–5111 CrossRef CAS PubMed; (g) D. Grassi, H. Li and A. Alexakis, Chem. Commun., 2012, 48, 11404–11406 RSC; (h) Y.-L. Liu, T.-D. Shi, F. Zhou, X.-L. Zhao, X. Wang and J. Zhou, Org. Lett., 2011, 13, 3826–3829 CrossRef CAS PubMed; (i) R. Smits, C. D. Cadicamo, K. Burger and B. Koksch, Chem. Soc. Rev., 2008, 37, 1727–1739 RSC; (j) W.-S. Huang, M.-L. Delcourt, X. Pannecoucke, A. B. Charette, T. Poisson and P. Jubault, Org. Lett., 2019, 21, 7509–7513 CrossRef CAS PubMed; (k) J. Liu, W. Ding, Q.-Q. Zhou, D. Liu, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2018, 20, 461–464 CrossRef CAS PubMed; (l) D. Bai, F. Wu, L. Chang, M. Wang, H. Wu and J. Chang, Angew. Chem., Int. Ed., 2022, 61, e202114918 CAS; (m) S. M. Banik, J. W. Medley and E. N. Jacobsen, Science, 2016, 353, 51–54 CrossRef CAS PubMed.
- T. Charvillat, P. Bernardelli, M. Daumas, X. Pannecoucke, V. Ferey and T. Besset, Chem. Soc. Rev., 2021, 50, 8178–8192 RSC.
- (a) P. G. Andersson and I. J. Munslow, Modern Reduction Methods, John Wiley & Sons, 2008 CrossRef; (b) J. Tsuji, Modern Rhodium-Catalyzed Organic Reactions, John Wiley & Sons, 2005 Search PubMed; (c) S.-I. Murahashi, Ruthenium in Organic Synthesis, John Wiley & Sons, 2006 Search PubMed.
- (a) J. J. Verendel, O. Pamies, M. Dieguez and P. G. Andersson, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed; (b) C. Margarita and P. G. Andersson, J. Am. Chem. Soc., 2017, 139, 1346–1356 CrossRef CAS PubMed.
- (a) K. Iseki, Y. Kuroki, T. Nagai and Y. Kobayashi, J. Fluorine Chem., 1994, 69, 5–6 CrossRef CAS; (b) Y. Kuroki, D. Asada, Y. Sakamaki and K. Iseki, Tetrahedron Lett., 2000, 41, 4603–4607 CrossRef CAS; (c) M. Z. Chen, N. W. Sach, P. Nuhant, O. O. Fadeyi, J. Trujillo, V. Lombardo, L. Bernier, R. Unwalla and A. C. Flick, Tetrahedron: Asymmetry, 2016, 27, 882–887 CrossRef CAS.
- (a) C. Benhaim, L. Bouchard, G. Pelletier, J. Sellstedt, L. Kristofova and S. Daigneault, Org. Lett., 2010, 12, 2008–2011 CrossRef CAS PubMed; (b) K. Dong, Y. Li, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 14191–14195 CrossRef CAS PubMed; (c) J. Jiang, W. Lu, H. Lv and X. Zhang, Org. Lett., 2015, 17, 1154–1156 CrossRef CAS PubMed.
- (a) F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 2850–2852 CrossRef CAS PubMed; (b) G. Szőllősi, T. Varga, K. Felföldi, S. Cserényi and M. Bartók, Catal. Commun., 2008, 9, 421–424 CrossRef; (c) A. Alimardanov, A. Nikitenko, T. J. Connolly, G. Feigelson, A. W. Chan, Z. Ding, M. Ghosh, X. Shi, J. Ren, E. Hansen, R. Farr, M. MacEwan, S. Tadayon, D. M. Springer, A. F. Kreft, D. M. Ho and J. R. Potoski, Org. Process Res. Dev., 2009, 13, 1161–1168 CrossRef CAS; (d) S. Feng, Y. Tang, C. Yang, C. Shen and K. Dong, Org. Lett., 2020, 22, 7508–7512 CrossRef CAS PubMed; (e) M.-W. Chen, Q. Yang, Z. Deng, Q. Ding and Y. Peng, J. Org. Chem., 2019, 84, 10371–10379 CrossRef CAS PubMed.
- (a) M. Engman, P. Cheruku, P. Tolstoy, J. Bergquist, S. F. Völker and P. G. Andersson, Adv. Synth. Catal., 2009, 351, 375–378 CrossRef CAS; (b) S. Kerdphon, S. Ponra, J. Yang, H. Wu, L. Eriksson and P. G. Andersson, ACS Catal., 2019, 9, 6169–6176 CrossRef CAS.
- (a) B. B. C. Peters, J. Zheng, N. Birke, T. Singh and P. G. Andersson, Nat. Commun., 2022, 13, 361 CrossRef CAS PubMed; (b) J. Yang, L. Massaro, S. Krajangsri, T. Singh, H. Su, E. Silvi, S. Ponra, L. Eriksson, M. S. G. Ahlquist and P. G. Andersson, J. Am. Chem. Soc., 2021, 143, 21594–21603 CrossRef CAS PubMed; (c) L. Massaro, J. Zheng, C. Margarita and P. G. Andersson, Chem. Soc. Rev., 2020, 49, 2504–2522 RSC.
- M. Harmata, X. Hong and C. L. Barnes, Tetrahedron Lett., 2003, 44, 7261–7264 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02685f |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |