
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization of a high-spin three-coordinate Fe(III) imidyl complex by radical delocalization†
Po-Chun
Yang
a,
Kuan-Po
Yu
a,
Chi-Tien
Hsieh
a,
Junjie
Zou
b,
Chia-Te
Fang
a,
Hsin-Kuan
Liu
 c,
Chih-Wen
Pao
d,
Liang
Deng
b,
Mu-Jeng
Cheng
a and
Chun-Yi
Lin
*a
c,
Chih-Wen
Pao
d,
Liang
Deng
b,
Mu-Jeng
Cheng
a and
Chun-Yi
Lin
*a
aDepartment of Chemistry, National Cheng Kung University, No. 1 University Road, Tainan 701014, Taiwan. E-mail: cylin@gs.ncku.edu.tw
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China
cCore Facility Center, National Cheng Kung University, No. 1 University Road, Tainan 701014, Taiwan
dNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 300092, Taiwan
First published on 21st July 2022
Abstract
High-spin, late transition metal imido complexes have attracted significant interest due to their group transfer reactivity and catalytic C–H activation of organic substrates. Reaction of a new two-coordinate iron complex, Fe{N(tBu)Dipp}2 (1, Dipp = 2,6-diisopropylphenyl), with mesitylazide (MesN3) afforded a three-coordinate Fe–imidyl complex, Fe{N(tBu)Dipp}2(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMes) (2). X-ray crystallographic characterization of single crystals of 2 showed a long Fe–N distance of 1.761(1) Å. Combined magnetic and spectroscopic (Mössbauer and X-ray absorption near edge structure spectroscopy, XANES) characterization of 2 suggests that it has an S = 2 ground state comprising an S = 5/2 Fe(III) center antiferromagnetically coupled to an S = 1/2 imidyl ligand. Reaction of 1 and 1-azidoadamantane (AdN3) generated a putative, transient Fe{N(tBu)Dipp}2(NAd) (3′) complex that yielded an intramolecular C–H amination product, Fe{N(tBu)Dipp}{κ2-N,N′
NMes) (2). X-ray crystallographic characterization of single crystals of 2 showed a long Fe–N distance of 1.761(1) Å. Combined magnetic and spectroscopic (Mössbauer and X-ray absorption near edge structure spectroscopy, XANES) characterization of 2 suggests that it has an S = 2 ground state comprising an S = 5/2 Fe(III) center antiferromagnetically coupled to an S = 1/2 imidyl ligand. Reaction of 1 and 1-azidoadamantane (AdN3) generated a putative, transient Fe{N(tBu)Dipp}2(NAd) (3′) complex that yielded an intramolecular C–H amination product, Fe{N(tBu)Dipp}{κ2-N,N′![[- with combining low line]](https://www.rsc.org/images/entities/char_002d002d_0332.gif) N(CMe2CH
N(CMe2CH![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) NHAd)Dipp} (3). Quantum mechanical calculations further confirmed the spectroscopic assignment of 2 and 3′, as well as the differences in their stability and reactivity. Importantly, imidyl radical delocalization onto the mesityl ring significantly increased the stability of 2 and reduced its reactivity toward potential hydrogen atom transfer (HAT) reagents. In contrast, quantum mechanical calculations of 3′ revealed that the radical was solely localized on the imidyl N, leading to a high reactivity toward the proximal C–H bond of the N(tBu)Dipp− ligand.
NHAd)Dipp} (3). Quantum mechanical calculations further confirmed the spectroscopic assignment of 2 and 3′, as well as the differences in their stability and reactivity. Importantly, imidyl radical delocalization onto the mesityl ring significantly increased the stability of 2 and reduced its reactivity toward potential hydrogen atom transfer (HAT) reagents. In contrast, quantum mechanical calculations of 3′ revealed that the radical was solely localized on the imidyl N, leading to a high reactivity toward the proximal C–H bond of the N(tBu)Dipp− ligand.
Introduction
Transition metal complexes with metal–ligand multiple bonds, such as those featuring oxo and imido ligands, have long been synthetic targets.1,2 Terminal imido complexes of Fe are of special interest, not only due to their fundamental importance in the understanding of structural features and spectroscopic data,3 but because of their use in transferring nitrene functional groups to organic substrates with inert C–H bonds.4,5 In addition, high-spin states are rare among these compounds, with most of them being in low- and intermediate-spin states.3,6–35 Many of the terminal imido complexes of Fe characterized to date contain traditional closed-shell imido ligands, represented as NR2− (R = alkyl, aryl).3 It has been shown that inducing a weak ligand field around the metal center enables the isolation of the open-shell imidyl ligand with a radical on its N, represented as ˙NR−.36–38 Crucially, imidyl ligands have been shown to be generally more reactive toward inert C–H bonds and other organic substrates.39–43 Clearly, the intrinsically high reactivity of transition metal imidyl complexes makes their isolation difficult and warrants further stability studies.The four-coordinate Fe imido complexes, (ArL)FeCl(NC6H4-p-tBu), (ArL)FeCl(NAd), and (tBuL)FeCl(NDipp) (ArL = 5-mesityl-1,9-bis(2,4,6-triphenylphenyl)dipyrrinato, Ad = 1-adamantyl, Dipp = 2,6-diisopropylphenyl, tBuL = 5-(2,6-dichlorophenyl)-1,9-bis(t-butyl)dipyrrinato, Fig. 1), reported by Betley and coworkers are the three reported examples of Fe imidyl complexes. Moreover, they all have high-spin Fe(III) centers that are antiferromagnetically coupled to an open-shell imidyl radical.30,39,40 Importantly, the authors showed that the Fe imidyl complexes could catalyze C–H amination reactions.44,45 Therefore, significant interest has shifted toward the synthesis of this unusual class of complexes and the characterization of their electronic structure and reactivity.41–43 However, despite these findings, comparative study on how this seemingly reactive ligand could be stabilized in a low-coordinate environment is relatively unexplored. Previously, Betley proposed that the stability of the aryl imidyl complex might be originated from the delocalized radical onto the aryl substituent.30,42 Here, the synthesis and characterization of stable aryl- and transient alkyl–imidyl complexes of three-coordinate Fe(III) are reported for the first time, with the transient alkyl–imidyl complexes shown to undergo fast intramolecular C–H amination. Quantum mechanical calculations provided insights into the divergent reactivity and stability and showed that the imidyl radical was indeed delocalized onto the aryl ring to provide additional stability.
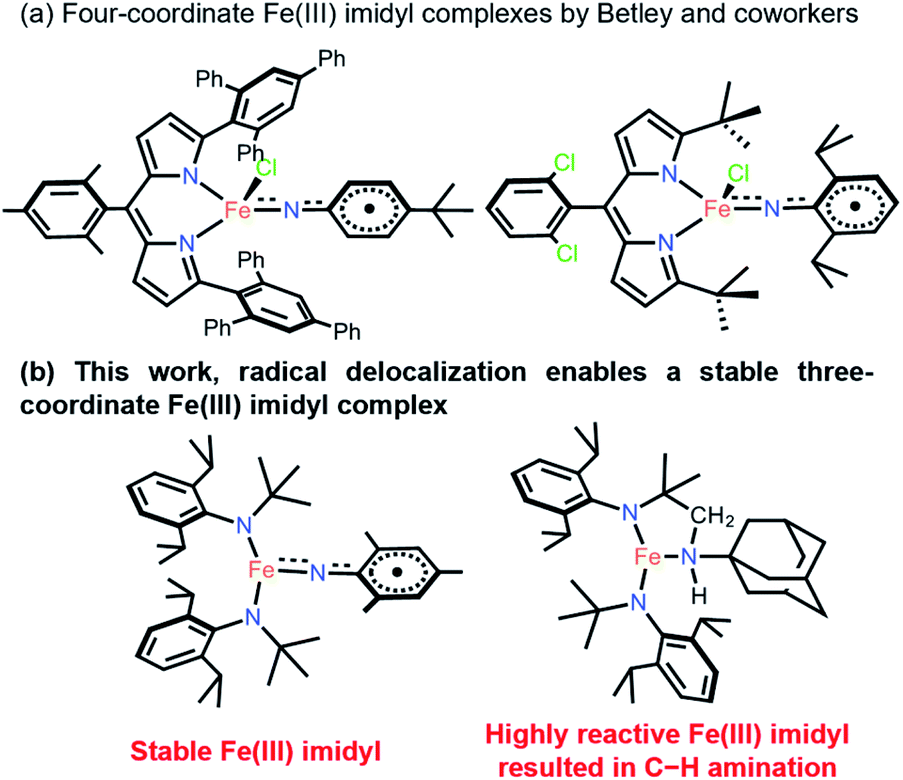 | ||
| Fig. 1 (a) Structurally characterized four-coordinate Fe(III) imidyl complexes reported by Betley and co-workers. (b) This work. | ||
Results and discussion
To stabilize a three-coordinate, high-oxidation state, and high-spin Fe imido complex, the monoanionic amido ligand, N(tBu)Dipp−,46,47 recently reported by the groups of Newhouse and Jones was selected. The synthesis of Fe{N(tBu)Dipp}2 (1) proceeded smoothly from the reaction of 1 equivalent (equiv.) of anhydrous FeCl2 and 2 equiv. of LiN(tBu)Dipp in diethyl ether (Et2O). 1 was isolated as a red crystalline solid in 66% yield, and its molecular structure was characterized by single-crystal X-ray diffractometry (SC-XRD). Multiple X-ray data sets of sufficient quality enabled the molecular structure of 1 to be identified as a near-linear two-coordinate Fe(II) amido complex (Fig. 2). Notably, unlike Fe{N(SiMe3)Dipp}2 (ref. 48 and 49) that has an exactly linear (180°) N–Fe–N angle, the N–Fe–N angle of 1 is 176.6(4)°. The dihedral angle of the two NR2 planes of the amido ligands is 29.6(4)°, reflecting the steric repulsion between the bulky Dipp and tBu substituents, which is similar to the related bent complex Fe{N(CH2tBu)Dipp}2.50 The structural difference between them may reflect the subtle interplay between steric repulsion and dispersion attraction.51,52 The room-temperature effective magnetic moment (μeff) of 1 (5.75(7) μB), measured using the Evans method in C6D6 solution, is consistent with an S = 2 ground state with a large orbital angular momentum (OAM) contribution, which has been observed in linear and near-linear Fe(II) complexes.48,49,53–571 was additionally characterized by nuclear magnetic resonance (NMR), vibrational, and electronic spectroscopies, as well as elemental analysis.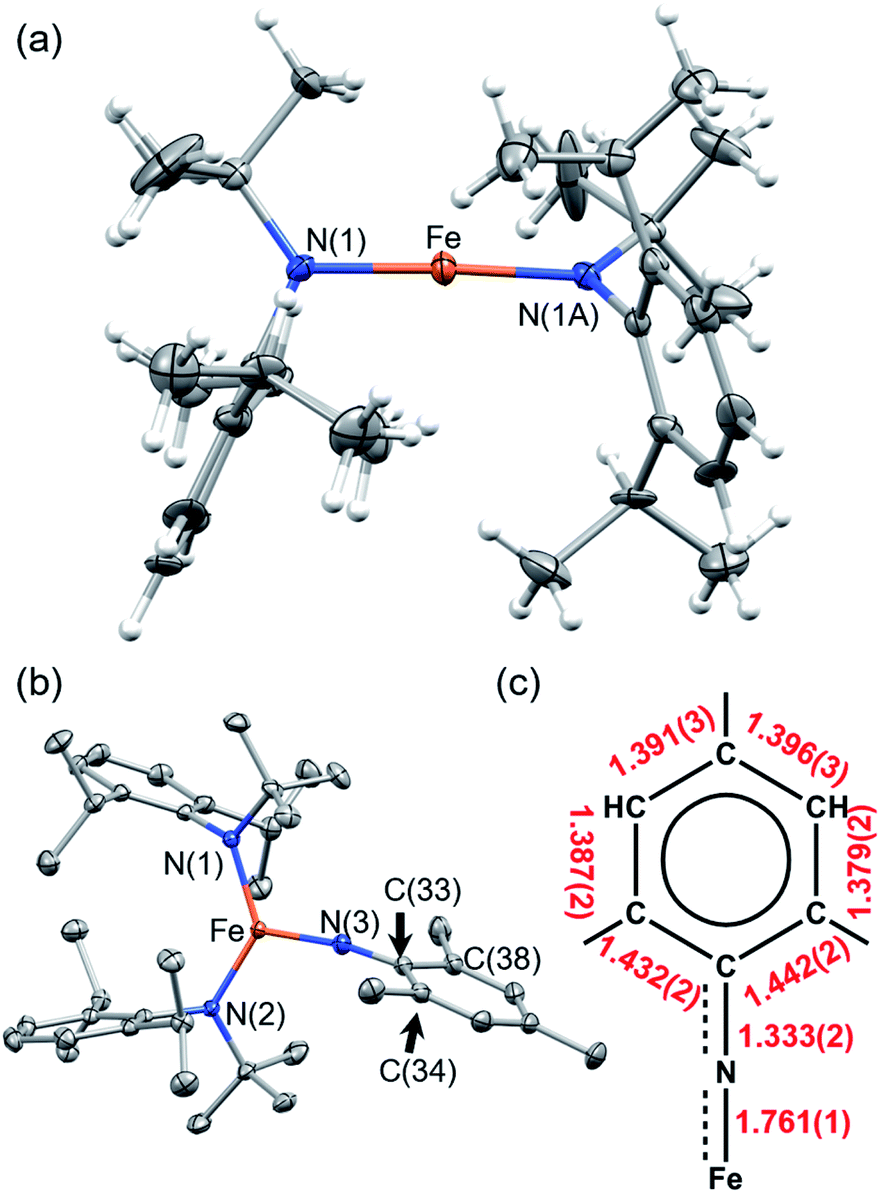 | ||
| Fig. 2 Crystallographically-determined structure of (a) 1 and (b) 2. Thermal ellipsoids are shown at 50% level. Gray ellipsoids and white spheres represent carbon and hydrogen, respectively. Selected bond lengths and angles (1): Fe–N(1), 1.859(5) Å; N(1)–Fe–N(1A) 176.6(4)°. (2): Fe–N(1), 1.909(1) Å; Fe–N(2), 1.910(1) Å; N(1)–Fe–N(2) 131.44(6)°. (c) Selected bond distances of the Fe imido unit of one molecule in the asymmetric unit. | ||
Reaction of 1 and mesitylazide (MesN3) in benzene for 1 h (Scheme 1), followed by work-up and crystallization, resulted in the formation of large black crystals suitable for SC-XRD in 69% yield. The product was characterized as the three-coordinate Fe–imido complex, Fe{N(tBu)Dipp}2(NMes) (2, Fig. 2). In the asymmetric unit of the crystal, there are two virtually indistinguishable but independent molecules. A notable feature is the long FeNMes distance [Fe–N(3)] of 1.761(1) Å, which is longer than those of most reported Fe mono-imido complexes (Table S6†). This distance is similar to those of the four-coordinate Fe(III) imidyl complexes reported by Betley, (ArL)FeCl(NC6H4-p-tBu) (1.768(2) Å)39 and (tBuL)FeCl(NDipp) (1.768(4) Å),40 and that of the imido–Fe4S4 cluster complex reported by Suess and co-workers, (IMes)3Fe4S4NDipp (1.763(2) Å, IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene).58 In addition, the short N(3)–C(33) distance and the elongation of the C(33)–C(34) and C(33)–C(38) distances of the mesityl ring (Fig. 2) indicated radical delocalization.42,43,59 The μeff values measured using the Evans method (4.9(1) μB, C6D6) and superconducting quantum interference device (SQUID) magnetometry (5.06 μB) suggested that 2 exhibits an S = 2 ground state at 300 K. Moreover, the χMT values obtained from SQUID measurements remained relatively stable at ca. 3.2 cm3 K mol−1 from 300 to 20 K, followed by a rapid drop to 1.67 cm3 K mol−1 at 1.8 K [Fig. 3(a)]. This low-temperature drop is likely a result of zero-field splitting (D). The variable-temperature magnetic susceptibility and variable-field magnetization data could be globally fit using PHI,60 with best-fit parameters of S = 2, g = 2.080 ± 0.006, and D = −5.15 ± 0.02 cm−1. The absence of X-band (ca. 9.5 GHz) electron paramagnetic resonance (EPR) signal in frozen toluene solution at 77 K may be the result of the integer S and large D, precluding the characterization of the radical character of the Mes substituent.43 To determine the chemical oxidation state of Fe, 57Fe Mössbauer and Fe K-edge X-ray absorption near edge structure (XANES) spectroscopy were employed. The Mössbauer spectrum of 2 recorded at 120 K is shown in Fig. 3(b), fit using an isomer shift (δ) of 0.23 mm s−1 and a quadrupole splitting (ΔEQ) of 3.27 mm s−1, which are in the typical range of those of high-spin (S = 5/2), Fe(III) ions.61 Crucially, the isomer shift was similar to those obtained for the four-coordinate Fe(III) imidyl complexes reported by Betley of 0.28 mm s−1 for (ArL)FeCl(NC6H4-p-tBu) and 0.26 mm s−1 for (ArL)FeCl(NAd),30 and the three-coordinate Fe(III) imido complex reported by Smith of 0.25 mm s−1 for Ph2B(tBuIm)2FeNDipp (Ph2B(tBuIm)2 = bis(3-tert-butylimidazol-2-ylidene-1-yl)(diphenyl)borate).32 In addition, it is smaller than those of the three-coordinate Fe(III) imido complexes reported by Betley of 0.44 mm s−1 for (ArL)FeN(C6H4-p-tBu), 0.47 mm s−1 for (ArL)FeNMes, and 0.37 mm s−1 for (ArL)FeNAd and by Holland of 0.47 mm s−1 for (Me,DippNacnac)FeNAd (Me,DippNacnac = N,N′-bis(2,6-di-isopropylphenyl)penta-2,4-diiminato).26,30,62 Notably, the isomer shift is greater than the structurally characterized Fe(IV) mono- and bis-imido complexes (Tables S6 and S7†).6,22–24,34 The normalized Fe K-edge XANES spectra of 1 (as an Fe(II) reference) and 2 are shown in Fig. 3(c). Here, the 1s to 3d pre-edge transition could be a useful indicator of the chemical oxidation state of Fe in different coordination environments. Importantly, the pre-edge signals at 7112.1 eV for 1 and 7113.3 eV for 2 are indicative of Fe-based oxidation. Typically, a ca. 1 eV increase in the pre-edge energy is a consequence of a unit oxidation state increase due to the increase in the ligand field. Furthermore, the 1.2 eV increase is similar to the values observed for the Fe- and Ni-dipyrrin systems reported by Betley.30,42 Finally, from the above results, it was speculated that the Fe spin state and oxidation state in 2 are S = 5/2 and +3, respectively, with antiferromagnetic coupling to an imidyl radical resulting in an overall S = 2 ground state.
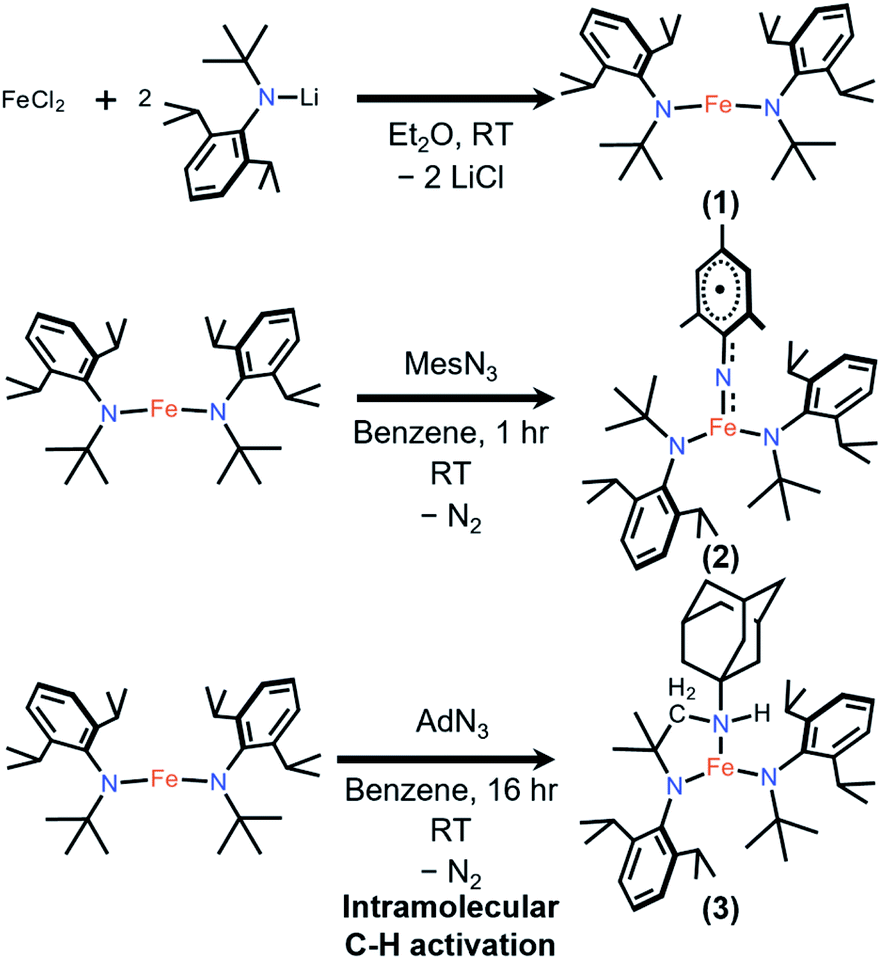 | ||
| Scheme 1 Synthesis of 1, 2, and 3. | ||
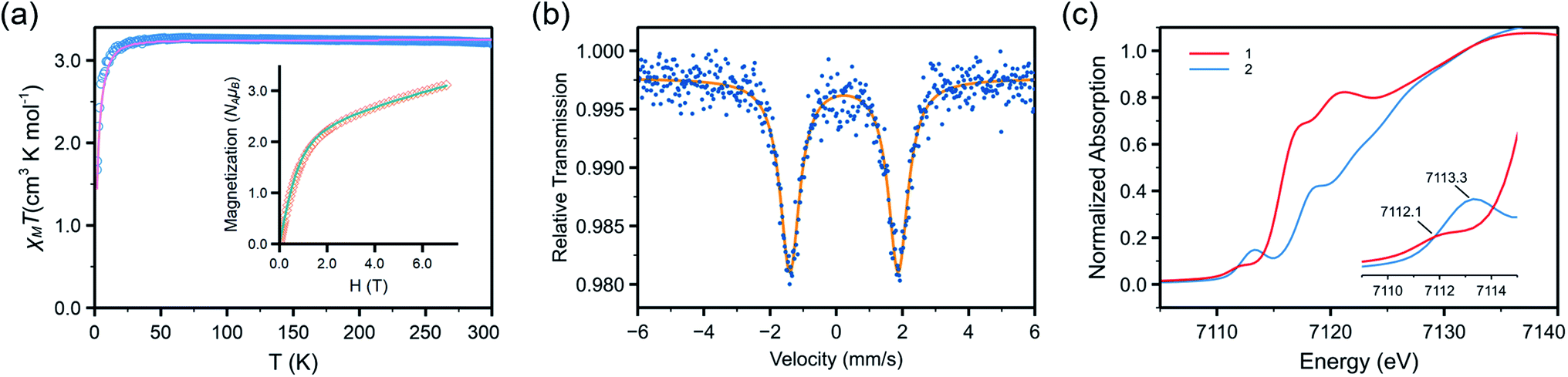 | ||
| Fig. 3 (a) Variable temperature magnetic susceptibility and variable field magnetization data of 2. Circles and squares are experimental data, and the solid lines represent fit described in text. (b) Zero-field 57Fe Mössbauer spectrum of solid 2 recorded at 120 K. Blue dots are experimental data, and the orange line represents the best fit. (c) Normalized Fe K-edge XANES spectra of 1 and 2. Inset represents the pre-edge region. | ||
The analogous reaction of 1 with AdN3 in benzene resulted in an immediate color change from red to brown with gas evolution (presumably N2). After work-up and crystallization, large pale green crystals formed (27% yield, Scheme 1), the molecular structure of which was shown by SC-XRD to be Fe{N(tBu)Dipp}{κ2-N,N′N(CMe2CHNHAd)Dipp} (3, Fig. 4). The H atom of the N–H group could be located on the Fourier difference map, indicating intramolecular C–H amination and formation of a new chelating bidentate amido/amine ligand. The room-temperature magnetic moment measured using the Evans method in C6D6 was 5.3(2) μB, indicating an S = 2 [Fe(II)] ground state with OAM contribution. Attempts to isolate the intermediate Fe{N(tBu)Dipp}2(NAd) (3′) were unsuccessful, likely the result of its highly reactive nature. Monitoring the reaction of 1 and AdN3 in C6D6 by 1H NMR spectroscopy only revealed proton signals of 1 and 3. Thus, we surmise that a three-coordinate, highly reactive intermediate 3′, was formed first, rapidly followed by intramolecular C–H amination of one of the tert-butyl groups of the amido ligand. An attempt was made to isolate the hydrogen atom transfer (HAT) product, Fe{N(tBu)Dipp}2{N(H)Ad} (3′–H), by reacting 1, AdN3, and 30 equiv. of 1,4-cyclohexadiene (1,4-CHD); however, only 3 was isolated, suggesting that intramolecular C–H amination was dominant in this reaction.
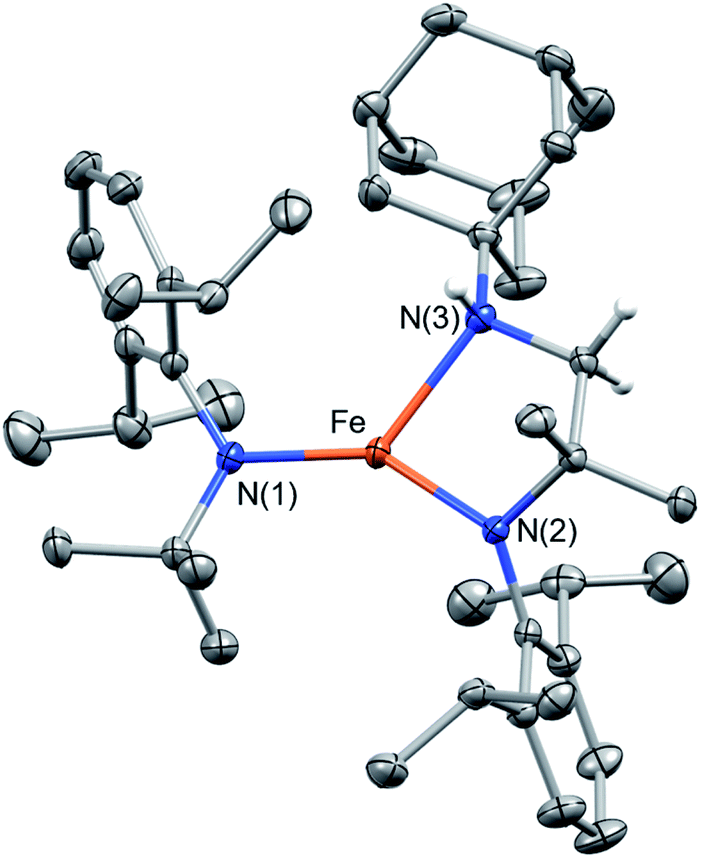 | ||
| Fig. 4 Crystallographically-determined structure of 3. Thermal ellipsoids are shown at 50% level. Hydrogen atoms, except for the C–H activated ones, are not shown for clarity. Gray ellipsoids and white spheres represent carbon and hydrogen, respectively. Selected bond lengths and angles: Fe–N(1), 1.894(2) Å; Fe–N(2), 1.919(2) Å; Fe–N(3), 2.173(2) Å; N(2)−Fe–N(3) 86.38(6)°. | ||
The successful isolation of 2 enabled the investigation of its reactivity for nitrene transfer or H atom abstraction (HAA). First, the stability of 2 was examined in common solvents to optimize the reaction conditions. 2 slowly decomposed in common non-coordinating (hexanes, benzene, and toluene) and coordinating (Et2O and tetrahydrofuran (THF)) solvents into the free HN(tBu)Dipp ligand and other unidentifiable products (see the ESI†). Next, 2 was reacted with molecules featuring weak aliphatic C–H bonds, such as 1,4-CHD [C–H bond dissociation energy (BDEC–H) = 76.0 ± 1.2 kcal mol−1]63 and triphenylmethane (HCPh3, BDEC–H = 81 ± 2 kcal mol−1).63 However, 2 showed no HAA reactivity, as benzene and the Gomberg dimer were not detected in its reactions with 1,4-CHD and HCPh3, respectively. Although a reaction was observed between 2 and TEMPO-H (TEMPO = (2,2,6,6-tetramethylpiperidin-1-yl)oxyl) that resulted in complete consumption of both TEMPO-H and TEMPO, the reaction mixture contained an intractable paramagnetic product that could neither be characterized nor crystallized. It was thus surmised that the TEMPO formed undergoes further reaction. In addition, attempts to transfer NMes to organic substrates such as PMe3, PPh3, and tBu-NCN-tBu, did not result in nitrene transfer (see the ESI†). The lack of HAA and nitrene transfer reactivity is in contrast to other reported late transition metal imidyl complexes.26,39,42,43 Yet, it is similar to the Ni(II) imidyl complex, (AdFL)Ni(NMes), reported by Betley, which was reported to exhibit no HAA reactivity.42 The large amido ligand and the mesityl imido substituent required to stabilize the three-coordinate environment had a detrimental effect on the imidyl reactivity. It may also be due to the radical character of the imidyl N, which could not guarantee a small reorganization energy because of the requirement of trigonal planar coordination on N of HAA in the transition state.64 Furthermore, quantum mechanical calculations (see below) indicated that the imidyl radical was delocalized onto the mesityl substituent, resulting in the decreased reactivity and increased stability of 2.
The different reactivities of 2 and 3′ toward intramolecular C–H activation prompted further atomistic insights to be gained using B3LYP-D3 DFT calculations (see the ESI†). Consistent with the experimental data, the ground state of 1 was found to be a quintet (S = 2), as well as 23.9 kcal mol−1 more stable than the triplet state. The reactions of 1 with MesN3 and AdN3 led to the formation of 2 and 3′, respectively. These reactions were very favorable, with a Gibbs free energy (ΔG) value of −42.2 kcal mol−1 for 1 → 2 and −22.5 kcal mol−1 for 1 → 3′ (Fig. 5). The ground states of 2 and 3′ were found to be quintets, consistent with the experimental data. It was noted that two quintet states were obtained using the broken-symmetry approach, and the converged wavefunctions were 〈S2〉 = 6.74 and 6.61 for 2 and 3′, respectively, deviating from the 〈S2〉 = 6.00 value for a pure quintet state. Using different functionals (i.e., M06, CAM-B3LYP, and ωB97XD) or switching to an all-electron basis set (def-TZVP) led to similar results (Table S8 in the ESI†). Mössbauer parameters of 2 were computed using the calibration constants and the procedure suggested by Neese and coworkers65 using SC-XRD-determined structure. The isomer shift and quadrupole splitting of Fe were calculated to be 0.27 and 3.06 mm s−1, respectively, both of which are in reasonable agreement with our experimental values of 0.23 and 3.27 mm s−1.
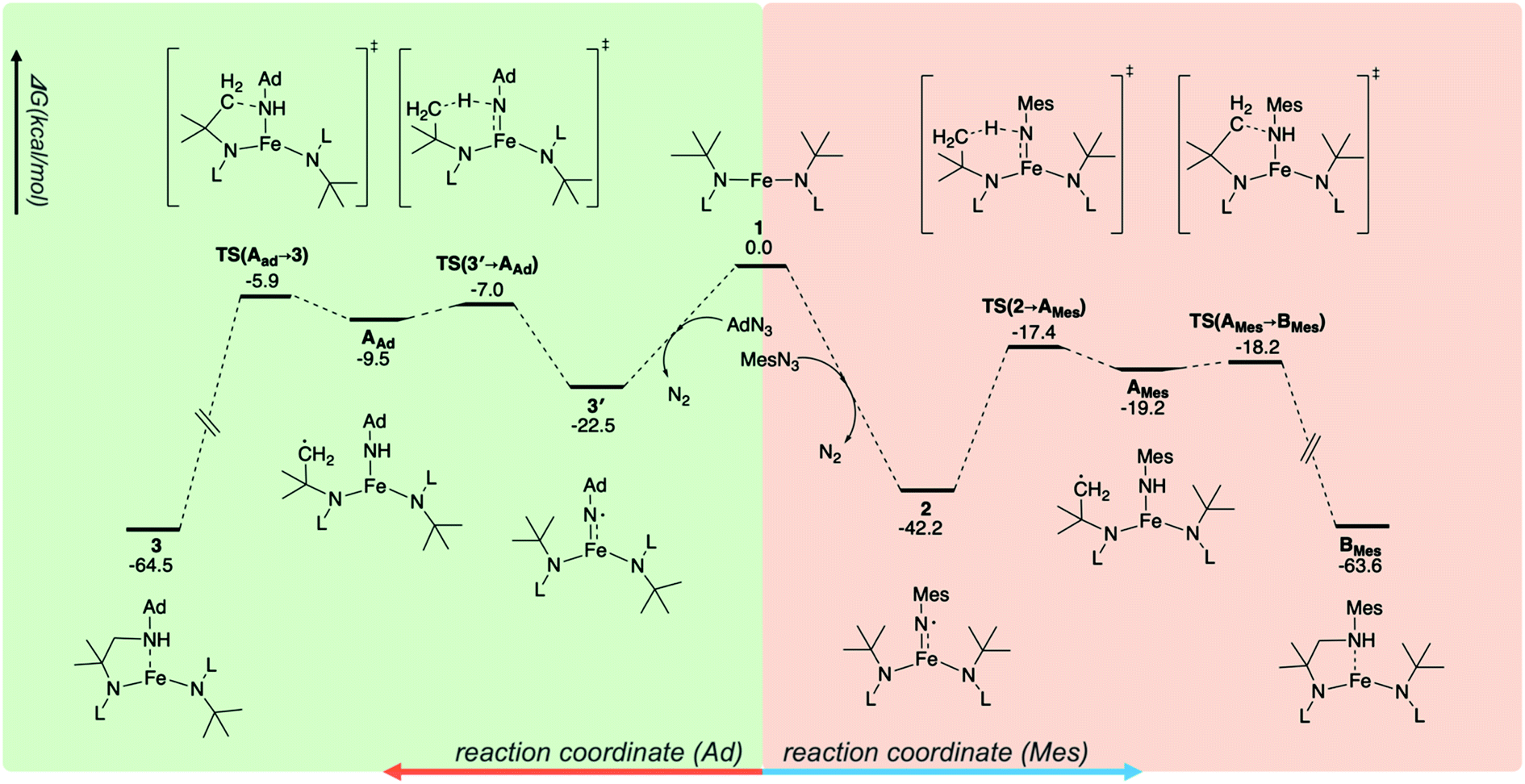 | ||
| Fig. 5 The reaction profiles for C–H amination. The green part is for the reaction of 1 with AdN3, whereas the orange part is for the reaction of 1 with MesN3. The unit of energy is kcal mol−1. The ground state for each stationary point along the reaction profiles is quintet. | ||
Interestingly, the analysis of the spin population based on the Mulliken scheme showed that in both 2 and 3′ the majority of the up spins are localized on Fe and the two N atoms of the N(tBu)Dipp ligands (4.30 e− for 2 and 4.25 e− for 3′), coupled antiferromagnetically with a low density of down spins localized on NMes (−0.38 e−, Fig. S27†) and NAd (−0.36 e−, Fig. S27†). Recalculating the electronic structures with a septet spin state led to flipping of the spins localized on NMes and NAd, with ΔG values of 5.1 and 7.6 kcal mol−1, respectively, higher than those of the corresponding quintet states. The sizable differences in the ΔG values between the two spin states indicates a significant exchange coupling between the spins on Fe and those on NMes and NAd. Fig. 6 shows spin density plots for 2 and 3′, both in the quintet and septet states. The electronic structure of 2 and 3′ were further corroborated by complete active space self-consistent field (CASSCF) calculation and subsequent localized orbital complete active space configuration interaction (CASCI) calculations to be a high-spin (S = 5/2) Fe(III) ion antiferromagnetically coupled to an imidyl radical (S = 1/2). The results can be found in the ESI.†
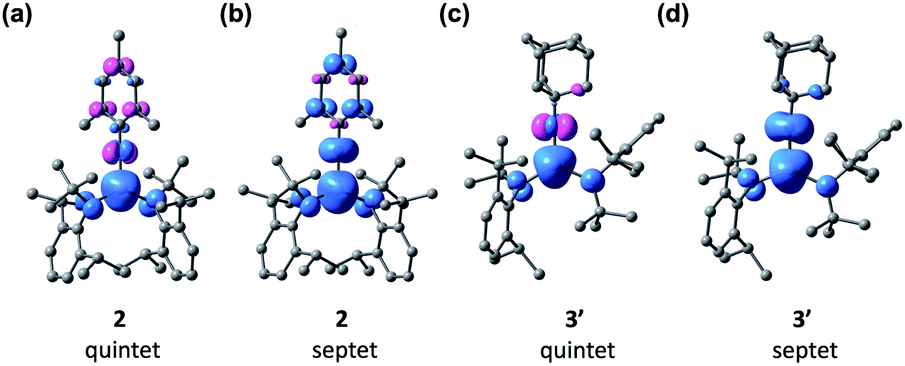 | ||
| Fig. 6 Spin density plots of the quintet and septet states of 2 [(a) and (b)] and 3′ [(c) and (d)] with isosurface values of 0.01 Å−3. | ||
Finally, the ΔG profiles of intramolecular C–H amination were examined. It was found that the reaction proceeds via the transfer of an H atom from one of the tert-butyl groups in N(tBu)Dipp to the N center of NMes and NAd, leading to the formation of AMes and AAd (Fig. 5). This elementary reaction is the rate determining step, with ΔG‡ of 24.8 and 15.5 kcal mol−1 for 2 → AMes and 3′ → AAd, respectively. Transferring H atom from the CH3 group of Mes in 2 to the imidyl N has an even higher ΔG‡ of 36.3 kcal mol−1. Thus, the present DFT calculations are qualitatively consistent with the experimental data, showing that 3′ is indeed more reactive toward intramolecular C–H amination than 2. The higher reactivity of 3′ is likely due to the higher localization of the down spin on the N center of NAd, increasing its reactivity, whereas for 2, the down spin is delocalized between N and Mes, making this complex inert. The connection between high spin density to high reactivity for metal imido and oxo systems is well documented in the literature.66–69 Alternatively, this trend can also be rationalized by the better thermodynamical driving force of 3′ (ΔG = 13.0 kcal mol−1, formation of a stronger N–H bond) compared to that of 2 (ΔG = 23.0 kcal mol−1) that leads to its higher reactivity.70 The reaction is completed by rebinding of the –CH2 radical generated after H atom abstraction with the N atom of NHMes or NHAd to form BMes or 3. This step is very facile and favorable: for the AMes → BMes reaction, ΔG‡ and ΔG are 1.0 and −44.4 kcal mol−1, respectively, whereas ΔG‡ is 3.6 kcal mol−1 and ΔG is −55.0 kcal mol−1 for AAd → 3.
Conclusions
A high-spin three-coordinate Fe imidyl complex 2 was isolated and characterized.71 Combined magnetic, spectroscopic, and computational (DFT, CASSCF, and CASCI) studies showed that 2 is an S = 2 Fe(III) imidyl complex, with an S = 5/2 ground state on Fe that is antiferromagnetically coupled to the imidyl radical on N. The reaction of 1 with AdN3 afforded the intramolecular C–H amination product 3, whereas the corresponding Fe imidyl intermediate (3′) could not be isolated. Quantum mechanical calculations shed light on the divergent reactivity and stability of 2 and 3′. In 2, the imidyl radical was found to be delocalized on the mesityl ring, making the N radical relatively inert. For 3, the aliphatic Ad substituent of the imidyl ligand resulted in the localization of the imidyl radical on N, making it highly reactive. These results highlight the intricate relationship between reactivity and stability and might pave the way for the design of highly active yet stable transition metal complexes.Data availability
Data for all new compounds and complexes (experimental and characterization details, 1H, vibrational, electronic absorption spectra) in this manuscript are provided in the ESI.† Computational details, including energies and xyz coordinates of the optimized structures, computed intermediates, and computed transition states are also available in the ESI.† Crystallographic data for LiN(tBu)Dipp (2122026), 1 (2119691), 2 (2119692), and 3 (2119693) have been deposited at the Cambridge Crystallographic Data Center (CCDC).Author contributions
C-YL and M-JC conceived this study and wrote the manuscript with inputs from all authors. C-TF performed the initial synthesis of 1 and 3. P-CY, K-PY, and C-TF performed the synthesis and characterization of all complexes, supervised by C-YL. C-TH performed computational studies supervised by M-JC. P-CY, K-PY, and C-WP performed XANES measurement. JZ performed Mössbauer spectroscopic studies supervised by LD. H-KL performed the SC-XRD studies of LiN(tBu)Dipp and 1–3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the Ministry of Science and Technology of Taiwan (Young Scholar Fellowship Program, MOST 108-2636-M-006-011, 109-2636-M-006-018, 110-2636-M-006-009, and 108-2113-M-006-015-MY2) and the National Cheng Kung University (through Higher Education Sprout Project of the Ministry of Education of Taiwan) for support of this work. We thank the Core Facility Center at the National Cheng Kung University (MOST 110-2731-M-006-001) for the use of the Bruker AV-500 NMR spectrometer (NMR000700), Quantum Design MPMS SQUID VSM (SQUID000200), and Elementar UNICUBE (EA000600), the Instrumentation Center at the National Tsing Hua University for the EPR measurement, and Professor Ho-Hsuan Chou for the use of Shimadzu UV-1800 spectrophotometer in his lab. We also thank anonymous reviewers for their invaluable comments and suggestions.Notes and references
- J. F. Berry, Comments Inorg. Chem., 2009, 30, 28–66 CrossRef CAS.
- J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed.
- C. T. Saouma and J. C. Peters, Coord. Chem. Rev., 2011, 255, 920–937 CrossRef CAS PubMed.
- C.-M. Che, C.-Y. Zhou and E. L.-M. Wong, in Iron Catalysis, ed. B. Plietker, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, vol. 33, pp. 111–138 Search PubMed.
- Y. Liu, T. You, T.-T. Wang and C.-M. Che, Tetrahedron, 2019, 75, 130607 CrossRef.
- A. K. Verma, T. N. Nazif, C. Achim and S. C. Lee, J. Am. Chem. Soc., 2000, 122, 11013–11014 CrossRef CAS.
- S. D. Brown, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 322–323 CrossRef CAS PubMed.
- T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2003, 125, 10782–10783 CrossRef CAS PubMed.
- S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed.
- M. P. Mehn, S. D. Brown, D. M. Jenkins, J. C. Peters and L. Que, Inorg. Chem., 2006, 45, 7417–7427 CrossRef CAS PubMed.
- C. M. Thomas, N. P. Mankad and J. C. Peters, J. Am. Chem. Soc., 2006, 128, 4956–4957 CrossRef CAS PubMed.
- S. C. Bart, E. Lobkovsky, E. Bill and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 5302–5303 CrossRef CAS PubMed.
- C. C. Lu, C. T. Saouma, M. W. Day and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 4–5 CrossRef CAS PubMed.
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed.
- J. J. Scepaniak, J. A. Young, R. P. Bontchev and J. M. Smith, Angew. Chem., Int. Ed., 2009, 48, 3158–3160 CrossRef CAS PubMed.
- R. E. Cowley, N. J. DeYonker, N. A. Eckert, T. R. Cundari, S. DeBeer, E. Bill, X. Ottenwaelder, C. Flaschenriem and P. L. Holland, Inorg. Chem., 2010, 49, 6172–6187 CrossRef CAS PubMed.
- M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063–2067 CrossRef CAS PubMed.
- A. C. Bowman, C. Milsmann, E. Bill, Z. R. Turner, E. Lobkovsky, S. DeBeer, K. Wieghardt and P. J. Chirik, J. Am. Chem. Soc., 2011, 133, 17353–17369 CrossRef CAS PubMed.
- R. E. Cowley and P. L. Holland, Inorg. Chem., 2012, 51, 8352–8361 CrossRef CAS PubMed.
- S. Kuppuswamy, T. M. Powers, B. M. Johnson, M. W. Bezpalko, C. K. Brozek, B. M. Foxman, L. A. Berben and C. M. Thomas, Inorg. Chem., 2013, 52, 4802–4811 CrossRef CAS PubMed.
- S. Kuppuswamy, T. M. Powers, B. M. Johnson, C. K. Brozek, J. P. Krogman, M. W. Bezpalko, L. A. Berben, J. M. Keith, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2014, 53, 5429–5437 CrossRef CAS PubMed.
- H. Zhang, Z. Ouyang, Y. Liu, Q. Zhang, L. Wang and L. Deng, Angew. Chem., Int. Ed., 2014, 53, 8432–8436 CrossRef CAS PubMed.
- K. Searles, S. Fortier, M. M. Khusniyarov, P. J. Carroll, J. Sutter, K. Meyer, D. J. Mindiola and K. G. Caulton, Angew. Chem., Int. Ed., 2014, 53, 14139–14143 CrossRef CAS PubMed.
- L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed.
- B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145–12148 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 12043–12049 CrossRef CAS PubMed.
- J. Cheng, J. Liu, X. Leng, T. Lohmiller, A. Schnegg, E. Bill, S. Ye and L. Deng, Inorg. Chem., 2019, 58, 7634–7644 CrossRef CAS PubMed.
- K. E. Aldrich, B. S. Fales, A. K. Singh, R. J. Staples, B. G. Levine, J. McCracken, M. R. Smith and A. L. Odom, Inorg. Chem., 2019, 58, 11699–11715 CrossRef CAS PubMed.
- M. R. Anneser, G. R. Elpitiya, J. Townsend, E. J. Johnson, X. B. Powers, J. F. DeJesus, K. D. Vogiatzis and D. M. Jenkins, Angew. Chem., Int. Ed., 2019, 58, 8115–8118 CrossRef CAS PubMed.
- M. J. T. Wilding, D. A. Iovan, A. T. Wrobel, J. T. Lukens, S. N. MacMillan, K. M. Lancaster and T. A. Betley, J. Am. Chem. Soc., 2017, 139, 14757–14766 CrossRef CAS PubMed.
- J. L. Martinez, S. A. Lutz, H. Yang, J. Xie, J. Telser, B. M. Hoffman, V. Carta, M. Pink, Y. Losovyj and J. M. Smith, Science, 2020, 370, 356–359 CrossRef CAS PubMed.
- Y. Gao, V. Carta, M. Pink and J. M. Smith, J. Am. Chem. Soc., 2021, 143, 5324–5329 CrossRef CAS PubMed.
- C. A. Richards, N. P. Rath and J. M. Neely, Organometallics, 2021, 40, 2945–2950 CrossRef CAS.
- Q. Liu, L. Long, P. Ma, Y. Ma, X. Leng, J. Xiao, H. Chen and L. Deng, Cell Rep. Phys. Sci., 2021, 2, 100454 CrossRef CAS.
- Y. Gao, M. Pink and J. M. Smith, J. Am. Chem. Soc., 2022, 144, 1786–1794 CrossRef CAS PubMed.
- A. I. O. Suarez, V. Lyaskovskyy, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2013, 52, 12510–12529 CrossRef CAS PubMed.
- K. Ray, F. Heims and F. F. Pfaff, Eur. J. Inorg. Chem., 2013, 3784–3807 CrossRef CAS.
- A. Grünwald, S. S. Anjana and D. Munz, Eur. J. Inorg. Chem., 2021, 4147–4166 CrossRef.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- D. A. Iovan and T. A. Betley, J. Am. Chem. Soc., 2016, 138, 1983–1993 CrossRef CAS PubMed.
- K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138–1143 CrossRef CAS PubMed.
- Y. Dong, J. T. Lukens, R. M. Clarke, S.-L. Zheng, K. M. Lancaster and T. A. Betley, Chem. Sci., 2020, 11, 1260–1268 RSC.
- A. Reckziegel, M. Kour, B. Battistella, S. Mebs, K. Beuthert, R. Berger and C. G. Werncke, Angew. Chem., Int. Ed., 2021, 60, 15376–15380 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS PubMed.
- Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166–1169 CrossRef CAS PubMed.
- J. A. Kelly, M. Juckel, T. J. Hadlington, I. Fernández, G. Frenking and C. Jones, Chem.–Eur. J., 2019, 25, 2773–2785 CrossRef CAS PubMed.
- J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125–138 RSC.
- C.-Y. Lin, J.-D. Guo, J. C. Fettinger, S. Nagase, F. Grandjean, G. J. Long, N. F. Chilton and P. P. Power, Inorg. Chem., 2013, 52, 13584–13593 CrossRef CAS PubMed.
- H. Y. Au-Yeung, C. H. Lam, C.-K. Lam, W.-Y. Wong and H. K. Lee, Inorg. Chem., 2007, 46, 7695–7697 CrossRef CAS PubMed.
- J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed.
- D. J. Liptrot and P. P. Power, Nat. Rev. Chem., 2017, 1, 0004 CrossRef.
- P. P. Power, Chem. Rev., 2012, 112, 3482–3507 CrossRef CAS PubMed.
- W. M. Reiff, A. M. LaPointe and E. H. Witten, J. Am. Chem. Soc., 2004, 126, 10206–10207 CrossRef CAS PubMed.
- W. M. Reiff, C. E. Schulz, M.-H. Whangbo, J. I. Seo, Y. S. Lee, G. R. Potratz, C. W. Spicer and G. S. Girolami, J. Am. Chem. Soc., 2009, 131, 404–405 CrossRef CAS PubMed.
- W. A. Merrill, T. A. Stich, M. Brynda, G. J. Yeagle, J. C. Fettinger, R. De Hont, W. M. Reiff, C. E. Schulz, R. D. Britt and P. P. Power, J. Am. Chem. Soc., 2009, 131, 12693–12702 CrossRef PubMed.
- A. M. Bryan, C.-Y. Lin, M. Sorai, Y. Miyazaki, H. M. Hoyt, A. Hablutzel, A. LaPointe, W. M. Reiff, P. P. Power and C. E. Schulz, Inorg. Chem., 2014, 53, 12100–12107 CrossRef CAS PubMed.
- A. Sridharan, A. C. Brown and D. L. M. Suess, Angew. Chem., Int. Ed., 2021, 60, 12802–12806 CrossRef CAS PubMed.
- Y. Park, S. P. Semproni, H. Zhong and P. J. Chirik, Angew. Chem., Int. Ed., 2021, 60, 14376–14380 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- P. Gütlich, E. Bill and A. X. Trautwein, in Mössbauer Spectroscopy and Transition Metal Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 73–135 Search PubMed.
- N. A. Eckert, S. Vaddadi, S. Stoian, R. J. Lachicotte, T. R. Cundari and P. L. Holland, Angew. Chem., Int. Ed., 2006, 45, 6868–6871 CrossRef CAS PubMed.
- Y.-R. Luo, Handbook of bond dissociation energies in organic compounds, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- J. M. Mayer, Acc. Chem. Res., 2011, 44, 36–46 CrossRef CAS PubMed.
- R. Bjornsson, F. Neese and S. DeBeer, Inorg. Chem., 2017, 56, 1470–1477 CrossRef CAS PubMed.
- D. Balcells, C. Raynaud, R. H. Crabtree and O. Eisenstein, Inorg. Chem., 2008, 47, 10090–10099 CrossRef CAS PubMed.
- M. D. Fayer, Acc. Chem. Res., 2012, 45, 3–14 CrossRef CAS PubMed.
- W. Lai, C. Li, H. Chen and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 5556–5578 CrossRef CAS PubMed.
- L. Hu and H. Chen, ACS Catal., 2017, 7, 285–292 CrossRef CAS.
- C. T. Saouma and J. M. Mayer, Chem. Sci., 2014, 5, 21–31 RSC.
- During submission and revision of this manuscript, similar three-coordinate imidyl complexes of Fe were reported: S. Reith, S. Demeshko, B. Battistella, A. Reckziegel, C. Schneider, A. Stoy, C. Lichtenberg, F. Meyer, D. Munz and C. G. Werncke, Chem. Sci., 2022, 13(26), 7907–7913 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. Computational details. Crystal data, details of data collection and refinements. CCDC 2119691–2119693 and 2122026. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02699f |
| This journal is © The Royal Society of Chemistry 2022 |