
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented pairs of uranium (IV/V) hydroxido and (IV/V/VI) oxido complexes supported by a seven-coordinate cyclen-anchored tris-aryloxide ligand†
Sascha T.
Löffler‡
 ,
Julian
Hümmer‡
,
Andreas
Scheurer
,
Frank W.
Heinemann
and
Karsten
Meyer
*
,
Julian
Hümmer‡
,
Andreas
Scheurer
,
Frank W.
Heinemann
and
Karsten
Meyer
*
Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Department of Chemistry and Pharmacy, Inorganic Chemistry, Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: karsten.meyer@fau.de
First published on 12th August 2022
Abstract
We present the synthesis and reactivity of a newly developed, cyclen-based tris-aryloxide ligand precursor, namely cyclen(Me)(t-Bu,t-BuArOH)3, and its coordination chemistry to uranium. The corresponding uranium(III) complex [UIII((OArt-Bu,t-Bu)3(Me)cyclen)] (1) was characterized by 1H NMR analysis, CHN elemental analysis and UV/vis/NIR electronic absorption spectroscopy. Since no single-crystals suitable for X-ray diffraction analysis could be obtained from this precursor, 1 was oxidized with methylene chloride or silver fluoride to yield [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(X)] (X = Cl (2), F (3)), which were unambiguously characterized and successfully crystallized to gain insight into the molecular structure by single-crystal X-ray diffraction analysis (SC-XRD). Further, the activation of H2O and N2O by 1 is presented, resulting in the U(IV) complex [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(OH)] (4) and the U(V) complex [(cyclen(Me)(t-Bu,t-BuArO)3)UV(O)] (6). Complexes 2, 3, 4, and 6 were characterized by 1H NMR analysis, CHN elemental analysis, UV/vis/NIR electronic absorption spectroscopy, IR vibrational spectroscopy, and SQUID magnetization measurements as well as cyclic voltammetry. Furthermore, chemical oxidation of 3, 4, and 6 with AgF or AgSbF6 was achieved leading to complexes [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5), [(cyclen(Me)(t-Bu,t-BuArO)3)UV(OH)][SbF6] (7), and [(cyclen(Me)(t-Bu,t-BuArO)3)UVI(O)][SbF6] (8). Finally, reduction of 7 with KC8 yielded a U(IV) complex, spectroscopically and magnetochemically identified as K[(cyclen(Me)(t-Bu,t-BuArO)3)UIV(O)].
Introduction
In low oxidation states, uranium complexes are immensely sensitive to even trace amounts of oxygen and/or water and, in uncontrolled reactions, tend to be fully oxidized to U(VI) compounds in the form of the thermodynamically stable uranyl dication [UVIO2]2+.1–7 Whereas a few reports of the formation and isolation of U(IV) hydroxido compounds exist,8–15 the controlled reaction with water – in one-electron oxidation processes with the concomitant release of dihydrogen – is only sparsely reported in the literature.16–19 The first uranium hydroxido complex was reported by Berthet and Ephritikhine in 1993. In a hydrolysis reaction of the uranium(IV) hydrido complex [(C5H4R)3UIVH] (R = t-butyl or SiMe3), with 1 equivalent of H2O in toluene, the hydroxide species [(C5H4R)3UIVOH] (R = t-butyl and SiMe3) were obtained.8 In 1996, Lukens et al. reported the formation and characterization of the dinuclear, bis-hydroxido-bridged complexes [{(Cp')2UIV}2(μ-OH)2] (Cp' = 1,3-(Me3Si)2C5H3) and [{(Cp′′)2UIV}2(μ-OH)2] (Cp′′ = 1,3-(Me3C)2C5H3) from the reaction of [UIII(Cp')3] and [{(Cp')3UIV}2(μ-H)2] with H2O, respectively.9 The first example of a crystallographically characterized terminal uranium(IV) hydroxide was given in 2002 by Scott and co-workers. The oxidation of [(NN′3)UIII(CH2PMe3)] (NN′3 = N(CH2CH2NSiMe2tBu)3) resulted in a non-reproducible structural characterization of [(NN′3)UIV(OH)(CH2PMe3)].10 Besides, only a few further examples of hydroxido compounds are structurally determined, but these examples were obtained from accidental and uncontrolled reactions, which can be traced back to minor impurities of water (moisture) and were not reproducible in most cases.11,14,15 Ariyaratne et al. reported the reaction of [(Cp*)2UIV(Cl)2(HNSPh2)] with 1 equivalent of HNSPh2 × H2O to give [(Cp*)2UIV(OH)(Cl)(HNSPh2)]. The terminal hydroxido complex is an intermediate in the formation of the tetra uranium oxido cluster [Cp*(Cl)(HNSPh2)U(μ3-O)(μ2-O)2U(Cl)(HNSPh2)2]2, which forms upon hydrolysis of [(Cp*)2U(Cl)2] with excess HNSPh2 × H2O.12Hence, up until now, reports of controlled one- or two-electron oxidations of uranium compounds to form defined terminal hydroxido and oxido complexes, even with excess amount of H2O, still remain scarce, and are of current interest to our group in the context of the electrocatalytic generation of dihydrogen from water. In 2016, we reported the electrocatalytic production of H2 mediated by [(mes(Ad,MeArO)3)UIV(OH)],16,20 and a series of uranium hydrochalcogenido (EH) and chalcogenido (E) (with E = O, S, Se, Te) uranium(IV) compounds.18 In the latter report, solutions of [UIII((OArAd,Me)3tacn)] were allowed to react with stoichiometric amounts of the hydrochalcogenides H2O, H2S, H2Se, and H2Te in THF to obtain the corresponding complexes [(tacn(Ad,MeArO)3)UIV(EH)]. While the reaction for the higher homologous H2S, H2Se and H2Te is relatively fast, the reaction with H2O is notably slow, takes at least three days, and the reaction outcome was observed to be the same even when an excess of H2O was used.17,18
In contrast to the stable uranyl dication [UVIO2]2+, complexes of uranium with reactive terminal oxido ligands – of which [(mes(Ad,MeArO)3)UV(O)] has been identified as an intermediate in the above-mentioned electrocatalytic production of H2 from H2O – require particular demands for their synthesis and spectroscopic characterization.5,20–29 Accordingly, the number of such complexes is comparably limited and insight into the understanding of the intrinsic bonding situation is still lacking behind their uranyl analogues. Consequently, compounds featuring uranium–oxygen bonds in the form of oxidos18,20,29–43 or hydroxidos10–16,18 are of particular interest, due to the varying degree of multiple bonding character in the U–O bonds and their resulting reactivity. In combination with a strictly retained ligand environment, essential information about the molecular and electronic structure of these U–O/OH species can be obtained. However, pairs of uranium oxido/hydroxido complexes – supported by an identical ligand system – remain exceedingly rare.
Here, we present the preparation, characterization, and redox-behavior of two uranium(IV/V) and uranium(IV/V/VI) hydroxido/oxido pairs, and closely related fluorido complexes, utilizing a low-valent uranium center surrounded by a newly developed chelate, the cyclen-anchored tris-aryloxide ligand precursor 1,4,7-tris-(3,5-di-tert-butyl-2-hydroxy-benzyl)-10-methyl-1,4,7,10-tetraazacyclododecane, abbreviated as cyclen(Me)(t-Bu,t-BuArOH)3 (Scheme 1, left).
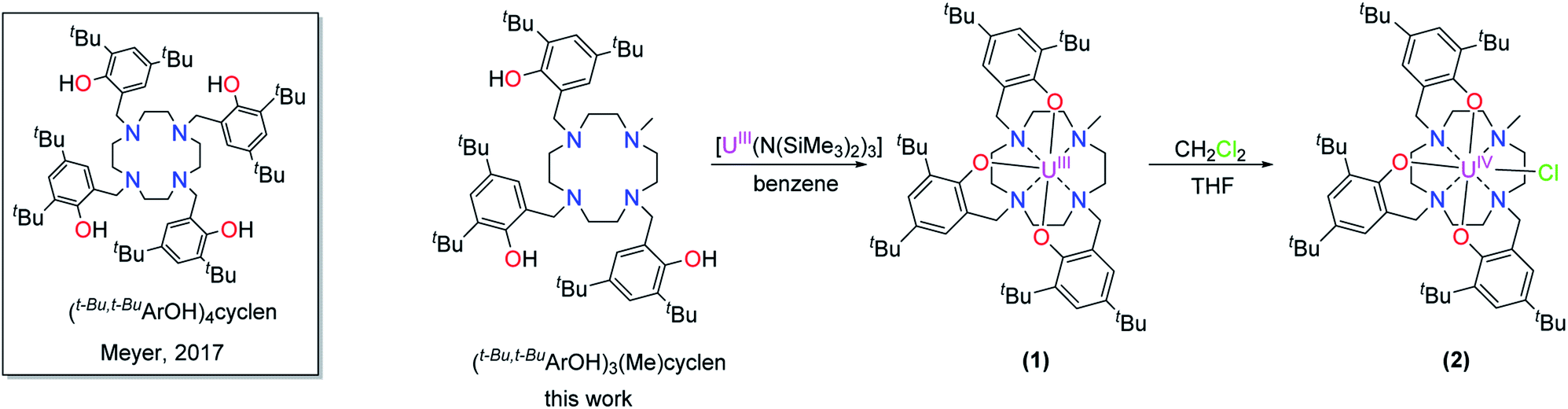 | ||
| Scheme 1 Synthesis of [UIII((OArt-Bu,t-Bu)3(Me)cyclen)] (1) and [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(Cl)] (2). | ||
Results and discussion
The new seven-coordinate tris-aryloxide ligand precursor, cyclen(Me)(t-Bu,t-BuArOH)3, represents a variation of the literature-known, cyclen-anchored, eight-coordinate tetrakis-aryloxide (cyclen(t-Bu,t-BuArOH)4) (Scheme 1). The corresponding U(IV) complex, [UIV((OArt-Bu,t-Bu)4cyclen)], possesses a remarkable stability with accompanied low reactivity towards small molecules; most notably, including H2O.44 Cyclen-anchored tetrakis-aryloxide chelates of the type cyclen(R1,R2ArO)4 (with R1 = Me, t-Bu; R2 = Me) were first coordinated to terbium.45,46 To increase their metal complexes' reactivity and enable the capability of small molecule activation, one aryloxide arm is substituted by a methyl (Me) group. This opens up coordination sites in axial and equatorial positions and engenders the formation of a more reactive trivalent precursor after complexation to uranium. For this purpose, the cyclen-derived anchoring unit 1-methyl-1,4,7,10-tetraazacyclododecane (methyl-cyclen) was synthesized by a modest variation of a literature-known procedure, adopted from Rodríguez–Rodríguez et al.47 A subsequent SN2-reaction with 2-bromomethyl-4,6-di-t-butylphenol as the electrophile yields the ligand precursor (1,4,7-tris-(3,5-di-tert-butyl-2-hydroxy-benzyl)-10-methyl-1,4,7,10-tetraazacyclododecane) ((t-Bu,t-BuArOH)3(Me)cyclen; yield: 93%).Reaction of cyclen(Me)(t-Bu,t-BuArOH)3 with [UIII(N(SiMe3)2)3]48 in thawing benzene generates the target precursor complex [UIII((OArt-Bu,t-Bu)3(Me)cyclen)] (1) in 65% yield (Scheme 1). 1H NMR analysis of 1 in benzene-d6 shows 12 of the 15 signals for the 85 hydrogen atoms, paramagnetically broadened and shifted, in the range from 15.17 to –13.29 ppm (Fig. S6†). The number of signals suggests CS symmetry in solution and the lack of some signals can be explained by strong broadening due to the coordination of the chelate to the paramagnetic U(III) f3 ion. The bulk purity of 1 was established by CHN combustion analysis, and its 3+ oxidation state is unambiguously supported by UV/vis/NIR electronic absorption spectroscopy (Fig. 3, top) and SQUID magnetic measurements (Fig. S47–S49†). We assume that the ligand's high flexibility prevents the formation of suitable single-crystals for X-ray diffraction (SC-XRD) analysis. Although the structural proof of 1 is missing, all characterizations, the subsequent reactivity, and structural studies support the formation and existence of the targeted uranium(III) precursor complex. For instance, oxidation of 1 with a few drops of methylene chloride yields the corresponding uranium(IV) complex, [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(Cl)] (2) (Scheme 1, 76% yield). For 2, suitable single-crystals for SC-XRD analysis were obtained by slow diffusion of pentane into a concentrated THF solution (2 × 2 THF), which crystallizes in the orthorhombic space group P212121. Better quality crystals of 2 were obtained from a concentrated benzene solution, which crystallized in the monoclinic space group P21/n. The molecular structures observed in the two determined crystal structures are very similar and only the latter (2 × 2.5 benzene) will be discussed here (Fig. 1, top; Table 1). Tetravalent 2 reveals a distorted square antiprismatic geometry with an N4O3Cl polyhedron, consisting of the cyclen's N4 and an O3Cl plane. Notably, the chloride ligand is not coordinated in an axial position but with a slight shift of 0.628 Å out of the plane of the three aryloxide ligands and with a dihedral angle of 47.2(3)° to the substituted cyclic amine's methyl group. The U–Cl bond distance is 2.734(1) Å and the average U–OArO (2.179(3) Å) and U–Ncyclen (2.732(4) Å) distances are in agreement with literature values of the cyclen- and tacn-based U(IV) complexes [U(OArt-Bu,t-Bu)4cyclen)]44 and [(tacn(R,R′ArO)3)UIV(Cl)] (R = tAmyl, DMB, Ad, neoP, t-Bu, Dia; R′ = tAmyl, DMB, t-Bu, Me) (Cl, axially bound, d(U–Cl) = 2.691(3)–2.875(5) Å).49–52
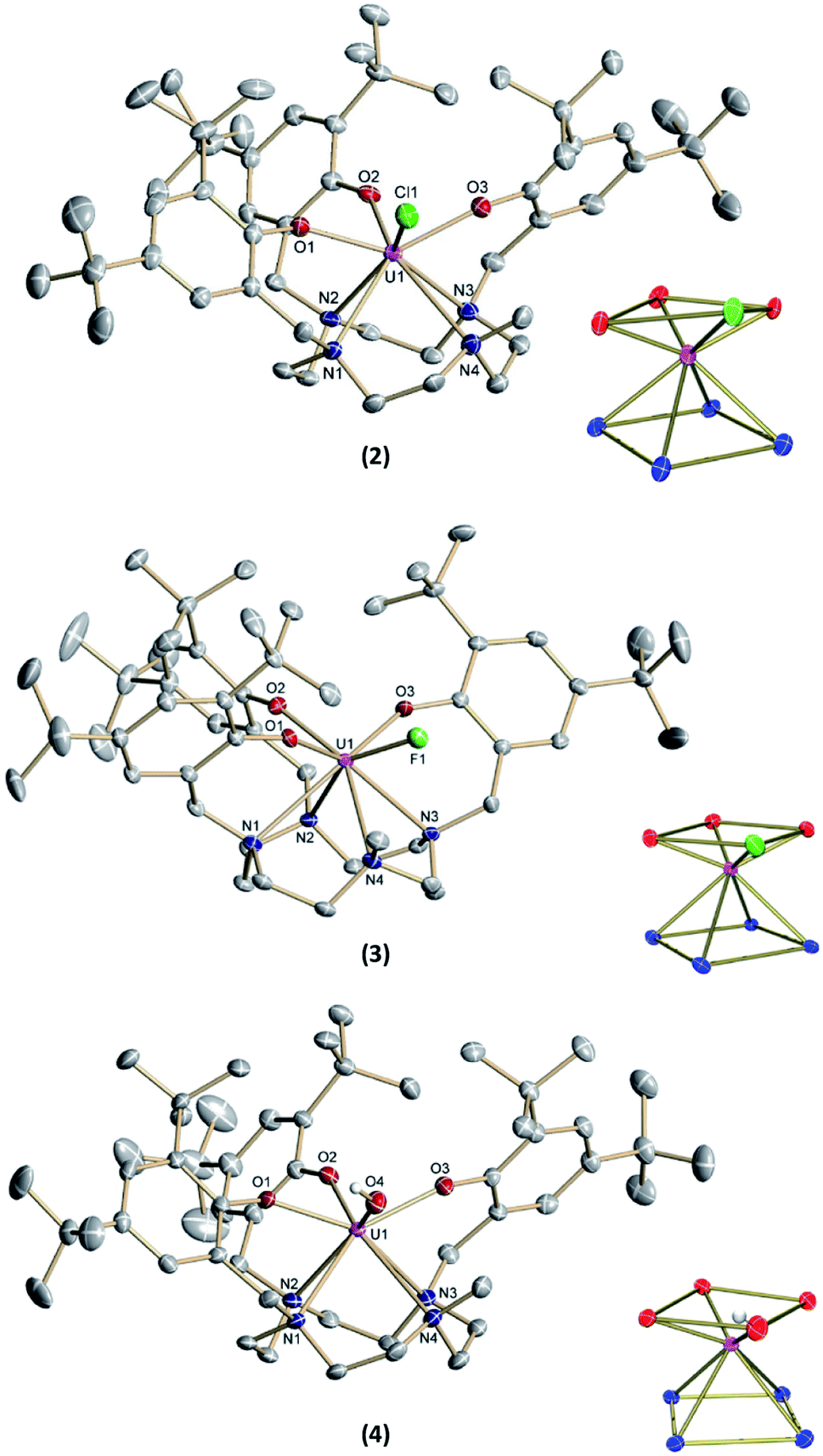 | ||
| Fig. 1 Solid-state molecular structures of 2 (top), 3 (middle), and 4 (bottom) in crystals of [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(Cl)] × 2.5 benzene, [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(F)] × 2 Et2O, and [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(OH)] × 2 THF, respectively, as well as the coordination polyhedra of the heteroatoms (each bottom right). Hydrogen atoms (except for the hydroxyl H), co-crystallized solvent molecules, and the second component (3) in the mixed crystal are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. | ||
Treatment of [(cyclen(Me)(t-Bu,t-BuArO)3)UIII] (1) with stoichiometric amounts of AgF induces a one-electron oxidation and leads to the formation of [(cyclen(Me)(t-Bu,t-BuArO)3)U(F)] (3) in 36% yield with concomitant precipitation of elemental silver. Similarly, oxidation of 1 with a slight excess of H2O (0.2 M solution in THF) results in the corresponding U(IV) hydroxido derivative [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(OH)] (4) in 23% yield (Scheme 2). 1H NMR spectroscopy reveals signals ranging from 60.78 to −65.59 ppm for 3 and 64.76 to −67.77 ppm for 4, respectively (Fig. S8 and S9†). As expected, the spectra of 3 and 4 are quite comparable and show the 35 signals originating from the 85 and 86 protons, respectively. This is in accordance with the expected CS symmetry in the solution. While the acidic OH proton of 4 has not been observed and likely is broadened into the baseline by rapid exchange or paramagnetism, the six most dominant peaks at 4.88, 4.04, 3.83, −6.52, −9.65 and −12.35 ppm for 3 and 5.16, 5.00, 3.82, −3.16, −12.26 and −13.35 ppm for 4 can be assigned to the t-butyl groups. Moreover, the methyl groups can be detected at 33.21 ppm for 3 and 20.14 ppm for 4. The OH group of 4 was detected via infrared vibrational spectroscopy centered at a wavenumber ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) of 3675 cm−1.
of 3675 cm−1.
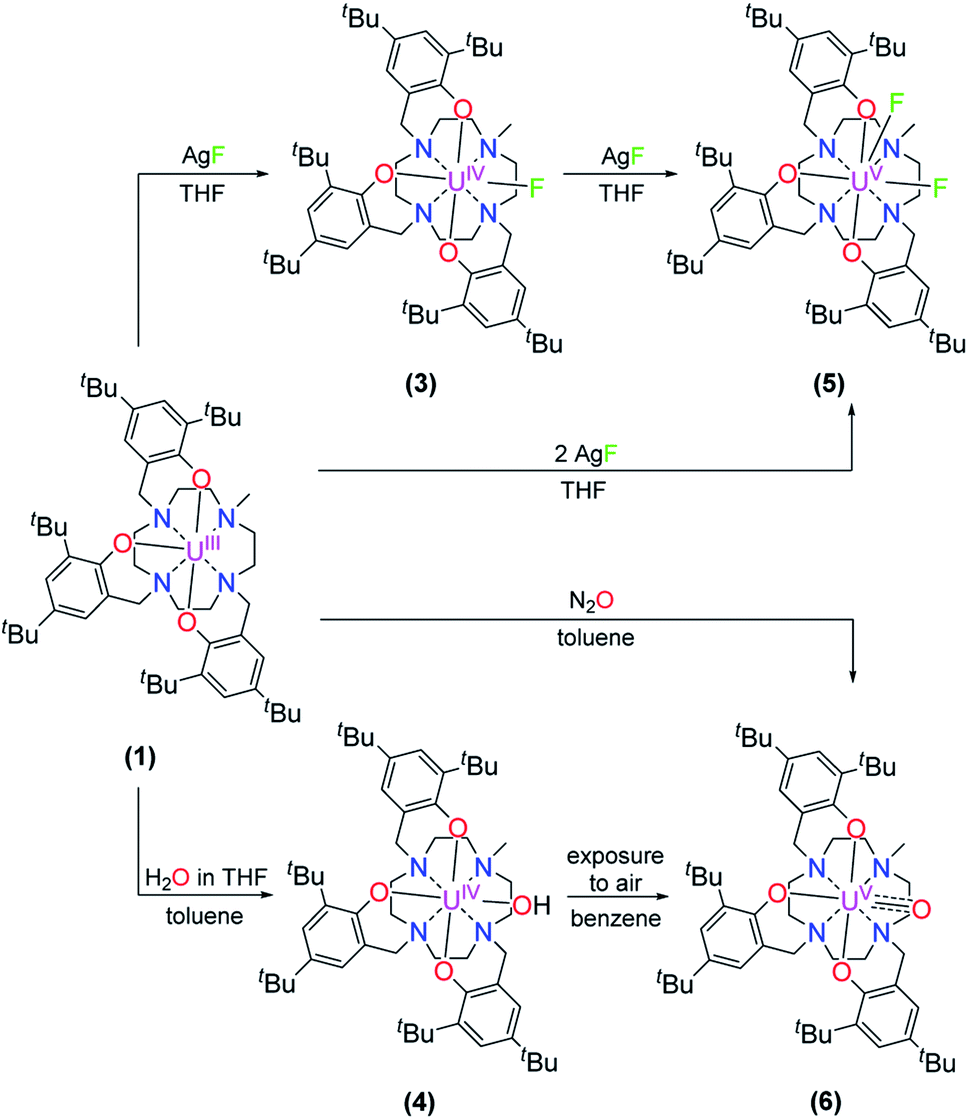 | ||
| Scheme 2 Synthesis of [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(F)] (3), [(cyclen(Me)(t-Bu,t-BuArO)3UIV(OH)] (4), [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5), and [(cyclen(Me)(t-Bu,t-BuArO)3)UV(O)] (6). | ||
| 2 × 2 THF | 2 × 2.5 benzene | 3 | 4 | 5 | 6 | [(cyclen(t-Bu,t-BuArO)4)U] | |
|---|---|---|---|---|---|---|---|
| U–O1ArO | 2.152(6) | 2.195(3) | 2.176(2) | 2.231(2) | 2.123(2) | 2.234(3) | 2.203(2) |
| U–O2ArO | 2.211(6) | 2.173(2) | 2.213(2) | 2.233(2) | 2.096(2) | 2.183(3) | 2.228(2) |
| U–O3ArO | 2.175(6) | 2.170(3) | 2.196(2) | 2.208(2) | 2.137(2) | 2.211(4) | 2.203(2) |
| U–O4ArO | — | — | — | — | — | — | 2.228(2) |
| U–OArO (av) | 2.179(6) | 2.179(3) | 2.195(2) | 2.224(2) | 2.119(2) | 2.209(6) | 2.216(2) |
| U–N1 | 2.717(8) | 2.715(3) | 2.770(2) | 2.730(2) | 2.895(3) | 2.688(4) | 2.801(2) |
| U–N2 | 2.675(7) | 2.708(3) | 2.778(2) | 2.784(3) | 2.851(3) | 2.759(4) | 2.755(2) |
| U–N3 | 2.765(8) | 2.772(3) | 2.708(2) | 2.817(2) | 2.983(3) | 2.850(4) | 2.801(2) |
| U–N4 | 2.788(8) | 2.810(3) | 2.805(2) | 2.817(3) | 3.018(3) | 2.743(4) | 2.755(2) |
| U–Ncyclen (av) | 2.736(8) | 2.751(3) | 2.765(2) | 2.787(3) | 2.937(3) | 2.760(4) | 2.778(2) |
| Uoop | −0.949(3) | −0.952(2) | −0.875(5) | −0.865(1) | −0.480(2) | −0.801(2) | −0.879(2) |
| U–Leq | 2.770(2) | 2.734(1) | 2.118(1) | 2.141(2) | 2.106(2) | 1.850(3) | — |
| U–Lax | — | — | — | — | 2.105(2) | — | — |
Suitable single-crystals of complexes 3 and 4 were grown from a concentrated THF solution and slow evaporation of a saturated THF solution, respectively (Fig. 1). Both complexes crystallize in monoclinic space groups, namely P21/c (3) and P21/n (4). The U–X (X = F (3) and OH (4)) bond distances are 2.118(2) Å and 2.141(2) Å, respectively. Notably, in the crystal structure of 4, the position of the O4 bound hydrogen atom H4 has been unequivocally identified in a difference Fourier map. The correctness of the determined H4 position is further supported by the involvement of this O–H group in a hydrogen bond to the oxygen atom of one of the located solvent tetrahydrofuran molecules (for details see ESI†). The average U–OArO und U–Ncyclen distances are in agreement with literature-reported values of the cyclen- and tacn-based U(IV) complexes [(cyclen(t-Bu,t-BuArO)4)U] and [(tacn(R,R′ArO)3)UIV(X)] (X = axially bound ligand) (Table 1). Both molecules reveal a distorted square antiprism-like geometry with an N4O3X polyhedron scaffold consisting of an N4 and an O3X plane. Thereby, the fluorido and hydroxido ligand fit well into the vacant site of the O3X plane with out-of-plane shifts of 0.040(3) Å for 3 and 0.165(5) Å for 4. Tetravalent 3 is conveniently oxidized with silver fluoride to yield the corresponding U(V) complex [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5) in 23% yield (Scheme 2). Compound 5 is also accessible by treating 1 with two equivalents of AgF. Suitable single crystals of 5 were obtained from slow diffusion of n-pentane into a concentrated Et2O solution at −30 °C, but always in the form of mixed crystals with an approximate ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20% of the components [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5) (Fig. 2, top) and [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(F)] (3). The compound crystallizes in the monoclinic space group P21/n, revealing the expected cis orientation of the two fluorido ligands for 5. The geometry of the complex is best described as capped, distorted square antiprismatic, with one fluorido ligand in the equatorial O3Feq plane and one axial fluorido ligand (Fax). The occupation of the axial position forces Feq to shift towards the cyclen methyl group. The resulting repulsion leads to a slightly longer U–N4 distance (3.018(3) Å with regard to complex 3 (2.805(2) Å). Complex 5 demonstrates that it is, in principle, possible to bind a ninth, axial ligand in cis-position to a monodentate equatorial ligand. This is an important prerequisite in water reduction catalysis, where a cis-hydroxido/hydrido intermediate is an imperative intermediate in the postulated reaction mechanism.20
:20% of the components [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5) (Fig. 2, top) and [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(F)] (3). The compound crystallizes in the monoclinic space group P21/n, revealing the expected cis orientation of the two fluorido ligands for 5. The geometry of the complex is best described as capped, distorted square antiprismatic, with one fluorido ligand in the equatorial O3Feq plane and one axial fluorido ligand (Fax). The occupation of the axial position forces Feq to shift towards the cyclen methyl group. The resulting repulsion leads to a slightly longer U–N4 distance (3.018(3) Å with regard to complex 3 (2.805(2) Å). Complex 5 demonstrates that it is, in principle, possible to bind a ninth, axial ligand in cis-position to a monodentate equatorial ligand. This is an important prerequisite in water reduction catalysis, where a cis-hydroxido/hydrido intermediate is an imperative intermediate in the postulated reaction mechanism.20
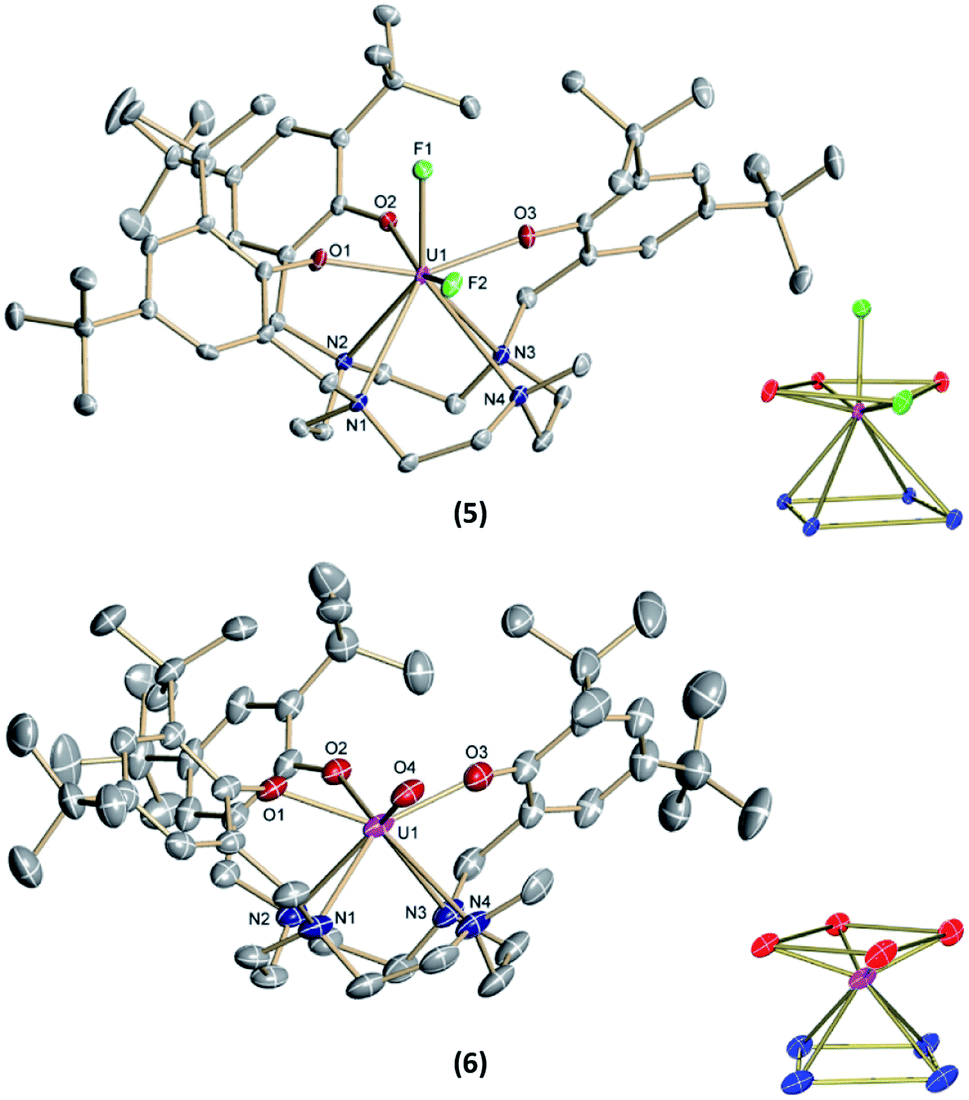 | ||
| Fig. 2 Solid-state molecular structures of 5 (top) and 6 (bottom) in crystals of [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2]0.8[(cyclen(Me)(t-Bu,t-BuArO)3)UIV(F)]0.2 × 2 Et2O and [(cyclen(Me)(t-Bu,t-BuArO)3)UV(O)] × 1.5 THF, respectively, as well as the coordination polyhedra of the heteroatoms (each bottom right). Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. | ||
The 1H NMR spectra of the U(V) complex 5 shows 26 resonances that range from 17.08 to −5.94 ppm (Fig. S10†). The spectrum shows a pattern consisting of six resonances in the aromatic region (10.51 to 8.04 ppm), one broad signal (5.94 ppm), two nearby sharp singlets (4.49 and 4.31 ppm), a broad singlet (2.04 ppm), three nearby sharp singlets (1.87, 1.79 and 1.70 ppm) and two further broad singlets (0.30 and −0.39 ppm).
Complex 1 also serves as a starting material for the oxido complex [(cyclen(Me)(t-Bu,t-BuArO)3UV(O)] (6) when treating the precursor with oxygen-atom transfer (OAT) reagents, such as N2O gas, 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) or trimethylamine N-oxide (Me3NO). Remarkably, pentavalent oxido 6 is also formed when a benzene solution of hydroxido 4 is exposed to air for several days. Regardless, the highest yield (61%) of 6 was accomplished by stirring 1 under an atmosphere of N2O in toluene solution overnight (Scheme 2).
Single-crystals suitable for SC-XRD analysis of 6 were obtained by slow evaporation of a concentrated THF solution at −30 °C (Fig. 2, bottom). Complex 6 crystallizes in the monoclinic space group C2/c. Despite the different oxidation states of the uranium center, the geometry, structure, and bond lengths of 6 are comparable to the U(IV) fluorido and hydroxido complexes 3 and 4, the main difference being the short U(V)–O bond distance of 1.850(3) Å. While the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length in 6 is in agreement with literature-known values for terminal U(V) oxo complexes (1.706–1.863 Å),11–18,45 it is noteworthy that the oxido atom is situated perfectly within the O3X plane and that the oxido's equatorial binding mode is quite uncommon, with merely two other comparable examples reported in the literature; namely, the tacn-based neo-pentyl-substituted complexes [(tacn(nP,MeArO)3)UV(O)(X)] (X = O–Py, O–NMe3). In the latter cases, the axial position is occupied by the donor ligands pyridine N-oxide or trimethylamine-N-oxide. In 6, however, the axial coordination site remains open. One possible reason for the preferred equatorial binding mode could be the inverse trans influence (ITI).19,53–56 Although the ∢ O4–U1–O2 angle is significantly bent (132.8(2) °), the ITI is reflected in the shorter uranium – aryloxide U–O2 bond distance (2.183(3) Å) trans to the oxido ligand. This bond is about 0.04 Å shorter than the U–OAr bonds in cis position (U–O1 = 2.234(3) Å and U–O3 = 2.211(4) Å). In this context, it is also worth mentioning that the U–O2 bond distances trans to the bound chlorido ligand in 2 (2.211(6) Å) and the fluorido ligand in 3 (2.213(1) Å) are slightly longer than the U–O1 and U–O3 bond distances (2.152(6) and 2.175(6) Å for 2, 2.176(1) and 2.196(1) Å for 3). This observation suggests a stronger ITI of the aryloxides than of the chloride and fluoride, which is in agreement with the literature.19 The fact that the newly employed, low-symmetry seven-coordinate chelate, plus the additional oxido/hydroxido ligands, enforce a geometry with four oxygen donors on one face and four nitrogen donors on the other, allows for a comparison of uranium complexes with our group's “classic” tris-aryloxide-tacn-based systems (N3O3) and a number of extended solids, including those with [UO2]2+ motifs.
O bond length in 6 is in agreement with literature-known values for terminal U(V) oxo complexes (1.706–1.863 Å),11–18,45 it is noteworthy that the oxido atom is situated perfectly within the O3X plane and that the oxido's equatorial binding mode is quite uncommon, with merely two other comparable examples reported in the literature; namely, the tacn-based neo-pentyl-substituted complexes [(tacn(nP,MeArO)3)UV(O)(X)] (X = O–Py, O–NMe3). In the latter cases, the axial position is occupied by the donor ligands pyridine N-oxide or trimethylamine-N-oxide. In 6, however, the axial coordination site remains open. One possible reason for the preferred equatorial binding mode could be the inverse trans influence (ITI).19,53–56 Although the ∢ O4–U1–O2 angle is significantly bent (132.8(2) °), the ITI is reflected in the shorter uranium – aryloxide U–O2 bond distance (2.183(3) Å) trans to the oxido ligand. This bond is about 0.04 Å shorter than the U–OAr bonds in cis position (U–O1 = 2.234(3) Å and U–O3 = 2.211(4) Å). In this context, it is also worth mentioning that the U–O2 bond distances trans to the bound chlorido ligand in 2 (2.211(6) Å) and the fluorido ligand in 3 (2.213(1) Å) are slightly longer than the U–O1 and U–O3 bond distances (2.152(6) and 2.175(6) Å for 2, 2.176(1) and 2.196(1) Å for 3). This observation suggests a stronger ITI of the aryloxides than of the chloride and fluoride, which is in agreement with the literature.19 The fact that the newly employed, low-symmetry seven-coordinate chelate, plus the additional oxido/hydroxido ligands, enforce a geometry with four oxygen donors on one face and four nitrogen donors on the other, allows for a comparison of uranium complexes with our group's “classic” tris-aryloxide-tacn-based systems (N3O3) and a number of extended solids, including those with [UO2]2+ motifs.
Electronic absorption spectroscopy
The electronic absorption spectra of complexes 1, 2, 3, and 4 (Fig. 3) clearly reflect the difference between the uranium(III) and the uranium(IV) oxidation states. In uranium(III) complexes, the 5f–6d energy gap is smaller than in uranium(IV) complexes. As a consequence, the Laporte-allowed 5f–6d transitions are shifted towards lower energy, viz. higher wavelength, for uranium(III), usually between 500–700 nm.57 Consequently, these chromophoric transitions of dark purple 1 are located at 537 nm (ε = 1220 M−1 cm−1), 515 nm (ε = 1215 M−1 cm−1), and 465 nm (ε = 1180 M−1 cm−1) and the pale green color of complexes 2, 3 and 4 seems to originate from absorption bands centered at 325 nm (ε = 8000 M−1 cm−1 for 2, ε = 6000 M−1 cm−1 for 3, ε = 4500 M−1 cm−1 for 4, see SI), starting to gain intensity from 400 nm (Fig. 3). The UV region of uranium(IV) complexes is characteristic for charge-transfer bands, likely originating from ligand-based π → π*, LMCT or MLCT and inter-configurational 5f–6d transitions.58 All further low-intensity absorption bands in the range from 400 to 2100 nm arise from Laporte-forbidden f–f transitions and, therefore, exhibit small extinction coefficients of ε ≈ 10–60 M−1 cm−1 for 2, 10–77 M−1 cm−1 for 3, and 10–64 M−1 cm−1 for 4 (Fig. 3, middle). The number and shape of the bands of 2, 3 and 4 is in accordance with the typical pattern observed for the 5f2 electronic configuration of uranium(IV) complexes.45,66,63–65 As expected from the similar molecular structure and geometry, the data for 3 and 4 are very comparable over the whole spectral range, even though the maximum intensities slightly vary (ε = 77 M−1 cm−1 for 3, ε = 64 M−1 cm−1 for 4).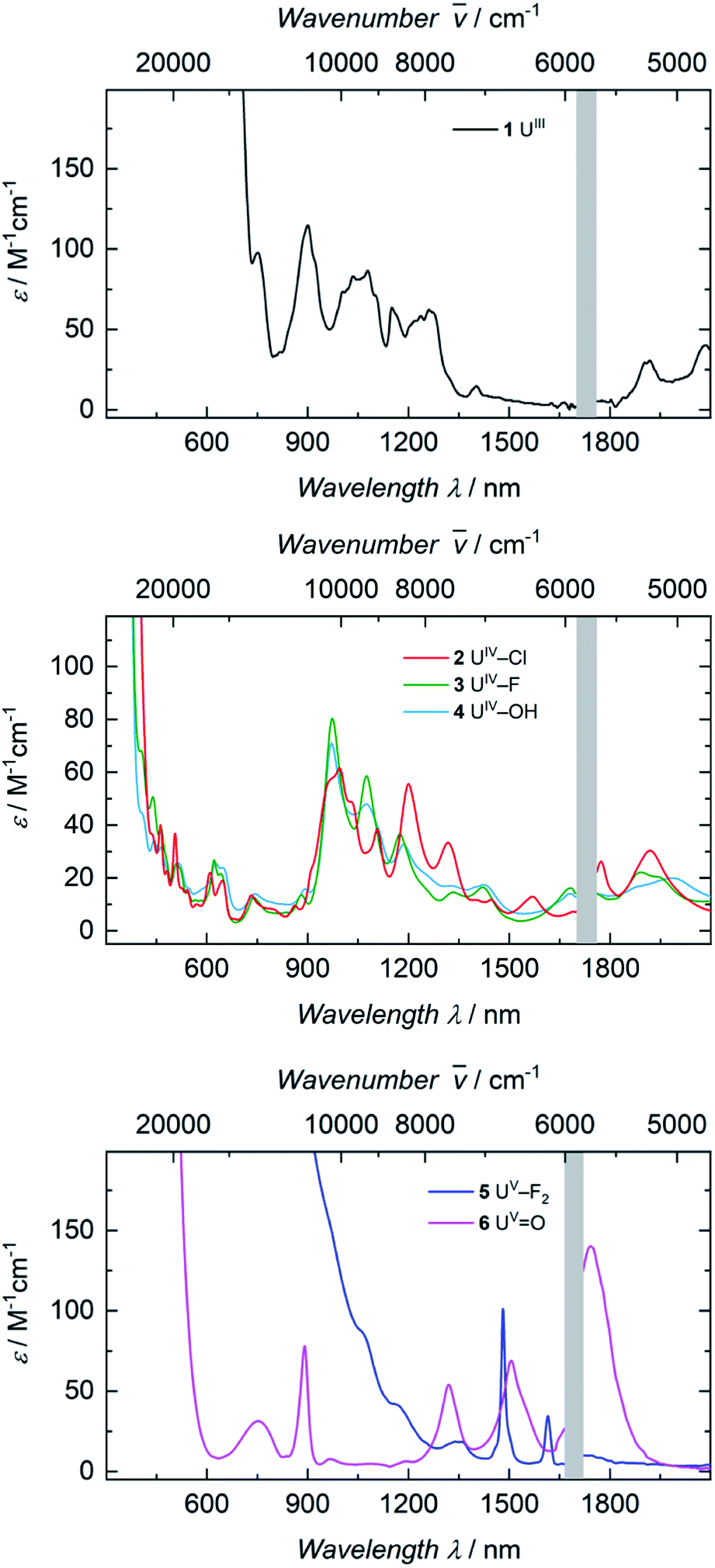 | ||
| Fig. 3 Top: Electronic absorption spectrum of complex 1 recorded at room temperature in benzene solution. Middle: Electronic absorption spectra of complexes 2, 3 and 4 recorded at room temperature in THF solution. Bottom: Electronic absorption spectra of complexes 5 and 6 recorded at room temperature in benzene solution (5) and THF solution (6). Data points from 1700 nm to 1760 nm (top and middle) and 1660 nm to 1720 nm (bottom) are omitted due to the change from a PbS to an In/Ga/As detector. | ||
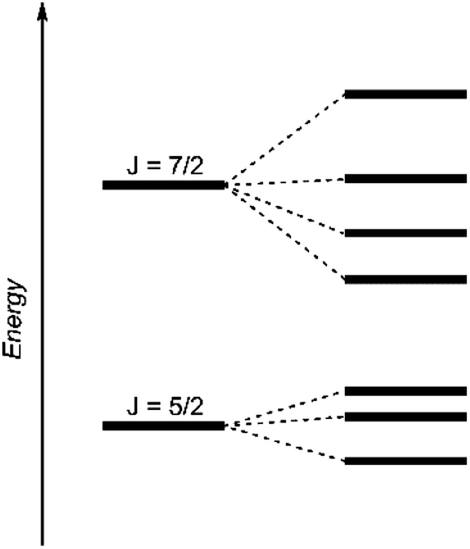 | ||
| Fig. 4 Schematic energy levels for a 5f1 system assuming the spin–orbit interaction is greater than the ligand field interaction.29 | ||
The dark black color of the U(V) complex 5 can be explained by the intense absorptions starting in the NIR region at 1020 nm (value given for ε = 100 M−1 cm−1) (Fig. 3, bottom), covering the entire visible light spectrum. This, in combination with the NIR region that is dominated by only a few, notably sharp Laporte forbidden f–f transitions, is in agreement with a U(V) metal center with its 5f1 electronic configuration and resulting 2F5/2 ground state with seven non-degenerate energetic states for both complexes (Fig. 4).2,43 As a consequence, only six f–f transitions are possible in U(V) complexes. For 5, two sharp (1480 and 1615 nm) and four broad transitions (1065 nm, 1160 nm and 1735 nm) are detected with the most intense band located at 1480 nm with ε = 98 M−1 cm−1. The strong absorption band of 6 starts at 527 nm in the visible region, explaining the bright orange color of this compound. This hypsochromic shift of the assumed 5f–6d transitions simplifies the detection of five f–f transitions in the case of 6. The transitions are detected at 760 nm (ε = 27 M−1 cm−1), 890 nm (ε = 65 M−1 cm−1), 1322 nm (ε = 46 M−1 cm−1), 1507 nm (ε = 59 M−1 cm−1), and 1760 nm (ε = 105 M−1 cm−1) (Fig. 3, bottom). The transitions of 5 are weaker than those of 6, presumably due to greater orbital mixing in the latter.
Magnetism
For the tetravalent complexes, variable temperature-dependent SQUID measurements reveal an effective magnetic moment, μeff, of 1.36 μB at 2 K and 2.81 μB at 300 K for the chlorido complex 2, 1.04 μB at 2 K and 2.91 μB at 300 K for fluorido 3, and 0.77 μB at 2 K and 2.72 μB at 300 K for hydroxido 4 (Fig. 5). Especially the low-temperature magnetic moment of 2 is relatively high for uranium(IV) complexes with its 5f2 electronic configuration and its, presumably, 3H4 non-magnetic ground state,59 suggests either a high contribution of temperature-independent paramagnetism (TIP) at low temperatures or an unusual pseudo-doublet ground state.60,61 Tetravalent complexes of the tacn-based tris-aryloxide ligand system, for instance, often possess low-temperature moments down to approx. 0.3 μB.52 In the case of 3 and 4, the magnetic data is comparable to that of [UIV((OArt-Bu,t-Bu)4cyclen)].44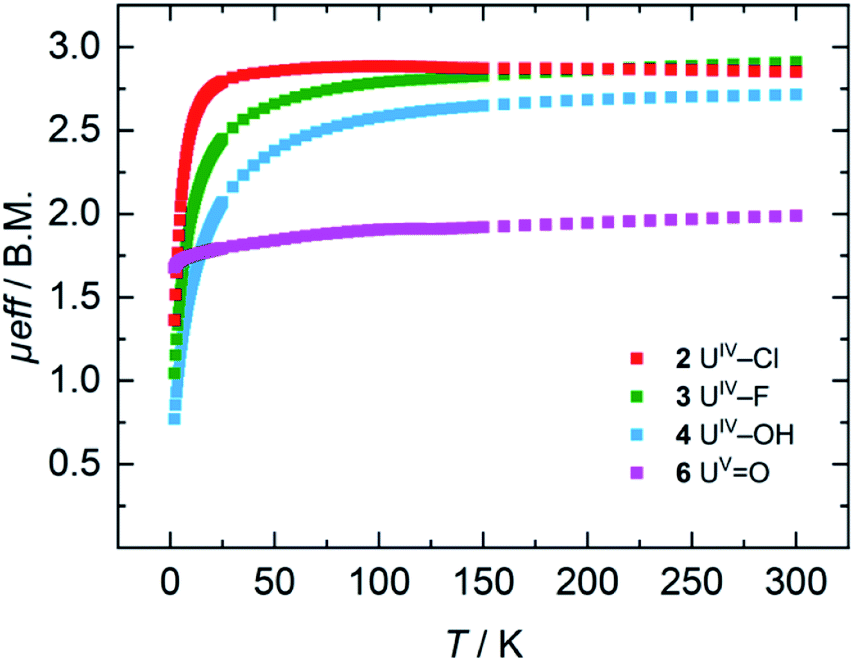 | ||
| Fig. 5 VT SQUID magnetization measurements of microcrystalline, solid samples of 2, 3, 4, and 6. The plot shows averaged values of two independently synthesized samples. | ||
Pentavalent 6 is nearly temperature-independent with a low-temperature value of μeff = 1.68 μB at 2 K, which increases to 1.99 μB at 300 K. This behavior is ordinary for a U(V) ion with a 5f1 electronic configuration and a 2F5/2 ground state.67 Furthermore, the magnetic data of 6 compare well with that of the tacn-based oxido complex [(tacn(t-BuArO)3)UV(O)] (1.61 μB at 5 K, and 1.98 μB at 300 K), despite the fact that the oxido ligand is bound in the axial position for [(tacn(t-BuArO)3)UV(O)].29 Interestingly, the tacn-based and neo-pentyl-substituted systems [(tacn(nP,MeArO)3)UV(O)(X)] (X = O–Py, O–NMe3), with the oxido ligand coordinated in the equatorial site and an additional neutral donor ligand in the axial site, show a strikingly different magnetic behavior.64 This leads to the assumption that the addition of another (neutral) ligand and, thus, a change from an 8-coordinated to a 9-coordinated complex, has a stronger influence on the magnetic properties than a change of the oxido ligand from an axial to an equatorial position.
Electrochemistry
Cyclic voltammetry studies of complexes 1, 2, 3, and 4 all show two major reversible redox-events for each complex (see SI). For 1, the events at a half-wave potential of E1/2 = −1.71 V (all values referenced to [FeCp2]0/+ at 0.0 V) and at E1/2 = 0.00 V can be attributed to the U(III/IV) and U(IV/V) redox couples according to linear sweep measurements. Accordingly, 1 is quite sensitive to electrolysis and decomposes to a U(IV) species over the course of several measurements. The main reversible redox events of 2, 3 and 4 occur at half-wave potentials of E1/2 = 0.12 V, E1/2 = −0.12 V and E1/2 = −0.59 V for the U(IV/V) couples, and E1/2 = 0.85 V, E1/2 = 1.06 V and E1/2 = 0.64 V for the U(V/VI) couple, respectively (see SI). The cyclic voltammogram of 6 shows one non-reversible redox event at E1/2 = −1.97 V and one reversible at E1/2 = −0.32 V. The more negative one probably accounts for the reduction to a U(IV) species when scanning in a cathodic direction. According to linear sweep measurements, the event at E1/2 = −0.32 V can be assigned to the U(V/VI) redox couple. The metal-centered oxidations of hydroxido 4 and oxido 6 (Fig. 6) are of particular interest and, indeed, it was possible to chemically oxidize these complexes while retaining their structural main features (vide infra).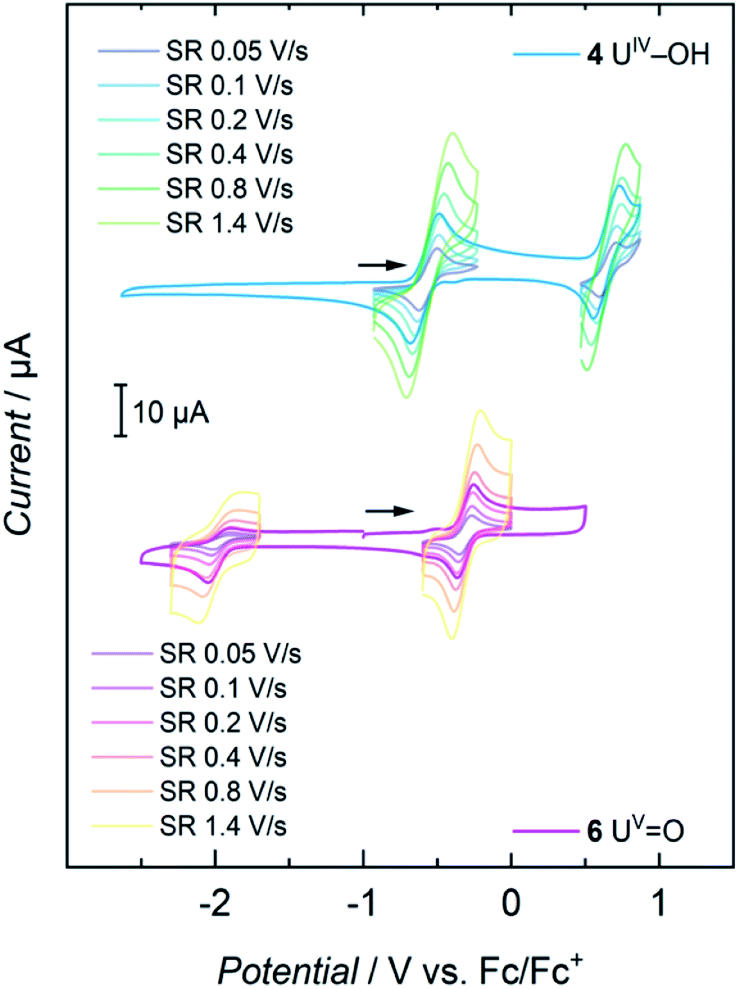 | ||
| Fig. 6 Cyclic voltammograms of 4 and 6 at scan rates varying from 0.05 to 1.4 V s−1, recorded in THF solution with 0.1 M TBAPF6 as supporting electrolyte. | ||
Redox chemistry of [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(OH)] and [(cyclen(Me)(t-Bu,t-BuArO)3)UV(O)]
Accordingly, one-electron oxidation of 4 and 6 with AgSbF6 yields the corresponding U(V) and U(VI) complexes [(cyclen(Me)(t-Bu,t-BuArO)3)UV(OH)][SbF6] (7) and [(cyclen(Me)(t-Bu,t-BuArO)3)UVI(O)][SbF6] (8) in 80% and 39% yield, respectively (Scheme 3). | ||
| Scheme 3 Synthesis of [(cyclen(Me)(t-Bu,t-BuArO)3)UV(OH)][SbF6] (7), [(cyclen(Me)(t-Bu,t-BuArO)3)UVI(O)][SbF6] (8), and K[(cyclen(Me)(t-Bu,t-BuArO)3)UIV(O)] (9). | ||
The 1H NMR spectrum of 7 shows a paramagnetic influence of the uranium(V) center with the resonances ranging from 36.04 to −25.41 ppm (Fig. S12†). By integration, the t-butyl groups can readily be attributed to the resonances at 4.48, 4.42, 1.04, −2.39, −4.75 and −4.85 ppm. The 1H NMR spectrum of 8 instead is characteristic for a uranium(VI) center with resonances in deuterated benzene in the diamagnetic region ranging from 8.11 to 1.12 ppm, with the resonances of the t-butyl groups at 1.80, 1.48, 1.43, 1.32 and 1.16 ppm, where the signal at 1.32 ppm accounts for two t-butyl groups according to the integration. Due to paramagnetic resonances in spectra of diamagnetic 8, even after crystallization, VT-VF 1H NMR studies were performed (see SI). The analyses suggest that the observed paramagnetic signals stem from trace amounts of 7 (ca. 3%) rather than magnetic excited states/TIP of 8.
Single-crystals suitable for SC-XRD analysis for 7 and 8 (Fig. 7) were grown from concentrated benzene solutions. Both complexes crystallize in the monoclinic space group P2/c. Due to the higher oxidation states of the uranium centers, the average U–OArO and U–Ncyclen bond distances decrease from 2.224(2) Å and 2.787(3) Å in 4 to 2.113(2) Å and 2.691(3) Å in 7 and from 2.179(6) and 2.760(4) in 6 to 2.116(2) and 2.698(3) in 8. Also, the U–OH bond distance decreases slightly from 2.141(2) Å in 4 to 2.052(2) Å in 7 (Table 2). It should be noted that in the crystal structure of 7 the position of the O4 bound hydrogen atom H4 could not be unequivocally derived from a difference Fourier synthesis. We have decided to use the O4 bound hydrogen atom in a calculated position and subsequently allowed it to ride on its carrier oxygen atom. Evidence for the presence of the hydroxyl group comes from the fact that the unit cell volume of 7 × 3.5 benzene is by 24.5 Å3 significantly larger than that of the oxido analogue 8 × 3.5 benzene, which crystallizes isostructurally to the hydroxido compound 7. Additionally, the U–O4 (hydroxido) bond distance in 7 × 3.5 benzene of 2.052(2) Å is significantly longer than the U–O4 (oxido) bond distance in 8 × 3.5 benzene of 1.949(3) Å (for further details see SI). Interestingly, the UO bond is elongated when going from pentavalent 6 (1.850(3) Å) to hexavalent 8 (1.949(3) Å), and is longer than the average bond length of U(VI) non-uranyl UO complexes, but still within the literature reported range (dU–O = 1.764(5)–1.98(3) Å).30,35,56,64–68 While we cannot rule out possible contamination of oxido complex 8 with minor impurities of the hydroxido 7 (vide supra), we were unable to detect these from the SC-XRD data. In the single-crystal structure determination of 8, we cannot find additional significant electron density at the location of the oxido ligand. While there is one minor electron density maximum (peak #31 in the list of residual electron density maxima, see ESI) with a height of 0.45 e Å−3 that is close to O4 and at a distance of 2.04 Å from the U center, attempts to refine a disorder between an oxido and a hydroxido ligand, including both the original position of O4 and Q31, remained unsuccessful. Eventually, all electron density was always located at the original O4 position.
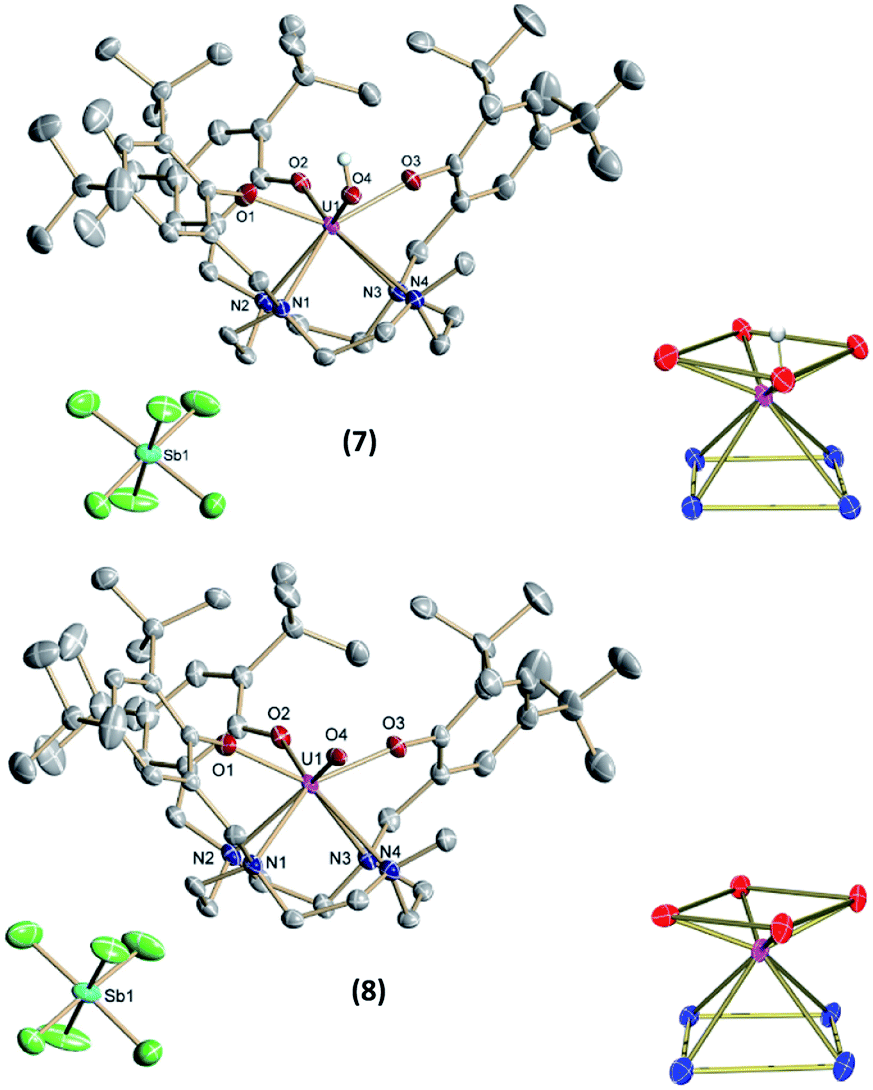 | ||
| Fig. 7 Solid-state molecular structure of 7 (top) and 8 (bottom) in crystals of [(cyclen(Me)(t-Bu,t-BuArO)3)UV(OH)][SbF6] × 3.5 benzene and [(cyclen(Me)(t-Bu,t-BuArO)3)UVI(O)][SbF6] × 3.5 benzene, as well as the coordination polyhedra of the heteroatoms (bottom right). Hydrogen atoms (except for the hydroxido H) and co-crystallized solvent molecules are omitted for clarity. Thermal ellipsoids are shown at the 50% probability level. | ||
| 4 | 6 | 7 | 8 | |
|---|---|---|---|---|
| U–O1 | 2.231(2) | 2.234(3) | 2.108(2) | 2.119(2) |
| U–O2 | 2.233(2) | 2.183(3) | 2.131(2) | 2.123(2) |
| U–O3 | 2.208(2) | 2.211(4) | 2.100(2) | 2.106(2) |
| U–OArO(av) | 2.224(2) | 2.179(6) | 2.113(2) | 2.116(2) |
| U–N1 | 2.730(2) | 2.688(4) | 2.646(2) | 2.643(3) |
| U–N2 | 2.784(3) | 2.759(4) | 2.652(2) | 2.662(3) |
| U–N3 | 2.817(2) | 2.850(4) | 2.706(2) | 2.729(3) |
| U–N4 | 2.817(3) | 2.743(4) | 2.759(3) | 2.759(3) |
| U–Ncyclen(av) | 2.787(3) | 2.760(4) | 2.691(3) | 2.698(3) |
| Uoop | −0.865(1) | −0.801(2) | −0.831(1) | −0.804(2) |
| U–Leq(O4) | 2.141 (2) | 1.850(3) | 2.052(2) | 1.949(3) |
Finally, treating 6 with KC8 in benzene solution results in the light green complex 9, with a yield of 97%, that is identified as a uranium(IV) species by vis/NIR absorption spectroscopy and SQUID magnetometry (Fig. 8). Even though single-crystals suitable for XRD analysis could not be obtained, especially the electronic absorption measurements suggest that, in THF solution, the molecular structure of 9 is similar to that of hydroxido 4 (Scheme 3 and Fig. 8).
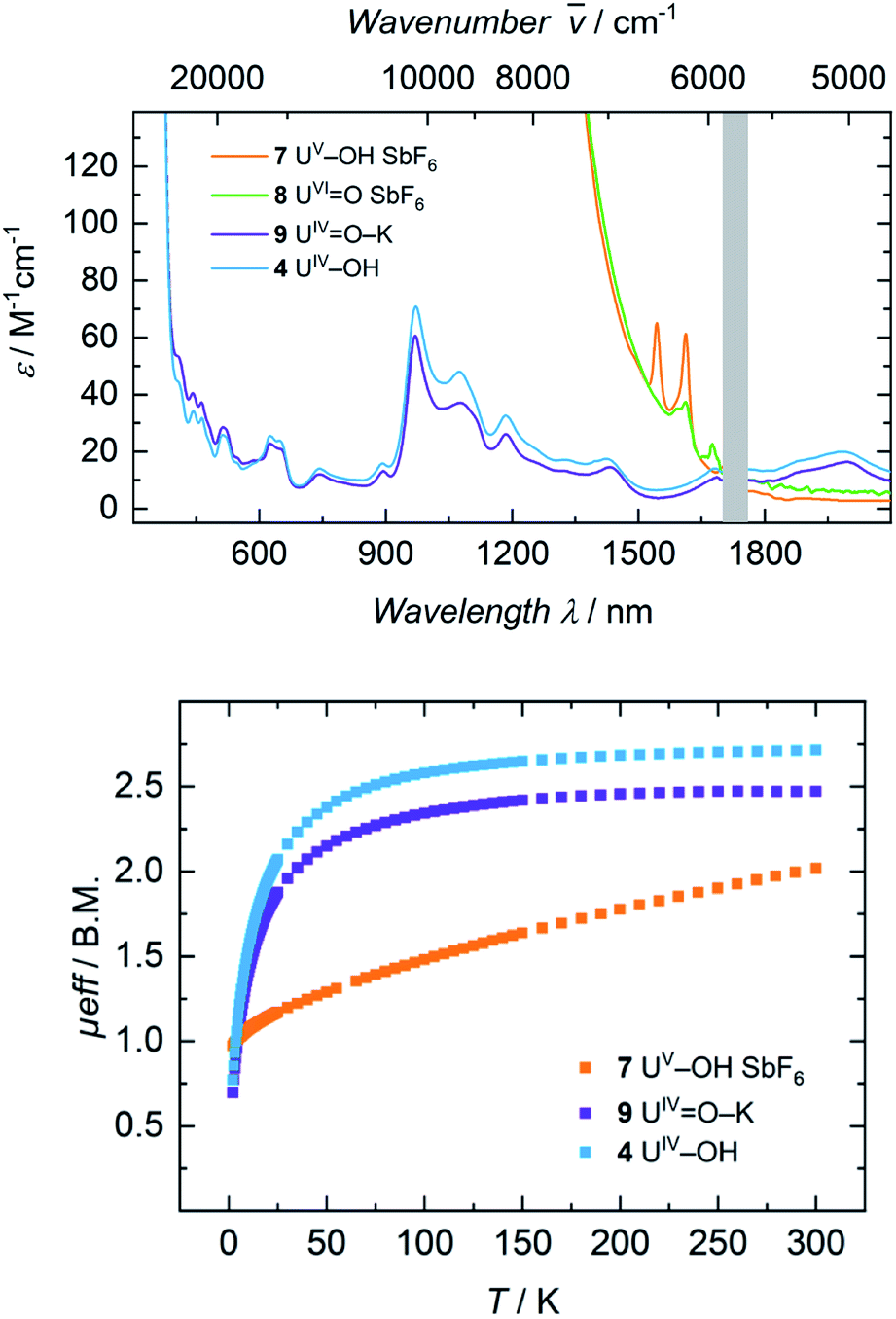 | ||
| Fig. 8 Top: Electronic absorption spectra of complexes 4, 7, 8, and 9 were recorded at room temperature in benzene (8) and THF (4, 7, 9) solution. Data points from 1700 nm to 1760 nm are omitted due to a detector change from a PbS to an In/Ga/As detector. Bottom: VT SQUID magnetization measurements of microcrystalline, solid samples of 4, 7 and 9. | ||
The 1H NMR spectrum of complex 9 shows resonances over a range from 91.50 to −72.47 ppm (Fig. S15†). The six t-butyl resonances can be attributed by integration to the signals at 11.93, −2.38, −3.02, −4.33, −9.16 and –20.24 ppm.
Electronic absorption spectra of 7 and 8 feature exceedingly intense absorptions below λ = 1400 nm (ε > 2000 M−1 cm−1), explaining the dark black color of both complexes (Fig. 8, top). Above 1500 nm, the uranium(V) hydroxido 7 reveals two sharp bands at 1545 nm (ε = 65 M−1 cm−1) and 1615 nm (ε = 60 M−1 cm−1), while the uranium(VI) oxido 8 shows two, very weak absorptions at 1610 nm with a shoulder at 1590 nm (ε = 35 M−1 cm−1) and 1673 nm (ε = 20 M−1 cm−1). Since there are no f–f-transitions expected in hexavalent uranium complexes, the origin of these bands is uncertain. It is assumed that these faint features origin from interactions of traces (3%) of paramagnetic hydroxido 7 with the oxido 8 in solution, since 7 is also present in the 1H NMR spectrum of 8. The spectrum of 9 in benzene looks like a typical uranium(IV) ion indicating a successful one-electron reduction of 6 with KC8 (Fig. S46,† top). It is noteworthy, that measuring 9 in THF leads to a different absorption. Now the spectrum of 9 and 4 are superimposable suggesting the solvation of the potassium ion and similar molecular and electronic structures of 4 and 9 in THF solution. This observed structural similarity is also supported by magnetic measurements (Fig. 8, bottom). The strong temperature dependence and the low magnetic moment μeff = 0.70 μB at 2 K rather fit to a uranium(IV) complex and is also comparable to that of 4 (0.77 μB). In contrast to that, complex 7 is clearly identified as a uranium(V) complex with μeff values ranging from 0.97 μB at 2 K to 2.02 μB at 300 K.
Conclusions
In summary, and motivated by our recently reported hydroxido and oxido uranium complexes as active catalysts for the production of H2 from water, we report the synthesis and coordination chemistry of a newly developed, hepta-dentate, cyclen-based tris(aryloxide) ligand precursor, namely cyclen(Me)(t-Bu,t-BuArOH)3, and its coordination chemistry to uranium. The resulting precursor complex [UIII((OArt-Bu,t-Bu)3(Me)cyclen)] (1) is more reactive than its previously reported, eight-coordinate, tetrakis-aryloxide derivative [U((OArt-Bu,t-Bu)4cyclen)], which could only be obtained in its tetravalent form.44 Since it was not possible to obtain suitable crystals of 1 for single-crystal X-ray diffraction analysis, trivalent 1 was oxidized to the corresponding uranium(IV) complexes [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(Cl)] (2) and [(cyclen(Me)(t-Bu,t-BuArO)3)U(F)] (3) to draw conclusions about the precursor's molecular structure.As expected, trivalent 1 is capable of reductively activating small molecules, such as H2O and N2O, yielding the tetravalent [(cyclen(Me)(t-Bu,t-BuArO)3)UIV(OH)] (4) and pentavalent [(cyclen(Me)(t-Bu,t-BuArO)3)UV(O)] (7) complexes, respectively. The solid-state molecular structures of these mononuclear complexes feature an N4O3X coordination mode, where the additional ligands (X = F, Cl, OH, O) bind in the equatorial position. Oxidation of 3 and 4 with AgF or AgSbF6 yields the corresponding uranium(V) complexes [(cyclen(Me)(t-Bu,t-BuArO)3)UV(F)2] (5) and [(cyclen(Me)(t-Bu,t-BuArO)3)UV(OH)][SbF6] (7). Oxidation and reduction of 6 was achieved by a reaction with AgSbF6 or potassium graphite; thus, providing access to hexavalent [(cyclen(Me)(t-Bu,t-BuArO)3)UVI(O)][SbF6] (8) and a tetravalent species, identified as [K][(cyclen(Me)(t-Bu,t-BuArO)3)U(O)] (9), respectively. Thus, the present study provides controlled and reproducible access to the first U(IV/V) hydroxido (4 and 7) and U(IV/V/VI) oxido (6, 8 and 9) complex pairs in a retained ligand environment. Regardless, these complexes could not be used for the electrocatalytic production of H2 from water.
Data availability
ESI† with general considerations, synthetic and spectroscopic details, NMR spectroscopy, IR vibrational spectroscopy, UV/Vis/NIR electronic absorption spectroscopy, VT SQUID magnetization spectroscopy, cyclic voltammetry, and single crystal X-ray structure determinations (PDF).Author contributions
J. H. synthesized the ligand, and J. H. and S. T. L. synthesized the complexes, collected most of the spectroscopic, magnetochemical and electrochemical data. A. S. collected and analyzed the VT-VF NMR spectroscopic data during the revision process. F. W. H. collected and analyzed the crystallographic data. K. M. supervised and directed the project, edited and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funds of the Bundesministerium für Bildung und Forschung (f-Char, BMBF support code 02NUK059E). We also thank the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) for generous financial support.Notes and references
- J. Selbin and J. D. Ortego, Chem. Rev., 1969, 69, 657–671 CrossRef CAS.
- G. Kniewald and M. Branica, Mar. Chem., 1988, 24, 1–12 CrossRef CAS.
- D. E. Morris, Inorg. Chem., 2002, 41, 3542–3547 CrossRef CAS PubMed.
- I. Korobkov and S. Gambarotta, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2005, pp. 321–348 Search PubMed.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831–3853 RSC.
- G. Wang, W. Um, Z. Wang, E. Reinoso-Maset, N. M. Washton, K. T. Mueller, N. Perdrial, P. A. O'Day and J. Chorover, Environ. Sci. Technol., 2017, 51, 11011–11019 CrossRef CAS PubMed.
- J. Berthet, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1993, 460, 47–53 CrossRef CAS.
- W. W. Lukens, S. M. Beshouri, L. L. Blosch and R. A. Andersen, J. Am. Chem. Soc., 1996, 118, 901–902 CrossRef CAS.
- P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423–1428 RSC.
- L. Karmazin, M. Mazzanti and J. Pécaut, Inorg. Chem., 2003, 42, 5900–5908 CrossRef CAS PubMed.
- K. A. N. S. Ariyaratne, R. E. Cramer, G. B. Jameson and J. W. Gilje, J. Organomet. Chem., 2004, 689, 2029–2032 CrossRef CAS.
- G. H. John, I. May, C. A. Sharrad, A. D. Sutton, D. Collison, M. Helliwell and M. J. Sarsfield, Inorg. Chem., 2005, 44, 7606–7615 CrossRef CAS PubMed.
- L. Natrajan, M. Mazzanti, J.-P. Bezombes and J. Pécaut, Inorg. Chem., 2005, 44, 6115–6121 CrossRef CAS PubMed.
- P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136, 10218–10221 CrossRef CAS PubMed.
- D. P. Halter, F. W. Heinemann, J. Bachmann and K. Meyer, Nature, 2016, 530, 317–321 CrossRef CAS PubMed.
- M. W. Rosenzweig, A. Scheurer, C. A. Lamsfus, F. W. Heinemann, L. Maron, J. Andrez, M. Mazzanti and K. Meyer, Chem. Sci., 2016, 7, 5857–5866 RSC.
- M. W. Rosenzweig, J. Hümmer, A. Scheurer, C. A. Lamsfus, F. W. Heinemann, L. Maron, M. Mazzanti and K. Meyer, Dalton Trans., 2019, 48, 10853–10864 RSC.
- S. T. Löffler and K. Meyer, in Comprehensive Coordination Chemistry III, Elsevier, 2021, pp. 471–521 Search PubMed.
- D. P. Halter, F. W. Heinemann, L. Maron and K. Meyer, Nat. Chem., 2018, 10, 259–267 CrossRef CAS PubMed.
- Z. Zhang and R. M. Pitzer, J. Phys. Chem. A, 1999, 103, 6880–6886 CrossRef CAS.
- G. Schreckenbach, P. J. Hay and R. L. Martin, J. Comput. Chem., 1999, 20, 70–90 CrossRef CAS.
- J. K. Gibson, R. G. Haire, M. Santos, J. Marçalo and A. Pires de Matos, J. Phys. Chem. A, 2005, 109, 2768–2781 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- N. Kaltsoyannis, Inorg. Chem., 2000, 39, 6009–6017 CrossRef CAS PubMed.
- N. Kaltsoyannis, Chem. Soc. Rev., 2003, 32, 9–16 RSC.
- M. Pepper and B. E. Bursten, Chem. Rev., 1991, 91, 719–741 CrossRef CAS.
- T. W. Hayton, Dalton Trans., 2018, 47, 1003–1009 RSC.
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed.
- B. Kosog, H. S. La Pierre, F. W. Heinemann, S. T. Liddle and K. Meyer, J. Am. Chem. Soc., 2012, 134, 5284–5289 CrossRef CAS PubMed.
- S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- C. J. Hoerger, H. S. La Pierre, L. Maron, A. Scheurer, F. W. Heinemann and K. Meyer, Chem. Commun., 2016, 52, 10854–10857 RSC.
- S. M. Franke, B. L. Tran, F. W. Heinemann, W. Hieringer, D. J. Mindiola and K. Meyer, Inorg. Chem., 2013, 52, 10552–10558 CrossRef CAS PubMed.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS.
- D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem, Int. Ed, 2013, 52, 4921–4924 CrossRef CAS PubMed.
- N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Polyhedron, 2004, 23, 2689–2694 CrossRef CAS.
- J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed.
- T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- P. Waldschmidt, C. J. Hoerger, J. Riedhammer, F. W. Heinemann, C. T. Hauser and K. Meyer, Organometallics, 2020, 39, 1602–1611 CrossRef CAS.
- J. Hümmer, F. W. Heinemann and K. Meyer, Inorg. Chem., 2017, 56, 3201–3206 CrossRef PubMed.
- H. Nakai, K. Nonaka, T. Goto, J. Seo, T. Matsumoto and S. Ogo, Dalton Trans., 2015, 44, 10923–10927 RSC.
- H. Nakai, J. Seo, K. Kitagawa, T. Goto, K. Nonaka, T. Matsumoto and S. Ogo, Inorg. Chem., 2016, 55, 6609–6615 CrossRef CAS PubMed.
- A. Rodríguez-Rodríguez, M. Regueiro-Figueroa, D. Esteban-Gómez, T. Rodríguez-Blas, V. Patinec, R. Tripier, G. Tircsó, F. Carniato, M. Botta and C. Platas-Iglesias, Chem.– Eur. J., 2017, 23, 1110–1117 CrossRef PubMed.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, Organometallics, 2011, 30, 2031–2038 CrossRef CAS.
- A.-C. Schmidt, F. W. Heinemann, C. E. Kefalidis, L. Maron, P. W. Roesky and K. Meyer, Chem.– Eur. J., 2014, 20, 13501–13506 CrossRef CAS PubMed.
- I. Castro-Rodríguez, H. Nakai and K. Meyer, Angew. Chem., Int. Ed., 2006, 45, 2389–2392 CrossRef PubMed.
- O. P. Lam, F. W. Heinemann and K. Meyer, C. R. Chim., 2010, 13, 803–811 CrossRef CAS.
- A. V. Nizovtsev, A. Scheurer, B. Kosog, F. W. Heinemann and K. Meyer, Eur. J. Inorg. Chem., 2013, 2013, 2538–2548 CrossRef CAS.
- O. P. Lam, S. M. Franke, H. Nakai, F. W. Heinemann, W. Hieringer and K. Meyer, Inorg. Chem., 2012, 51, 6190–6199 CrossRef CAS PubMed.
- H. S. La Pierre, M. Rosenzweig, B. Kosog, C. Hauser, F. W. Heinemann, S. T. Liddle and K. Meyer, Chem. Commun., 2015, 51, 16671–16674 RSC.
- M. Gregson, E. Lu, D. P. Mills, F. Tuna, E. J. L. McInnes, C. Hennig, A. C. Scheinost, J. McMaster, W. Lewis, A. J. Blake, A. Kerridge and S. T. Liddle, Nat. Commun., 2017, 8, 14137 CrossRef CAS PubMed.
- H. S. La Pierre and K. Meyer, Inorg. Chem., 2013, 52, 529–539 CrossRef CAS PubMed.
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- E. Hashem, A. N. Swinburne, C. Schulzke, R. C. Evans, J. A. Platts, A. Kerridge, L. S. Natrajan and R. J. Baker, RSC Adv., 2013, 3, 4350–4361 RSC.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed.
- J. A. Seed, L. Birnoschi, E. Lu, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem, 2021, 7, 1666–1680 CAS.
- F. Delano and S. Demir, Chem, 2021, 7, 1686–1688 CAS.
- Z. Gajek, J. Mulak and J. C. Krupa, J. Solid State Chem., 1993, 107, 413–427 CrossRef CAS.
- R. D. Fischer, R. V. Ammon and B. Kanellakopulos, J. Organomet. Chem., 1970, 25, 123–137 CrossRef CAS.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 511–518 CrossRef CAS PubMed.
- N. T. Rice, K. McCabe, J. Bacsa, L. Maron and H. S. La Pierre, J. Am. Chem. Soc., 2020, 142, 7368–7373 CrossRef CAS PubMed.
- T. W. Hayton, J. M. Boncella, B. L. Scott and E. R. Batista, J. Am. Chem. Soc., 2006, 128, 12622–12623 CrossRef CAS PubMed.
- E. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 6696–6700 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: [DETAILS]. CCDC 2080557, 2150855–2150860. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02736d |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |