
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting halogen-bonding catalyzed living radical polymerization through ion-pair strain†
Shiwen
Huang
a,
Xinjian
Su
a,
Yanzhen
Wu
a,
Xiao-Gen
Xiong
 c and
Yiliu
Liu
*ab
c and
Yiliu
Liu
*ab
aSouth China Advanced Institute for Soft Matter Science and Technology, School of Emergent Soft Matter, South China University of Technology, Guangzhou 510640, China. E-mail: liuyiliu@scut.edu.cn
bGuangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices, South China University of Technology, Guangzhou 510640, China
cSino-French Institute of Nuclear Engineering and Technology, Sun Yat-sen University, Zhuhai 519082, China
First published on 13th September 2022
Abstract
Discovering efficient catalysts is highly desired in expanding the application of halogen-bonding catalysis. We herein report our findings on applying triaminocyclopropenium (TAC) iodides as highly potent catalysts for halogen-bonding catalyzed living radical polymerization. Promoted by the unique effect of ion-pair strain between the TAC cation and the iodide anion, the TAC iodides showed high catalytic efficiency in the halogen-bonding catalysis toward radical generation, and surpassed the previously reported organic iodide catalysts. With the TAC iodide as catalyst, radical polymerization with a living feature was successfully realized, which shows general applicability with a variety of monomers and produced block copolymers. In addition, the TAC-iodides also showed promising feasibility in catalyzing the radical depolymerization of iodo-terminated polymethacrylates. Noteworthily, the catalytic capacity of the TAC iodides is demonstrated to be closely related to the electronic properties of the TAC cation, which offers a molecular platform for further catalyst screening and optimization.
Introduction
Halogen bonding (XB) is defined as the noncovalent interaction between a polarized halogen substituent and a neutral or charged Lewis base.1–4 Taking the advantage of its high directionality, hydrophobicity, and strength tunability, XB has found usages in a wide variety of aspects, ranging from mesomorphic materials to drug discovery.5–10 Among them, XB-based organo-catalysis has seen an upsurge of interest in recent years, with a growth in the number of organic reactions reported that are catalyzed through XB.11,12 In many of these cases, the halide substituent on the substrate forms XB with the catalyst, which induces a polarization and elongation of the R–X bond, thereby facilitating nucleophilic substitution reactions.13Although the XB-based organo-catalysis is undergoing a rapid development, only a few examples of polymerization reactions catalyzed by XB have been reported. Takagi and coworkers employed XB to activate alkyl halide dormant species, which generates carbon-centered cations and initiates cationic polymerization.14 Coulembier and coworkers applied XB-based catalysis in the ring-opening polymerization of lactides.15 The catalyst functions as XB donor to coordinate with the carbonyl group of the monomer and facilitates its ring-opening. In particular, the application of XB-based catalysis in radical polymerization was demonstrated by the seminal work of Goto and coworkers, in which alkyl iodides and electron-donating XB-acceptors were used as dormant initiators and catalysts, respectively.16–21 Uniquely, the initiator and the catalyst form XB and reversibly generates radical species, which mediates living radical polymerization. This polymerization approach, namely reversible complexation mediated polymerization (RCMP), is attractive in its non-metal catalysts, broad monomer scope, as well as mild conditions. Further developments of efficient XB-catalysts are highly desired for increasing the feasibility for industrial applications.
Cyclopropenium, firstly synthesized by Breslow in 1957, are the smallest member of the Hückel aromatic systems.22,23 Despite the apparent ring strain, the cyclopropeniums possess considerable thermodynamic stability owing to the Hückel aromaticity. Triaminocyclopropeniums (TACs) in particular, show remarkable stability against high temperature and strongly alkaline conditions.24,25 The synthesis and application of TACs have gained revived interest, and the TACs have found broad uses as ionic liquids,26,27 electrolytes,28,29 nitrogen-based ligands,30 fluorophores,31 biomaterials,32 and catalysts.33–36 An unusual feature of TACs is their electron-rich π systems despite being positively charged cations. As a result of this, there exists an electronic ion-pair strain between TACs and their electron-rich counter-anions, which leads to highly “naked” counter-anions with unprecedented coordinative power in forming strong noncovalent complexes.37–39 Such ion-pair strain-promoted noncovalent interactions intrigued us to consider if this unique effect can be applied in XB-based catalysis. To our knowledge, although the TACs have been applied as catalysts in several cases, e.g., phase transfer catalysis and electro-photo catalysis,40–43 its application in XB-based catalysis, especially for radical polymerization reactions has yet to be explored.
In the present work, we report our recent progress in exploring TAC iodides as efficient XB catalysts in catalyzing radical polymerization reactions. To this end, pyrrolidine-substituted cyclopropenium iodide (TACP-I, Scheme 1) was synthesized and used as a catalyst. Promoted by the effect of ion-pair strain, TACP-I can catalyze the radical-generation reaction of 2-cyanopropyl iodide (CP-I) and mediate the polymerization reactions of methacrylates with unprecedented efficiency. Its catalytic performance significantly surpasses the previously reported catalysts, such as quaternary alkyl phosphonium iodide and quaternary alkyl ammonium iodide. Besides being efficient in catalyzing polymerization, TACP-I also shows enhanced capacity in catalyzing the depolymerization of iodo-terminated polymethacrylates.
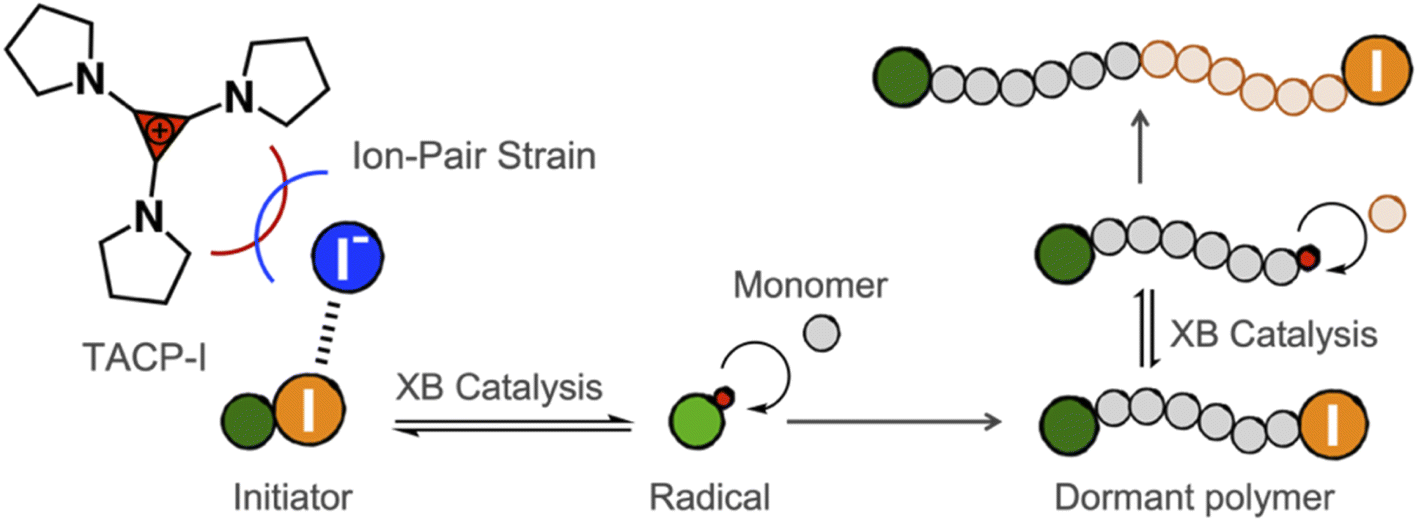 | ||
| Scheme 1 Promoting halogen-bonding catalyzed radical polymerization through ion-pair strain. | ||
Results and discussion
XB catalysis in the radical-generation reaction
Before performing polymerization reactions, the catalytic capacity of TACP-I in catalyzing the radical-generation reaction of CP-I was investigated through radical-trapping experiments. It is expected that the iodide anion of TACP-I can coordinate with the iodo-substituent of CP-I through XB, and reversibly generate CP radicals (Fig. 1a). 2,2,6,6-Tetramethylpiperidinyl-1-oxy (TEMPO) was used to trap the generated radicals, yielding CP-TEMPO. In a typical experiment, TACP-I (80 mM) was dissolved together with CP-I (10 mM) and TEMPO (20 mM) in a mixed solvent of toluene-d8 and acetonitrile-d3, and then the mixture was heated at 60 °C. The conversion of CP-I to CP-TEMPO was monitored using 1H NMR spectroscopy. A representative 1H NMR spectrum of the solution of TAC-I, CP-I, and TEMPO before and after heating is shown in Fig. 1b. After 2 h reaction, signals belonging to CP-TEMPO appeared with a significant decrease in the signal of CP-I. The nearly quantitative conversion demonstrates the high efficiency of TACP-I in XB-catalyzed radical generation.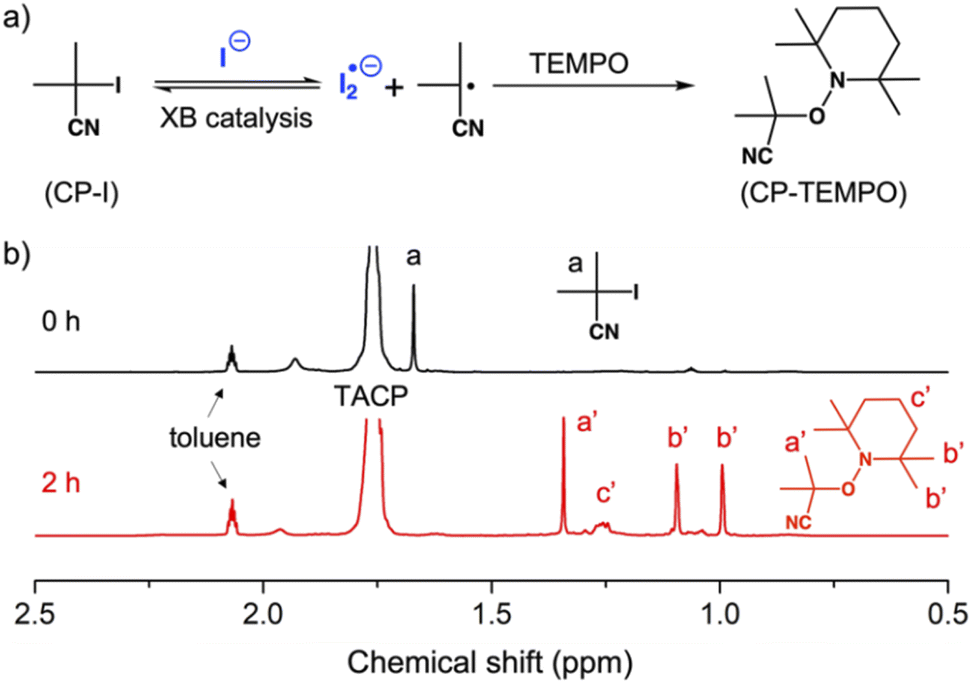 | ||
| Fig. 1 Trapping the radicals generated from CP-I by TEMPO. (a) The chemical equation of the radical-trapping reaction; (b) the 1H NMR spectra of the reaction mixture after 0 and 2 h of heating. | ||
The importance of XB for the catalysis was confirmed using an inhibition experiment. A tridentate XB donor TPFI (Scheme 2), which can strongly compete with CP-I for iodide anions, was added into the reaction mixture.44 As expected, the radical generation was fully inhibited by TPFI, no CP-TEMPO product was detected after 2 h of reaction (Fig. S1†). In addition, TACP-Cl with chloride anion as counterion was also applied as a catalyst under the same reaction condition. Compared with the iodide anion, the chloride anion is much less potent in XB formation, thus being less effective in catalysis. The elimination of hydrogen iodide to produce methacrylonitrile prevailed as a side-reaction, leading to the low efficiency of radical generation (Fig. S2†).
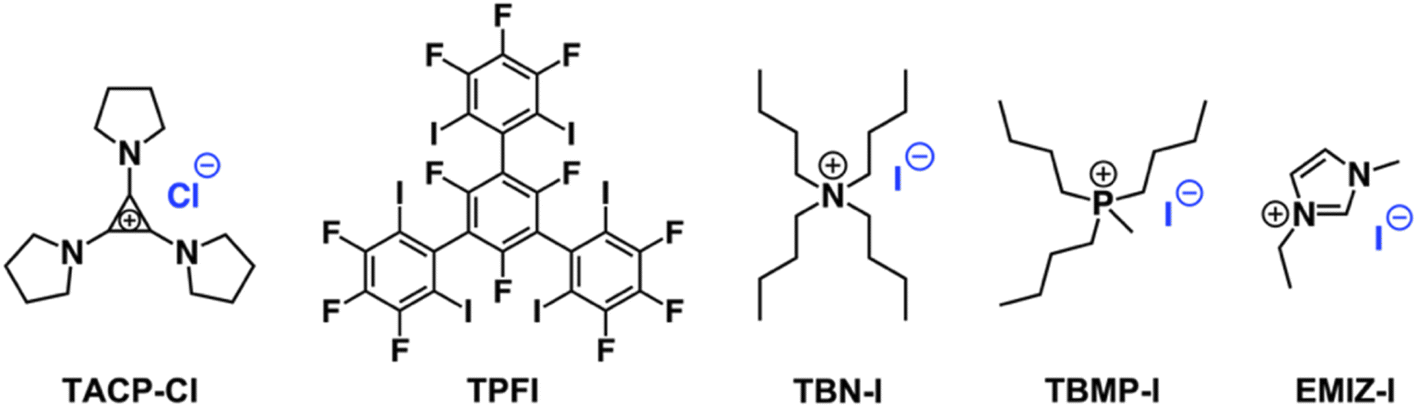 | ||
| Scheme 2 Chemical structures of the involved catalysts and the inhibitor. | ||
Organic iodide salts, including ammonium iodide TBN-I, phosphonium iodide TBMP-I, and imidazolium iodide EMIZ-I (Scheme 2), were reported as efficient catalysts for the radical-generation reaction of CP-I.18 For comparison, these iodide salts were applied as control catalysts under the same reaction conditions as TACP-I. The time–conversion plots for different temperatures are shown in Fig. 2. At 40 °C, the conversion of CP-I to CP-TEMPO reached 94% in 5 h with TACP-I as the catalyst. In remarkable contrast, none of the reactions catalyzed by TBN-I, TBMP-I, or EMIZ-I gave conversions over 10%. In all cases, increasing the reaction temperature led to profound increases in reaction rates. Noteworthily, the reaction catalyzed by TACP-I reached a plateau with a conversion over 95% within 3 h at 50 °C and 2 h at 60 °C, respectively. Under all reaction temperatures, the catalytic capacity of TACP-I was significantly higher than that of the three other organic iodide salts. Remarkably, only variations in the non-catalytic cations lead to distinct differences in the catalytic performances, which shows the prominent effect of ion-pair strain in promoting the XB-based catalysis.
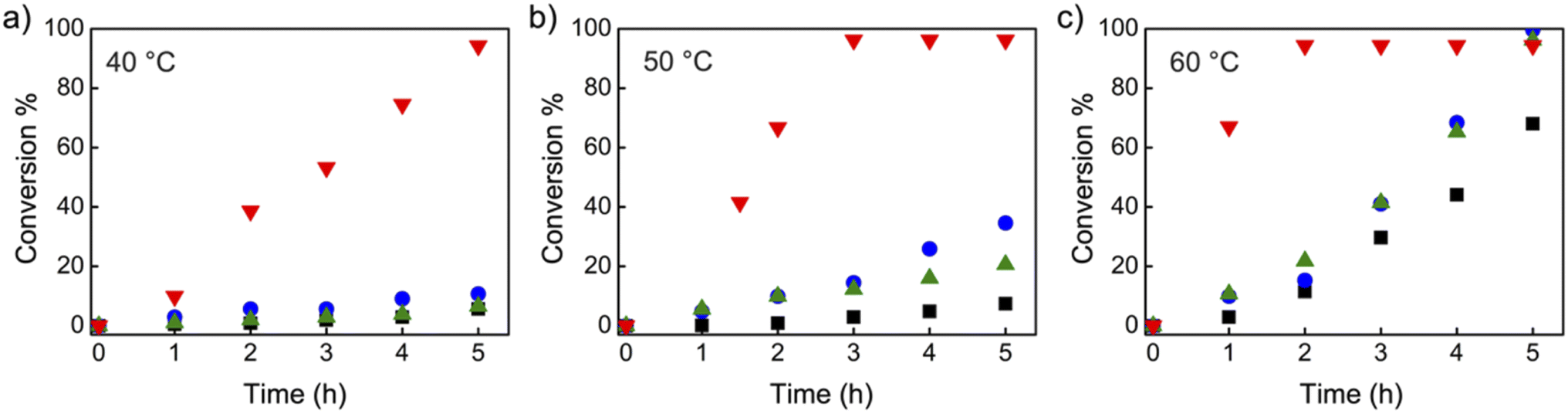 | ||
| Fig. 2 The time–conversion plots of the radical-trap experiments catalyzed by TACP-I (red triangles), TBN-I (blue dots), TBMP-I (green triangles) and EMIZ-I (black squares) at different temperatures (a: 40 °C; b: 50 °C; c: 60 °C). | ||
Because the ion-pair strain is derived from the orbital repulsion between the high-lying highest occupied molecular orbital (HOMO) of the TAC cation and the HOMO of the counter-anion,25 it is legitimate to speculate that tuning the electronic properties of the TAC cation could affect the strength of the ion-pair strain, thus leading to different catalytic performances. To this end, TACE-I, which contains isopropyl, ethyl, and methyl groups as substituents, was designed and synthesized (Fig. 3a). The cyclic voltammetry analysis showed that oxidation of the TACP cation occurred at 92.5 mV lower in potential than that of the TACE cation (Fig. 3b). The lower oxidation potential of the TACP cation suggests a higher energy of its HOMO than that of the TACE cation. This was further supported by density functional (DFT) calculation, which shows that the HOMO of the TACP cation is 0.16 eV higher in energy than that of the TACE cation (Fig. 3a). In principle, a higher HOMO corresponds to a higher ion-pair strain effect, which suggests that TACP-I should be a better XB catalyst than TACE-I. The radical-generation reaction catalyzed by TACE-I was performed with identical conditions to TACP-I, and the time–conversion plot is shown in Fig. 3c. Indeed, the catalytic efficiency of TACE-I is relatively lower than that of TACP-I, but still considerably higher than that of TBN-I, TBMP-I, and EMIZ-I at all three reaction temperatures. These results not only confirmed the effect of ion-pair strain in XB catalysis, but also showed the possibility of further optimization of the TAC-based catalysts in a controllable manner.
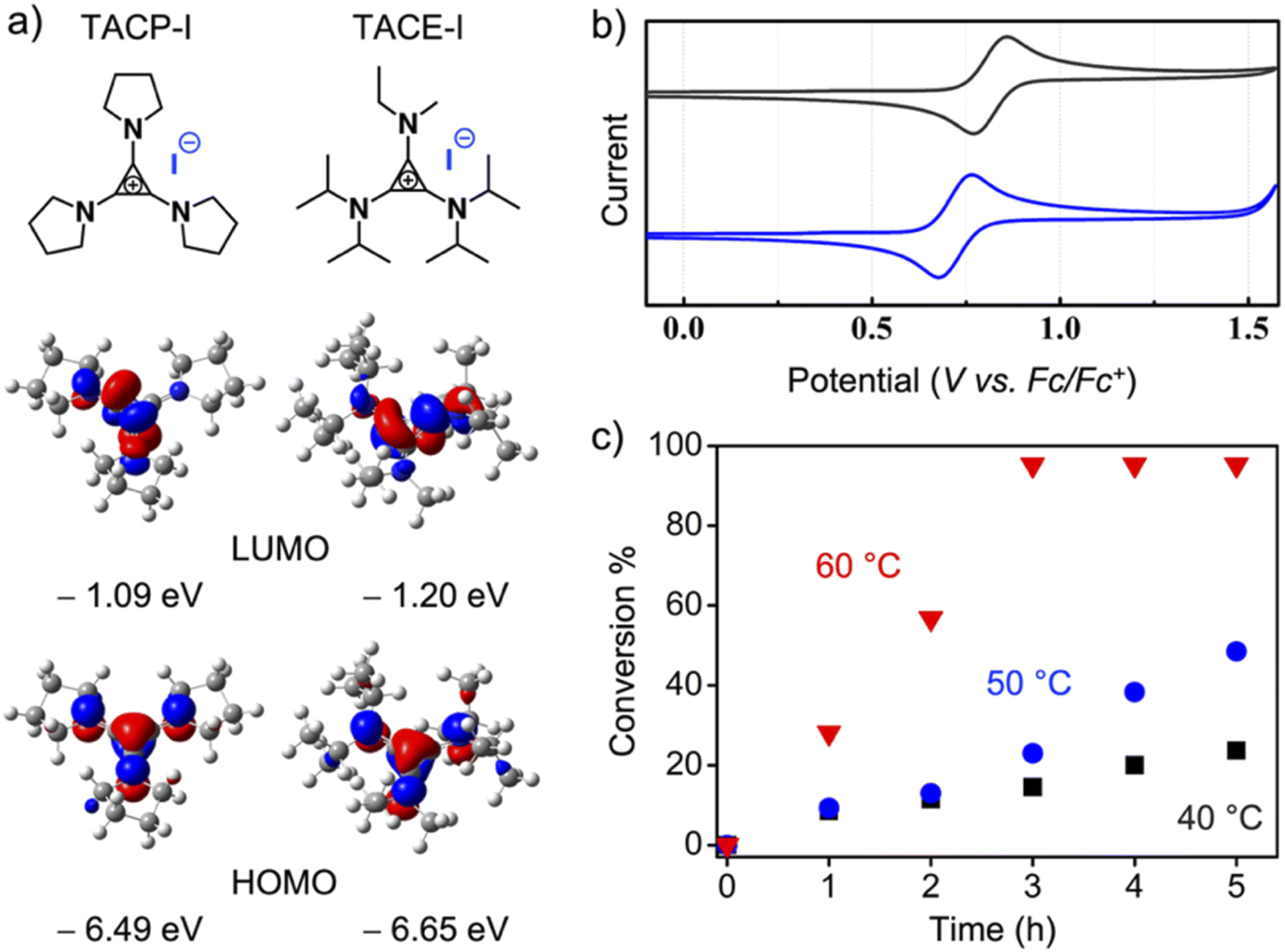 | ||
| Fig. 3 (a) The chemical structures and the calculated frontier molecular orbitals of TACP-I and TACE-I; (b) cyclic voltammograms of TACP-PF6 (blue) and TACE-PF6 (black), Fc/Fc+: ferrocene/ferrocenium; (c) the time–conversion plot of the radical trapping catalyzed by TACE-I. | ||
XB-catalyzed living radical polymerization
Without the radical trapping by TEMPO, the radical-generation reaction under XB catalysis is reversible. We expected that TACP-I can act as a highly potent catalyst for radical polymerization as well, to provide the polymerization reaction with efficient initiation and fast activation–deactivation cycles, thus achieving controlled/living polymerization (Fig. 4a). Bulk polymerization of methyl methacrylate (MMA) was chosen as the model reaction, and the four organic iodide salts (TACP-I, TBN-I, TBMP-I, and EMIZ-I) were applied as catalysts. In general, a mixture of CP-I and the catalyst in MMA with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5:200 was heated at 60 °C for 5 h. The polymerization reaction catalyzed by TACP-I was chosen as an example. The 1H NMR spectrum of the reaction mixture was obtained and shown in Fig. S3,† in which the signals belonging to the monomer MMA and the formed polymer PMMA were coexisted with a calculated conversion of 66%. The obtained PMMA was further characterized using gel permeation chromatography (GPC), which gave a number-average molecular weight (Mn) of 19.3 kD with a dispersity value (Đ) of 1.10 (Fig. 4b). The first–order plot of the monomer concentration [M] against reaction time is shown in Fig. 4c, which is linear in the studied time range. The Mn of the resulted polymers also increased linearly with the monomer conversion, and the molecular weight distributions maintained narrow (with Đ < 1.20) even at high conversions (Fig. 4d).
:0.5:200 was heated at 60 °C for 5 h. The polymerization reaction catalyzed by TACP-I was chosen as an example. The 1H NMR spectrum of the reaction mixture was obtained and shown in Fig. S3,† in which the signals belonging to the monomer MMA and the formed polymer PMMA were coexisted with a calculated conversion of 66%. The obtained PMMA was further characterized using gel permeation chromatography (GPC), which gave a number-average molecular weight (Mn) of 19.3 kD with a dispersity value (Đ) of 1.10 (Fig. 4b). The first–order plot of the monomer concentration [M] against reaction time is shown in Fig. 4c, which is linear in the studied time range. The Mn of the resulted polymers also increased linearly with the monomer conversion, and the molecular weight distributions maintained narrow (with Đ < 1.20) even at high conversions (Fig. 4d).
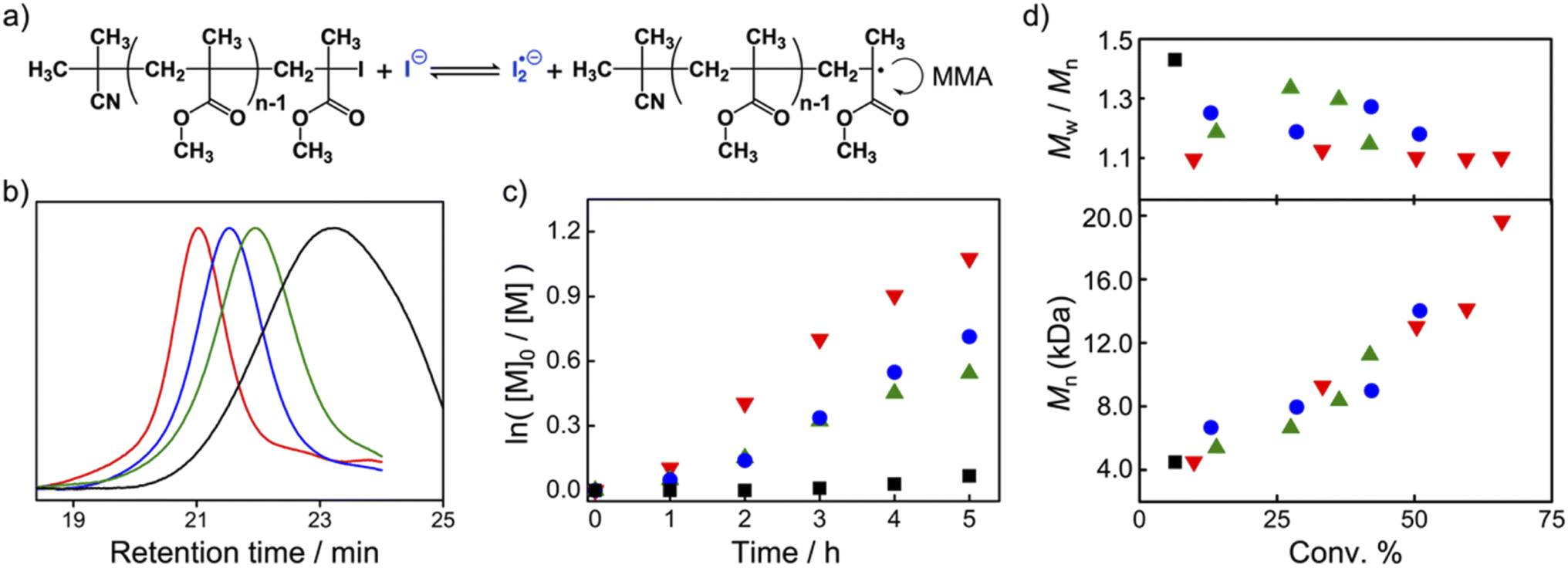 | ||
| Fig. 4 XB-catalyzed radical polymerization of MMA. (a) General scheme of MMA polymerization; (b) GPC chromatograms of the obtained PMMA polymers from the reactions catalyzed by different catalysts; (c) the plots of ln([M]0/[M]) against reaction time; (d) the plots of Mn and Đ against conversion (red: TACP-I, blue: TBN-I, green: TBMP-I, black: EMIZ-I). | ||
The corresponding results from the three other catalysts are also included in Fig. 4 and Table 1. In general, the polymerization reaction proceeded more efficiently under the catalysis by TACP-I, which is not just reflected in the reaction extent but also in the polymer dispersity control (Fig. 4c and d). The performances of the catalysts in polymerization agree well with their capacity in catalyzing the radical-generation reaction, with the order of TACP-I > TBN-I > TBMP-I > EMIZ-I. Noteworthily, the superiority of TACP-I over the other organic iodide salts in catalysis is even more distinct at lower reaction temperatures. The performances of different catalysts in the polymerization reaction of MMA at 50 °C and 40 °C were studied, and the results are included in Table 1 (entry 12–18). At the low reaction temperatures, TACP-I remained effective in catalyzing the polymerization, whereas the catalytic efficiency of TBN-I and TBMP-I significantly decreased and resulted in polymers of low Mn with broad dispersity. Other than the reaction temperature, the effect of catalyst loading on the polymerization was also studied. As shown in entry 9–11 of Table 1, the decrease in the ratio of TACP-I led to slower reaction rate, but high conversions could be realized by prolonging the reaction. Polymers with growing Mn and narrow Đ were obtained, demonstrates a controlled polymerization process. Moreover, the initiator efficiency under different reaction conditions was evaluated by comparing the ratio of Mn,cal. to Mn,GPC. It revealed that the decrease in reaction temperature and catalyst loading improved the initiator efficiency, although at the expense of the rate of polymerization.
| Entry | Monomer | Catalyst | [M]0/[I]/[cat.] | T (°C) | t (h) | Conv.% | M n,GPC (kDa) | M n,cal. (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Note: the conv.% values were calculated from 1H NMR; Mn,GPC and Đ were obtained from GPC (DMF, calibrated using PMMA as standard); Mn,cal. was the theoretical molecular weight calculated from the conversion; CP-I was used as initiator in all experiments. All polymerization reactions were performed in bulk, except AN was polymerized in ethylene carbonate (50 wt% solution). | |||||||||
| 1 | MMA | TACP-I | 200/1/0.5 | 60 | 5 | 66 | 19.2 | 13.2 | 1.10 |
| 2 | MMA | TBN-I | 200/1/0.5 | 60 | 5 | 51 | 14.0 | 10.2 | 1.18 |
| 3 | MMA | TBMP-I | 200/1/0.5 | 60 | 5 | 42 | 11.2 | 8.4 | 1.15 |
| 4 | MMA | EMIZ-I | 200/1/0.5 | 60 | 5 | 7 | 4.5 | 1.4 | 1.43 |
| 5 | MMA | TACP-I | 200/1/0.5 | 60 | 10 | 75 | 26.8 | 15.0 | 1.15 |
| 6 | MMA | TBN-I | 200/1/0.5 | 60 | 10 | 64 | 20.3 | 12.8 | 1.17 |
| 7 | MMA | TBMP-I | 200/1/0.5 | 60 | 10 | 59 | 17.2 | 11.8 | 1.15 |
| 8 | MMA | EMIZ-I | 200/1/0.5 | 60 | 10 | 25 | 7.4 | 5.0 | 1.36 |
| 9 | MMA | TACP-I | 200/1/0.3 | 60 | 5 | 55 | 12.5 | 11.0 | 1.17 |
| 10 | MMA | TACP-I | 200/1/0.3 | 60 | 10 | 71 | 19.5 | 14.2 | 1.13 |
| 11 | MMA | TACP-I | 200/1/0.3 | 60 | 24 | 84 | 22.5 | 16.8 | 1.20 |
| 12 | MMA | TACP-I | 200/1/1 | 50 | 10 | 65 | 18.9 | 13.0 | 1.11 |
| 13 | MMA | TACP-I | 200/1/1 | 50 | 6 | 54 | 13.3 | 10.8 | 1.14 |
| 14 | MMA | TBN-I | 200/1/1 | 50 | 6 | 22 | 6.0 | 4.4 | 1.23 |
| 15 | MMA | TBMP-I | 200/1/1 | 50 | 6 | 23 | 6.2 | 4.6 | 1.34 |
| 16 | MMA | TACP-I | 200/1/1 | 40 | 24 | 60 | 16.1 | 12.0 | 1.18 |
| 17 | MMA | TBN-I | 200/1/1 | 40 | 24 | 21 | 6.6 | 4.2 | 1.29 |
| 18 | MMA | TBMP-I | 200/1/1 | 40 | 24 | 25 | 5.6 | 5.0 | 1.31 |
| 19 | BzMA | TACP-I | 200/1/0.5 | 60 | 5 | 78 | 24.6 | 27.5 | 1.16 |
| 20 | OMA | TACP-I | 200/1/1 | 60 | 5 | 64 | 34.4 | 18.2 | 1.30 |
| 21 | TFMA | TACP-I | 200/1/0.5 | 60 | 5 | 73 | 40.4 | 24.5 | 1.06 |
| 22 | MEMA | TACP-I | 200/1/2 | 60 | 10 | 60 | 23.8 | 17.3 | 1.25 |
| 23 | BzA | TACP-I | 100/1/4 | 100 | 20 | 52 | 6.8 | 8.4 | 1.38 |
| 24 | BA | TACP-I | 100/1/4 | 110 | 48 | 50 | 2.9 | 6.4 | 1.24 |
| 25 | AN | TACP-I | 200/1/4 | 100 | 24 | 94 | 47.6 | 10.0 | 1.37 |
Besides MMA, several other monomers, such as benzyl methacrylate (BzMA), oxiran-2-ylmethyl methacrylate (OMA), 2,2,2-trifluoroethyl methacrylate (TFMA), 2-methoxyethyl methacrylate (MEMA), benzyl acrylate (BzA), butyl acrylate (BA), and acrylonitrile (AN) were also well polymerized by applying TACP-I as the catalyst, proving the general applicability of TACP-I (Scheme 3, Table 1 entry 19–25). It is to be noted that, owing to the stronger iodine–carbon bond in the acrylate polymer than in methacrylate polymer, an increased temperature is necessary for the polymerization of acrylate monomers. In addition, an acrylate monomer that contains TAC moiety (TACA) was designed and synthesized, which can act as monomer and catalyst at the same time.45 Self-catalyzed copolymerization of TACA and BzA was successfully achieved, produced copolymer PTACA-co-BzA with Mn of 6.4 kDa and Đ of 1.32 (Fig. S8†). This provides a facile approach to prepare TAC-containing polymers for biomedical or energy-related applications.
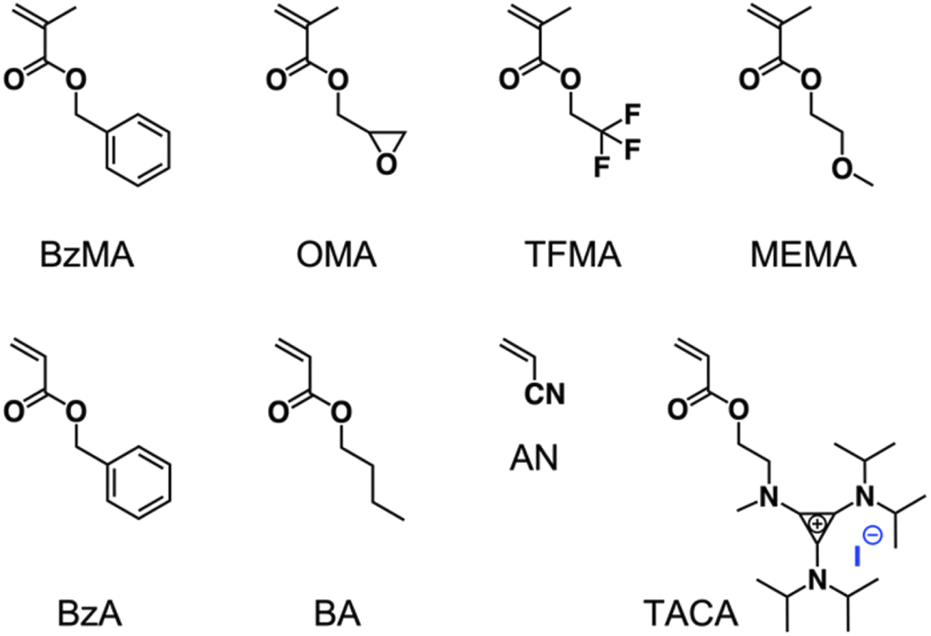 | ||
| Scheme 3 Chemical structures of the monomers used in polymerization. | ||
To exploit the living character of the polymerization reaction, chain-extension experiments were conducted. Premade PMMA-I with Mn of 15.3 kDa and Đ of 1.11 was used as a macroinitiator in the polymerization of MMA with TACP-I as the catalyst. The GPC chromatograms at different reaction times are shown in Fig. 5a. A large fraction of the PMMA-I macroinitiator gradually extended to higher molecular weights, which confirms the remaining activity of the iodide terminal group of PMMA-I. On this basis, block polymerizations were also performed. Using the same PMMA-I as macroinitiator and BzMA or TFMA as the monomer, block polymers of PMMA-b-PBzMA (Mn = 40.7 kDa, Đ = 1.23) and PMMA-b-PTFMA (Mn = 71.3 kDa, Đ = 1.09) were successfully obtained (Fig. 5b and S9–S12†). As an example, the GPC chromatograms of PMMA-I and PMMA-b-PBzMA are shown in Fig. 5b, the fraction of the PMMA-I macroinitiator chains mainly moved to block copolymers, indicating the high efficiency of block polymerization.
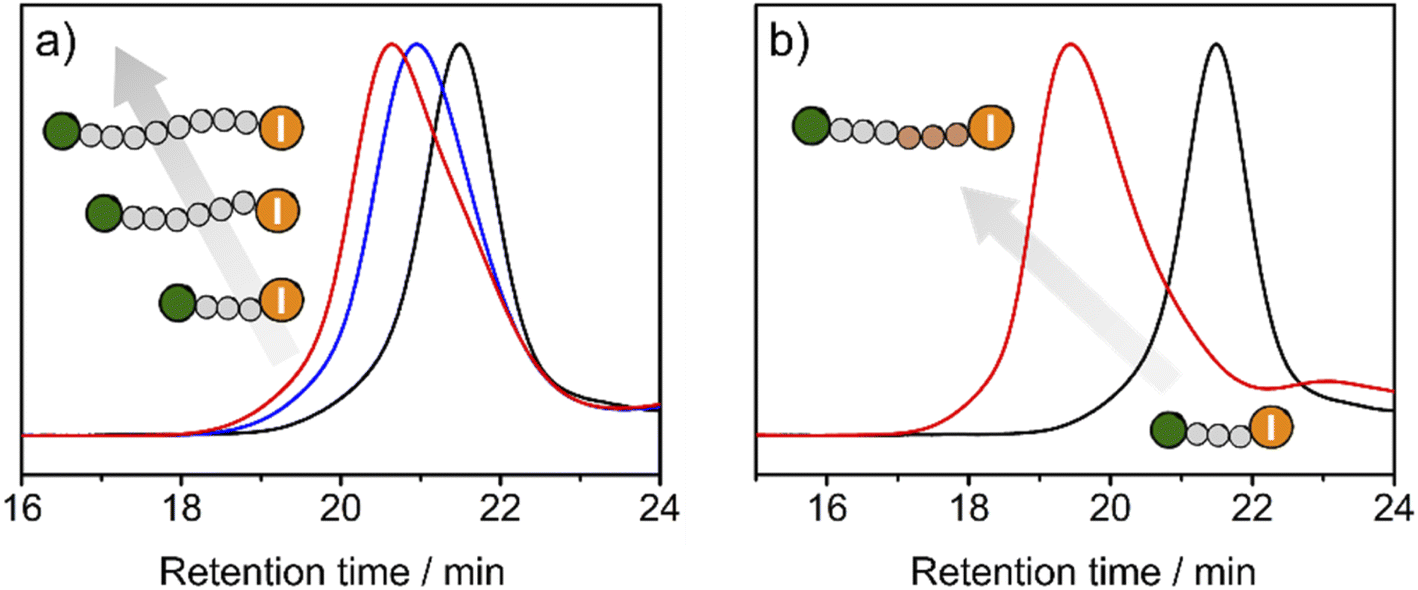 | ||
| Fig. 5 The GPC chromatograms of chain-extension polymerization (0 min: black; 40 min: blue; 60 min: red) and block polymerization (macroinitiator: black; PMMA-b-PBzMA: red). Characterized using GPC with DMF as eluent and PMMA as standard. | ||
XB-catalyzed depolymerization
Reversing polymerization to regenerate the monomer is attracting increased research interests.46–49 Cleaving the end-groups of the polymers to generate radicals and increasing the reaction temperature above the ceiling temperature of polymerization are the two prerequisites to reversing the radical polymerization.50–52 Because TACP-I shows high catalytic efficiency in radical-generation reactions, we wondered that if it can also be applied in catalyzing the depolymerization reaction. To this end, premade PMMA-I was used as a test polymer for the depolymerization reaction. In a typical reaction, PMMA-I was dissolved in tetraglyme with the catalysts (100 eq. relative to polymer chains), and the reaction mixtures were heated at 120 °C for 24 h. The GPC chromatograms of the PMMA-I before and after the reaction are shown in Fig. 6. With TACP-I as the catalyst, the PMMA-I underwent considerable depolymerization from Mn of 10.2 kDa to 6.4 kDa. The generation of the MMA monomer was confirmed using 1H NMR and gas chromatography (Fig. S13 and S14†). In contrast, the depolymerization of PMMA-I with TBN-I or TBMP-I as catalysts were less effective; only slight decreases in molecular weight were achieved. Moreover, the molecular weight distributions of the remaining polymers were clearly broadened with high Mn species generated, which indicates side reactions occur along with chain degradation. In general, TACP-I surpassed other organic iodide salts in catalyzing the depolymerization reaction. Even though only moderate conversions of depolymerization were achieved in the current system, it is expected that optimization of the reaction conditions, could further promote the depolymerization. For example, increasing the reaction temperature to 150 °C led to further depolymerization (Fig. S17†), although reaction pathways other than the radical mechanism may co-existed.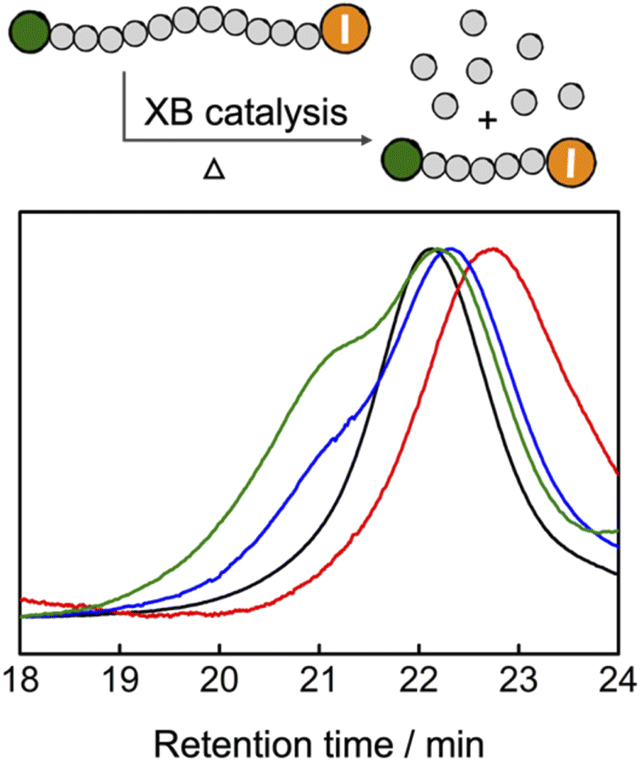 | ||
| Fig. 6 GPC characterization of the PMMA depolymerization reaction (PMMA-I: black; TACP-I catalyzed: red; TBN-I catalyzed: blue; TBMP-I catalyzed: green). | ||
Conclusions
Here, we have investigated the catalytic capacity of TAC iodides for the first time in catalyzing radical polymerization. Promoted by the unique effect of ion-pair strain between the TAC cation and the iodide anion, the TAC iodides showed high catalytic efficiency in the XB-catalyzed radical-generation reaction and surpassed other reported organic iodide catalysts, such as quaternary alkyl phosphonium iodide and quaternary alkyl ammonium iodide. TACP-I, which bears pyrrolidine as substituents, was further successfully applied in catalyzing the radical polymerization of methacrylate monomers. The living feature of this polymerization approach was demonstrated by chain-extension polymerization and block copolymerization experiments. Besides catalyzing the polymerization reactions, TACP-I was also applied as a catalyst for depolymerizing the iodo-terminated PMMA, which showed better performance than the other organic iodide catalysts. Noteworthily, the catalytic capacity of the TAC iodides was demonstrated to be closely related to the electronic properties of the TAC cation, which could be facilely tuned by varying the substituents. Our current efforts are directed toward the further optimization of the TAC-based catalysts in controlled radical polymerization, as well as the application of these catalysts in other XB-catalyzed organic reactions.Data availability
All experimental data is available within the article and the ESI.†Author contributions
S. Huang and Y. Liu conceived and designed the study. S. Huang, X. Su and Y. Wu performed the experiments. X. Xiong performed the DFT calculation. Y. Liu supervised the work. All authors contributed to the finalization of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from National Key R&D Program of China (grant no. 2021YFA1501600), National Natural Science Foundation of China (No. 21901077), The Recruitment Program of Guangdong (2016ZT06C322), and the Research Fund Program of Guangdong Provincial Key Laboratory of Functional and Intelligent Hybrid Materials and Devices (2019B121203003).References
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. Erdelyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- H. Wang, H. K. Bisoyi, A. M. Urbas, T. J. Bunning and Q. Li, Chem.–Eur. J., 2019, 25, 1369–1378 CrossRef CAS PubMed.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281–9290 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- D. Bulfield and S. M. Huber, Chem.–Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- R. L. Sutar and S. M. Huber, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- J. Bamberger, F. Ostler and O. G. Mancheno, ChemCatChem, 2019, 11, 5198–5211 CrossRef CAS PubMed.
- S. Guha, I. Kazi, A. Nandy and G. Sekar, Eur. J. Org. Chem., 2017, 2017, 5497–5518 CrossRef CAS.
- K. Takagi, K. Yamauchi and H. Murakata, Chem.–Eur. J., 2017, 23, 9495–9500 CrossRef CAS PubMed.
- O. Coulembier, F. Meyer and P. Dubois, Polym. Chem., 2010, 1, 434–437 RSC.
- C.-G. Wang, A. M. L. Chong, H. M. Pan, J. Sarkar, X. T. Tay and A. Goto, Polym. Chem., 2020, 11, 5559–5571 RSC.
- A. Goto, N. Hirai, K. Nagasawa, Y. Tsujii, T. Fukuda and H. Kaji, Macromolecules, 2010, 43, 7971–7978 CrossRef CAS.
- A. Goto, A. Ohtsuki, H. Ohfuji, M. Tanishima and H. Kaji, J. Am. Chem. Soc., 2013, 135, 11131–11139 CrossRef CAS PubMed.
- C.-G. Wang and A. Goto, J. Am. Chem. Soc., 2017, 139, 10551–10560 CrossRef CAS PubMed.
- H. Xu, C.-G. Wang, Y. Lu and A. Goto, Macromolecules, 2019, 52, 2156–2163 CrossRef CAS.
- W. Mao, C.-G. Wang, Y. Lu, W. Faustinelie and A. Goto, Polym. Chem., 2020, 11, 53–60 RSC.
- R. Breslow, J. Am. Chem. Soc., 1957, 79, 5318 CrossRef CAS.
- K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371–1427 CrossRef CAS PubMed.
- Z. Yoshida and Y. Tawara, J. Am. Chem. Soc., 1971, 93, 2573–2574 CrossRef CAS.
- J. S. Bandar and T. H. Lambert, Synthesis, 2013, 45, 2485–2498 CrossRef CAS.
- O. J. Curnow, D. R. MacFarlane and K. J. Walst, Chem. Commun., 2011, 47, 10248–10250 RSC.
- P. J. Griffin, J. L. Freyer, N. Han, N. Geller, X. Yin, C. D. Gheewala, T. H. Lambert, L. M. Campos and K. I. Winey, Macromolecules, 2018, 51, 1681–1687 CrossRef CAS.
- Y. Jiang, J. L. Freyer, P. Cotanda, S. D. Brucks, K. L. Killops, J. S. Bandar, C. Torsitano, N. P. Balsara, T. H. Lambert and L. M. Campos, Nat. Commun., 2015, 6, 6950 CrossRef PubMed.
- C. S. Sevov, S. K. Samaroo and M. S. Sanford, Adv. Energy Mater., 2017, 7, 1602027 CrossRef.
- H. Bruns, M. Patil, J. Carreras, A. Vazquez, W. Thiel, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2010, 49, 3680–3683 CrossRef CAS PubMed.
- M. Guest, R. Mir, G. Foran, B. Hickson, A. Necakov and T. Dudding, J. Org. Chem., 2020, 85, 13997–14011 CrossRef CAS PubMed.
- J. L. Freyer, S. D. Brucks, G. S. Gobieski, S. T. Russell, C. E. Yozwiak, M. Sun, Z. Chen, Y. Jiang, J. S. Bandar, B. R. Stockwell, T. H. Lambert and L. M. Campos, Angew. Chem., Int. Ed., 2016, 55, 12382–12386 CrossRef CAS.
- H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS PubMed.
- C. M. Vanos and T. H. Lambert, Chem. Sci., 2010, 1, 705–708 RSC.
- I. Smajlagic, R. Duran, M. Pilkington and T. Dudding, J. Org. Chem., 2018, 83, 13973–13980 CrossRef CAS PubMed.
- R. Mirabdolbaghi, T. Dudding and T. Stamatatos, Org. Lett., 2014, 16, 2790–2793 CrossRef CAS PubMed.
- R. Weiss, T. Brenner, F. Hampel and A. Wolski, Angew. Chem., Int. Ed., 1995, 34, 439–441 CrossRef CAS.
- R. Weiss, M. Rechinger, F. Hampel and A. Wolski, Angew. Chem., Int. Ed., 1995, 34, 441–443 CrossRef CAS.
- J. R. Butchard, O. J. Curnow, D. J. Garrett and R. G. A. R. Maclagan, Angew. Chem., Int. Ed., 2006, 45, 7550–7553 CrossRef CAS PubMed.
- J. S. Bandar, A. Tanaset and T. H. Lambert, Chem.–Eur. J., 2015, 21, 7365–7368 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS.
- H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed.
- T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed.
- S. H. Jungbauer, S. Schindler, E. Herdtweck, S. Keller and S. M. Huber, Chem.–Eur. J., 2015, 21, 13625–13636 CrossRef CAS PubMed.
- C.-G. Wang, X. Y. Oh, X. Liu and A. Goto, Macromolecules, 2019, 52, 2712–2718 CrossRef CAS.
- M. R. Martinez and K. Matyjaszewski, CCS Chem., 2022, 4, 2176–2211 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS.
- X. Yang, Q. Ji, J. Liu and Y. Liu, CCS Chem., 2022 DOI:10.31635/ccschem.022.202201936.
- C. Jehanno, M. M. Perez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- M. R. Martinez, F. D. L. Bossa, M. Olszewski and K. Matyjaszewski, Macromolecules, 2022, 55, 78–87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis of chemical compounds, and other supplementary results. See https://doi.org/10.1039/d2sc04196k |
| This journal is © The Royal Society of Chemistry 2022 |