
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversal linker-free assembly of core–satellite hetero-superstructures†
Yanfang
Hu
a,
Yonglong
Li
a,
Linfeng
Yu
a,
Yuying
Zhang
 b,
Yuming
Lai
c,
Wei
Zhang
a and
Wei
Xie
*a
b,
Yuming
Lai
c,
Wei
Zhang
a and
Wei
Xie
*a
aKey Lab of Advanced Energy Materials Chemistry (Ministry of Education), Tianjin Key Lab of Molecular Recognition & Biosensing, Renewable Energy Conversion and Storage Center, College of Chemistry, Nankai University, Weijin Rd. 94, Tianjin 300071, China. E-mail: wei.xie@nankai.edu.cn
bSchool of Medicine, Nankai University, Weijin Rd. 94, Tianjin 300071, China
cNational Center for Materials Service Safety, University of Science and Technology Beijing, Beijing 100083, China
First published on 20th September 2022
Abstract
Colloidal superstructures comprising hetero-building blocks often show unanticipated physical and chemical properties. Here, we present a universal assembly methodology to prepare hetero-superstructures. This straightforward methodology allows the assembly of building block materials varying from inorganic nanoparticles to living cells to form superstructures. No molecular linker is required to bind the building blocks together and thus the products do not contain any unwanted adscititious material. The Fourier transform infrared spectra, high resolution transmission electron microscopic images and nanoparticle adhesion force measurement results reveal that the key to self-organization is stripping surface ligands by adding non-polar solvents or neutralizing surface charge by adding salts, which allow us to tune the balance between van der Waals attraction and electrostatic repulsion in the colloid so as to trigger the assembling process. As a proof-of-concept, the superior photocatalytic activity and single-particle surface-enhanced Raman scattering of the corresponding superstructures are demonstrated. Our methodology greatly extends the scope of building blocks for superstructure assembly and enables scalable construction of colloidal multifunctional materials.
Introduction
Assembly of diverse nanoparticles (NPs) into superstructures has emerged as an important strategy toward generating new functional materials.1–6 The obtained hetero-superstructures integrate distinct components and often exhibit unanticipated properties with potential application in photonics,7–12 magnetics,13,14 catalysis,15,16 and biomedicine.17–21 However, the lack of a scalable assembly method limits broad application of hetero-superstructures. Current routes to hetero-superstructures are molecular linker-mediated NP conjugation and electrostatic attraction-induced NP assembly. For example, molecules with surface-seeking groups such as –SH and –NH2 are usually used as linkers to assemble core–satellite superstructures.22–25 DNA hybridization17,19,26–30 and metal–phenolic coordination31,32 are also well-established linker-mediated NP conjugation methods to assemble superstructures. In electrostatic assembly, Coulomb forces between oppositely charged NPs are the driving forces of forming superstructures.25,33–36 Besides, entropy-driven assembly could generate NP superlattices that consist of different types of NP building blocks,37–39 but has not yet been exploited for core–satellite superstructures.Despite the progressive assembly methods, the following 3 limitations have restricted the general assembly of hetero-superstructures using arbitrary building block materials: (1) highly specific assembly principles, such as thiol–metal bonding and complementary DNA strand hybridization, result in a limited assembly scope. A general approach with the driving force based on common properties of all NPs has been lacking. (2) Retained assembly additives in the obtained superstructures mask the original nature of the building blocks. The linkers which bring together the different building blocks could not be removed after assembly. Otherwise, the assembled material would be dissociated. (3) Complex assembly procedures seriously decrease the yield of assembly. The washing steps of NPs in the assembly, typically multiple centrifugations and resuspensions, result in aggregation of NPs in the sediment and loss of NPs in the discarded supernatant.
Here, we manipulate the interparticle balance between attraction and repulsion toward a simple and versatile methodology for universal core–satellite superstructure assembly. The key step is using colloid destabilizers such as a nonpolar solvent or inorganic salt to reduce the stability (Coulomb repulsion) of one building block. Then the other building block acts as a new stabilizer to approach the destabilized building block via van der Waals (vdW) attraction (Fig. 1a). In contrast to previously reported methods, our assembly does not need any molecular linkers to bind the different building blocks together. Thus, the assembly process is extremely simple (only one step) and no additional substance is retained in the obtained core–satellite superstructures. Because vdW forces ubiquitously exist in all colloids, this methodology could in principle be used for assembly of any kinds of NPs in a colloidal suspension (Fig. 1b–d).
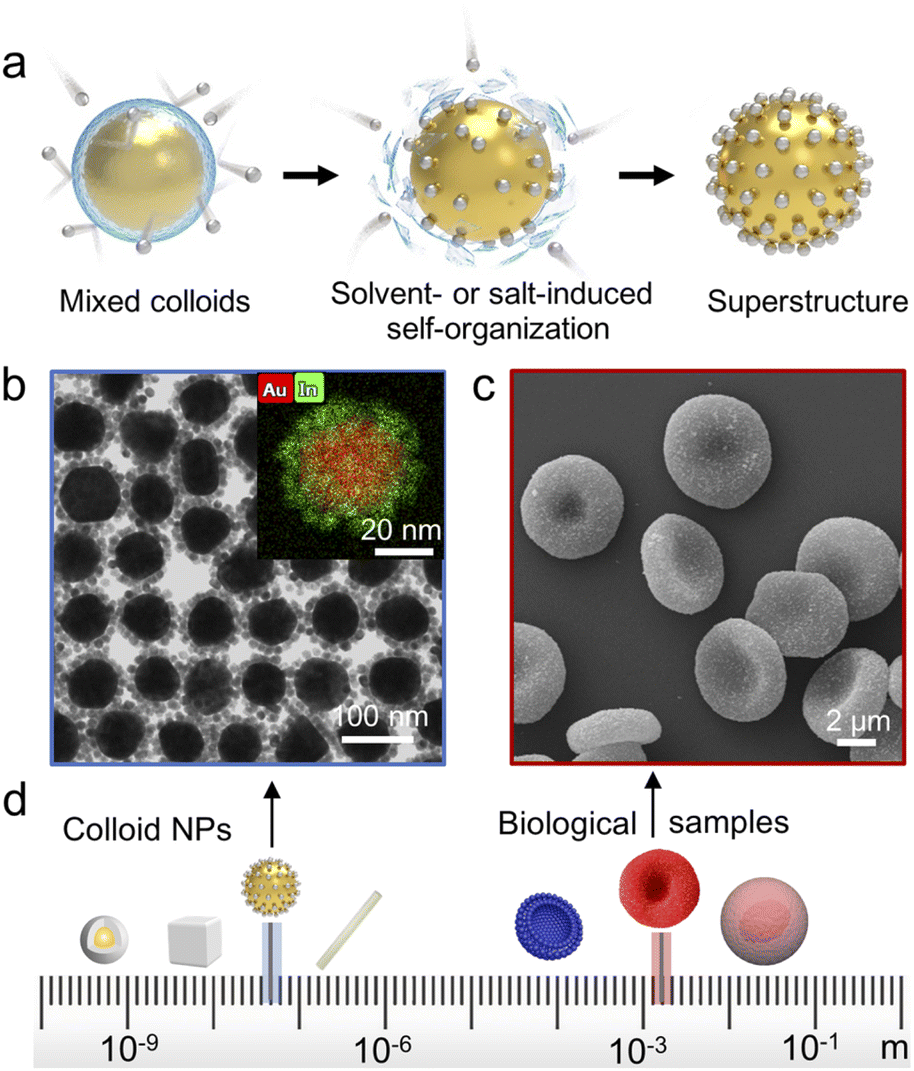 | ||
| Fig. 1 Preparation of superstructures. (a) Schematic illustration of the core–satellite superstructure assembly. (b) TEM image and EDS mapping of Au@In2O3 core–satellite superstructures. (c) SEM image of the red blood cell (RBC)@Au core–satellite superstructures. (d) Schematic of building blocks with various shapes, sizes, compositions and functionalities. | ||
Results and discussion
Citrate-stabilized Au NPs40 maintain colloidal stability via the strong interparticle repulsive forces (Fig. S1a and b†). When a nonpolar solvent such as hexane or toluene is added into the Au NPs dispersed in ethanol/isopropanol, a broad extinction band appears at ∼800 nm (Fig. S1d†) and the hydrodynamic diameter gradually increases due to partial coalescence of the Au NPs (Fig. S2†). The nonpolar solvent unambiguously breaks down colloidal stability by reducing repulsive forces and thus triggers NP self-organization. As a proof of concept, the citrate-stabilized Au NPs suspended in isopropanol/ethanol and oleylamine-stabilized In2O3 NPs suspended in hexane were selected as our model building blocks. By simply mixing the 2 colloids together, highly monodispersed binary Aucore–In2O3satellite superstructures were obtained. We observed an obvious Au plasmon band shift after In2O3 approached (Fig. S3†) because of the dielectric environment change.This assembly methodology is generally applicable and superstructures can be rationally designed using distinct building blocks. Fig. 2 shows diverse examples of superstructured nanomaterials. On quasi-spherical 80 nm Au NPs, different metal-based materials, including monometallic, alloy, oxide, sulfide, and perovskite NPs (Fig. 2a–d, j and S4–S11†), could easily be assembled as satellites. NPs having different morphologies such as Au–Ag core–shells, Au nanopeanuts, CdS nanowires, and Ag nanocubes could also be used as cores for superstructure assembly (Fig. 2f–i and S12–S15†).
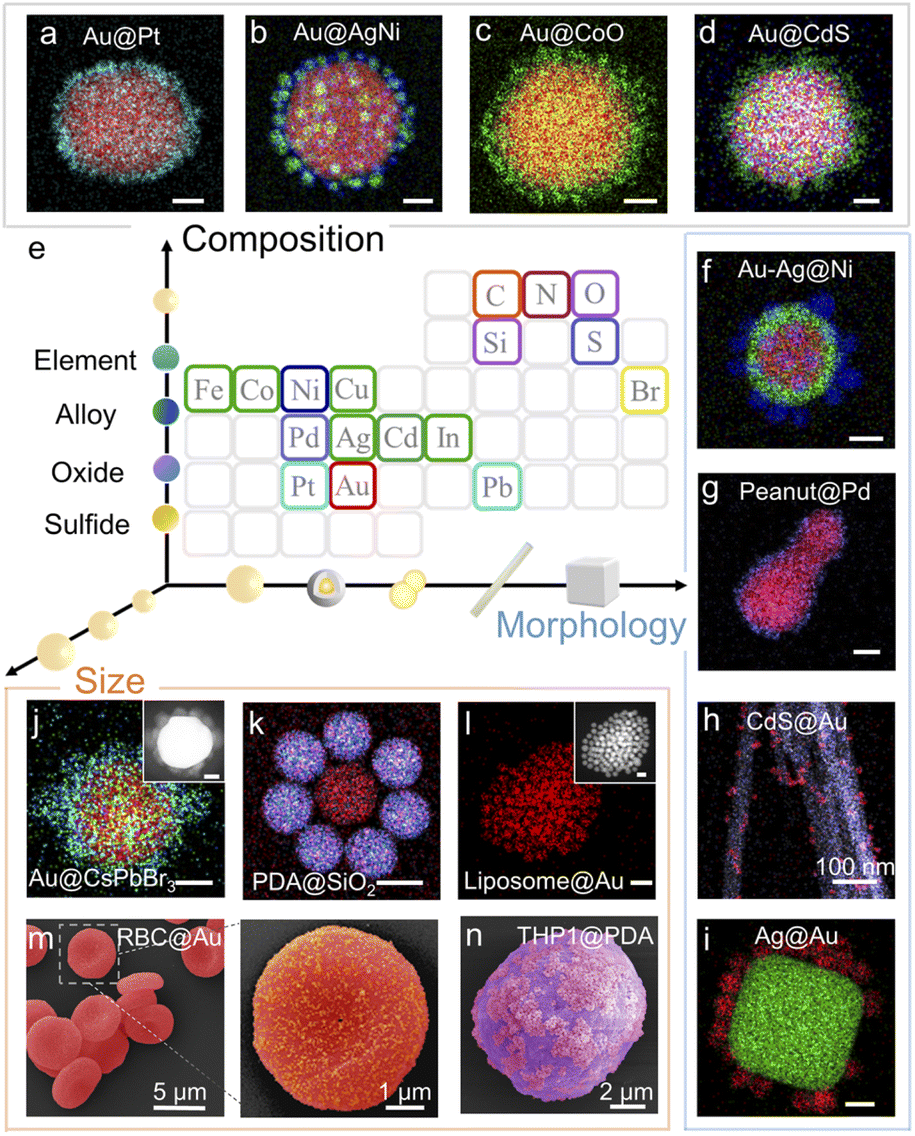 | ||
| Fig. 2 Diversity of superstructured materials. (a–d) Elemental mapping images of Au@Pt (a), Au@AgNi (b), Au@CoO (c), and Au@CdS core–satellite superstructures (d). Scale bar, 20 nm. (e) Illustration of assembly using building blocks of diverse compositions, morphologies and sizes. (f–l) Elemental mapping images of Au–Ag@Ni (f), Au peanut@Pd (g), CdS@Au (h), Ag nanocube@Au(I), and Au@CsPbBr3 perovskite (j) core–satellite superstructures. Scale bar, 20 nm. (k) Elemental mapping image of the PDA@SiO2 core–satellite superstructure. Scale bar, 200 nm. (l) Elemental mapping image of the liposome@Au core–satellite superstructure. Scale bar, 20 nm. Insets of J and L are high-angle annular dark-field (HAADF) images of the corresponding superstructures. Scale bar, 20 nm. (m and n) Colored SEM images of RBC@Au (m) and living THP1 cell@PDA (n) biological core–satellite superstructures. The colors of the elements in the EDS mapping are marked on the periodic table in (e). | ||
Greatly encouraged by these experimental results, we extended this methodology to the assembly of organic and even biological nanoarchitectures. Polydopamine (PDA) NPs have been widely used in catalysis, medical imaging, cancer therapy, and antibacterial coating because of their antioxidant activity and strong metal-ion chelation.41 We mixed PDA NPs with SiO2 NPs suspended in ethanol and obtained organic–inorganic hybrid core–satellite superstructures by adding hexane (Fig. 2k and S16†). However, in the assembly of biological nanoarchitectures, nonpolar solvents cannot be used because they typically denature the biological building blocks. Thus, we used biocompatible phosphate buffer solution (PBS) to trigger self-organization. The ions in PBS neutralize the surface charge of colloidal NPs, instead of detaching the surface ligand, to weaken the repulsive forces and reduce their colloidal stability (Fig. S17†).42,43 We obtained nanoarchitectures such as liposomecore@Ausatellite, red blood cell (RBC)core@Ausatellite and living THP1 cellcore@PDAsatellite superstructures (Fig. 2l–n and S18–S20†). This process is very gentle and the living mammalian cells maintained their structural stability after assembly (Fig. S20†). Therefore, our methodology shows good applicability and can realize the assembly of superstructures with satellite numbers ≥6 and a core/satellite size ratio from 1 to ∼200. The products are very robust and the core–satellite structure remains stable after vigorous ultrasonication, solvent-replacement and even 1 year aging at room temperature (Fig. S21†).
As a demonstration of preparing high-diversity superstructured materials, a co-assembly strategy was used to assemble superstructures with multiple types of satellites on the same core. We obtained a series of superstructures using the same batch of Au NP cores, including Aucore@Agsatellite, Aucore@(Ag + Ni)satellite, Aucore@(Ag + Ni + In2O3)satellite, and Aucore@(Ag + Ni + In2O3 + Pt)satellite superstructures (Fig. 3a–d). Alternatively, multiple satellites can be assembled via a super-assembly strategy. For example, when RBCcore@PDAsatellite superstructures were further used as the core in a second-round assembly, Au NPs could be loaded as the second satellites (Fig. 3e–g). Any change in the type and sequence of satellites will result in new core–satellite superstructures.
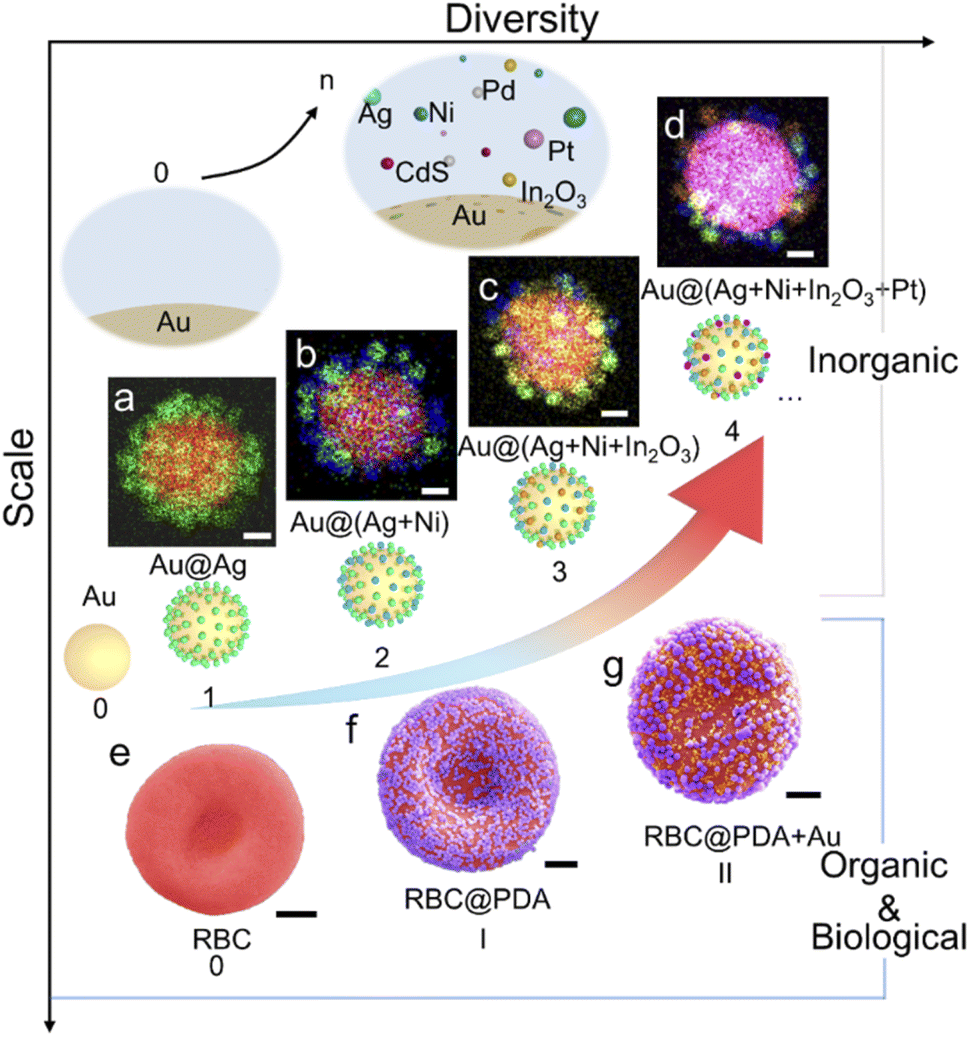 | ||
| Fig. 3 High-diversity superstructure assembly. (a–d) Elemental mapping images of core–satellite superstructures with up to 4 types of satellites co-assembled on an 80 nm Au core: Au@Ag (a), Au@Ag + Ni (b), Au@Ag + Ni + In2O3 (c), and Au@ Ag + Ni + In2O3 + Pt (d). Scale bar, 20 nm. (e–g) Colored SEM images of a red blood cell (e) and corresponding superstructures with PDA NPs (f) and PDA + Au NPs (g) as the satellites (scale bar, 1 μm). | ||
To explore the mechanism behind this methodology, we further investigated the driving force of the solvent-induced superstructure assembly. In our control experiments, cores and satellites with the same charge could also form superstructures on the addition of hexane (Fig. S23†). In contrast, when hexane was absent, even the oppositely charged Au and In2O3 NPs could not form superstructures (Fig. S22 and S24†), which confirms that the assembly is not driven by electrostatic attraction. In the Fourier transform infrared (FTIR) spectra (Fig. 4a), the absorption band assigned to the asymmetric stretching of carboxylic groups of the citrate ligand redshifts from 1585 to 1558 cm−1,44 indicating that the addition of hexane induces the attenuation of the intermolecular hydrogen bonds and disrupts the solvation of citrate molecules with isopropanol (Fig. S25†). As shown in Fig. 4b the citrate ligand forms approximately four molecular layers on the Au NP surface after drying on a TEM grid;45 in contrast, no obvious ligand shell is observed on hexane-treated NPs (Fig. 4c). Although the results were not measured from NPs in a colloidal suspension, it is clear that the hexane solvent removes most of the ligand molecules on the NP surface. Since non-polar solvents such as hexane are aimed at destroying citrate molecules, this methodology is suitable for the assembly of citrate-capped NPs. We further used density functional theory (DFT) calculations (see the ESI for the detailed calculation process†) to evaluate the adsorption energy (Eads = −0.77 eV) of a citrate ligand on the Au(111) surface (Fig. 4d). While ethanol solvent makes the surface ligand more stable (Eads = −0.84 eV), the presence of hexane obviously weakens the adsorption of citrate (Eads = −0.51 eV) on the Au(111) surface. The surface ligand has an important bearing on the colloidal stability of NPs.46,47ζ-Potential change of the Au NPs from −32.2 to −7.04 mV was observed within 12 min upon the addition of hexane (Fig. S2†).
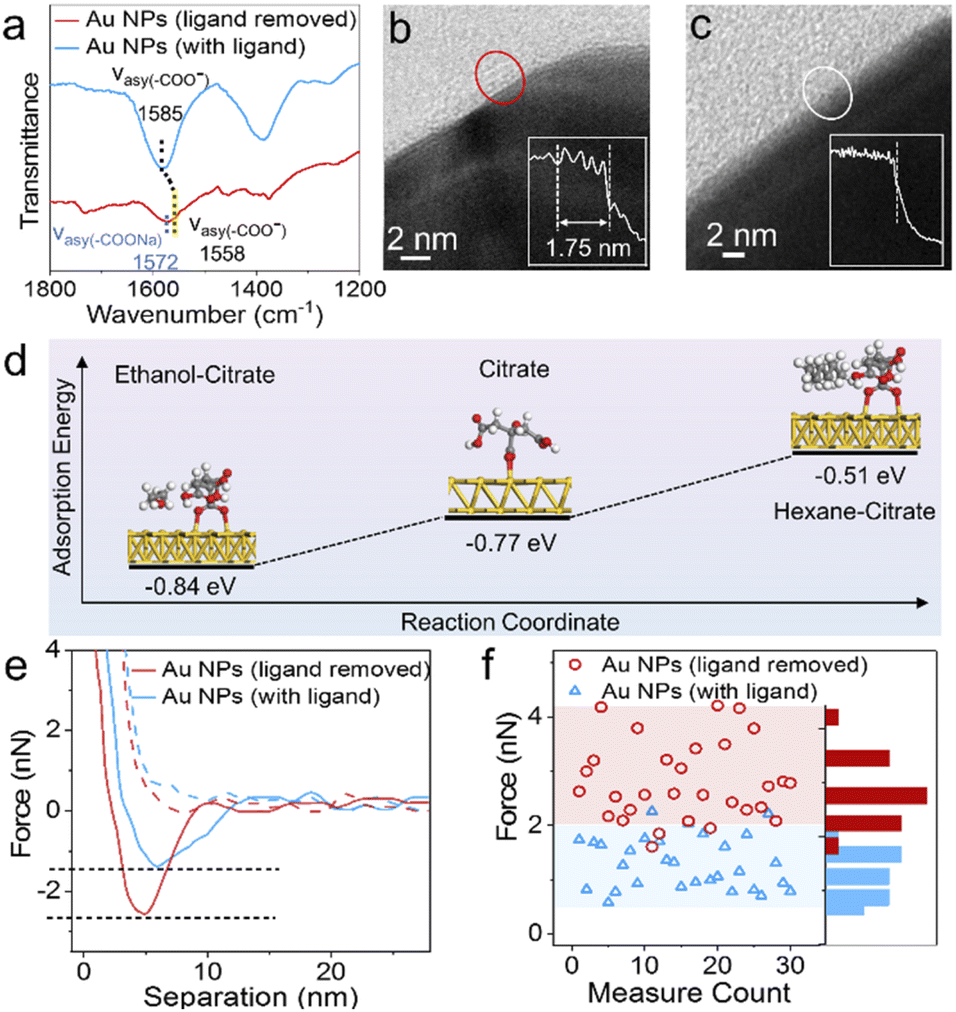 | ||
| Fig. 4 Driving force of superstructure assembly. FTIR spectra (a), high-resolution transmission electron microscope (HRTEM) images and line scans of a citrate ligand adsorbed on the surface of Au NPs before (b) and after (c) treatment with hexane. (d) Adsorption energy of citrate on the Au NP surface in different solvent environments. (e) Force-distance curves measured using a Pt coated AFM tip on Au NPs before (cyan) and after (red) hexane treatment. The approach and retraction are presented with dashed and solid lines, respectively. (f) Scatter plot of adhesive force measurements on 60 Au NPs. | ||
The assembly process of the above core–satellite superstructures comprises two steps. First, one of the colloidal building blocks is destabilized. Second, the other building block acts as a new stabilizing agent and self-organizes around the first building block, which is likely driven by vdW forces. We used atomic force microscopy (AFM) to examine the vdW forces between the core and satellite.48 A Pt tip (Fig. S27†) interacts with a Au core to simulate the Pt satellite of the Aucore@Ptsatellite superstructure. An approach-retraction loop test was performed in a liquid environment. As shown in Fig. 4e, the Au NP surface treated with hexane exhibited twice the attractive force (2.6 nN) during tip retraction compared with its untreated counterpart (1.3 nN), corresponding to a higher energy of adhesion work (E = 1.17 × 10−2vs. 6.85 × 10−3 fJ). The 60 tests on different Au NPs (Fig. 4f and S29†) suggested that the addition of hexane resulted in an appreciable attraction between the core and satellite NPs. Hexane acts as a ligand detacher and this role could also be played by other nonpolar solvents such as toluene, benzene and cyclohexane (Fig. S30†).
In classic Derjaguin–Landau–Verwey–Overbeek (DLVO) theory,49 two major interactions (electrostatic repulsion and vdW attraction) determine the colloidal stability of a NP suspension. In our assembly, a third interaction of short-range repulsion resulting from steric hindrance of interfacial molecules is involved.50,51Eqn (1) describes the total interaction potential energy (Vtot) between Au and Pt NPs, which consists of electrostatic energy (Velec), vdW potential energy (VvdW) and short-range repulsion energy (VBorn):
| Vtot = Velec + VvdW + VBorn | (1) |
As shown in Fig. 5a, for Au and Pt NPs suspended in a hexane–ethanol mixture, the total potential energy minimum appears at ∼0.23 nm (see the ESI for the detailed calculation process†). This value is consistent with the measured distance of ∼0.30 nm between the Au core and Pt satellite (Fig. 5b). In contrast, the two building blocks maintain their colloidal state when hexane is absent due to the very shallow potential energy well.
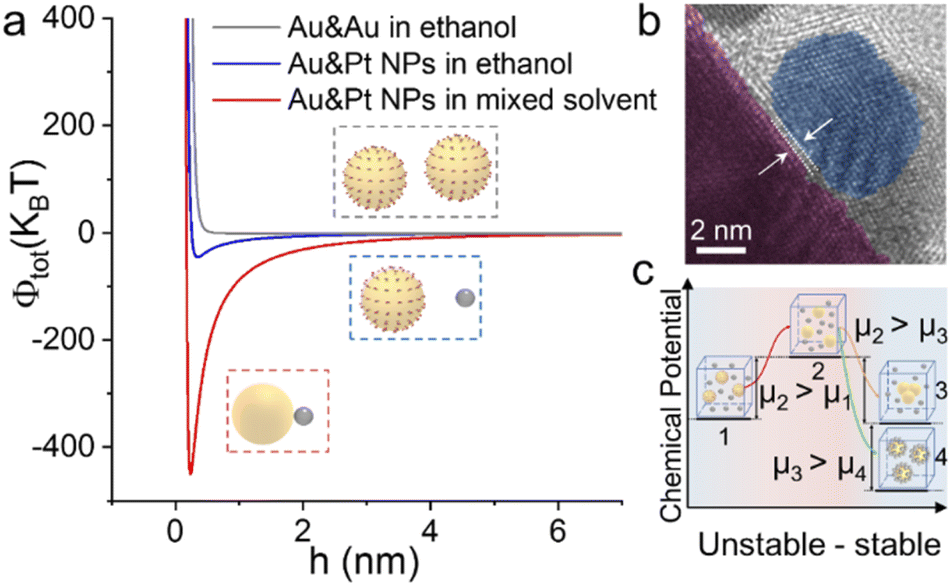 | ||
| Fig. 5 (a) Total colloidal potential energy of Au + Pt NPs suspended in ethanol (blue) and an ethanol–hexane mixture (red) as a function of interparticle distance. The gray curve is the energy of the Au colloid before assembly. (b) HRTEM image of the Aucore@Ptsatellite superstructure showing the distance between the Au core and Pt satellites. (c) Illustration of the chemical potential change of the colloidal mixture from the original steady state to the final steady state after assembly. | ||
Another important issue is the selective collision between the hetero-building blocks. In a binary colloid comprising two building blocks (A and B), there are 3 different collision directions, i.e. A–A, B–B and A–B collisions, among which only the third favors hetero-assembly. For example, in a mixed colloid of 80 nm Au and 10 nm Pt spheres, ∼70 completely inelastic collisions between Au and Pt result in the formation of a core–satellite superstructure (Fig. S4†) and the kinetic energy loss of the whole system is converted to heat. We can estimate the Gibbs free energy change according to the equation ΔG = ΔH — TΔS, in which ΔH and ΔS are enthalpy and entropy changes, respectively. The formation of a core–satellite superstructure (A–B) results in a lower Gibbs free energy than that in the formation of A–A or B–B clusters, indicating that the ligand-stripped Au core NPs intend to adsorb Pt NPs rather than self-aggregate (see the ESI for the detailed calculation process†). In view of thermodynamics, the unstable Au NPs after hexane treatment increased the chemical potential of the system from μ1 to μ2 (Fig. 5c). After this, the colloidal mixture reached a new steady state via self-organization of NPs. The selective collision of hetero-building blocks to form a superstructure is favored because of the lower chemical potential.
Finally, as a proof of concept, we showed the specific properties of superstructures based on assembly. Surface-enhanced Raman spectroscopy (SERS) is a sensitive analytical technique in the detection of trace chemical species. Normally, an SERS signal can only be detected from highly localized areas such as edges, tips of nanostructures and gaps between NPs. As shown in Fig. 6a, the Au@Au superstructure exhibits single-particle SERS sensitivity due to the plasmonic coupling between building blocks. In contrast, the corresponding 80 nm Au core does not enhance the Raman signal of reporter molecules under otherwise same conditions (Fig. S31†). Based on the sensitivity test, the detection limit of 4-nitrothiophenol and dye molecules (crystal violet) on the Au@Au superstructure can be acheived at 10−7 M (Fig. S32†). We further used Au@Pt superstructures with SERS activity to detect trace adsorbed H species in Pt-catalyzed hydrogen evolution reactions. The Pt–H band at around ∼2060 cm−1 red-shifts with the decreasing potential (Fig. S33†), showing the exciting potential of studying electrocatalysis using core–satellite superstructures. In another example, we demonstrated enhanced photocatalytic activity enabled by assembly of semiconductor@metal (CdS@Au) superstructures. Due to the efficient charge transfer at the metal–semiconductor interfaces, the superstructures exhibit enhanced photocatalytic performance with high stability in benzyl alcohol oxidation compared with the un-assembled building blocks (Fig. 6b and S34†).
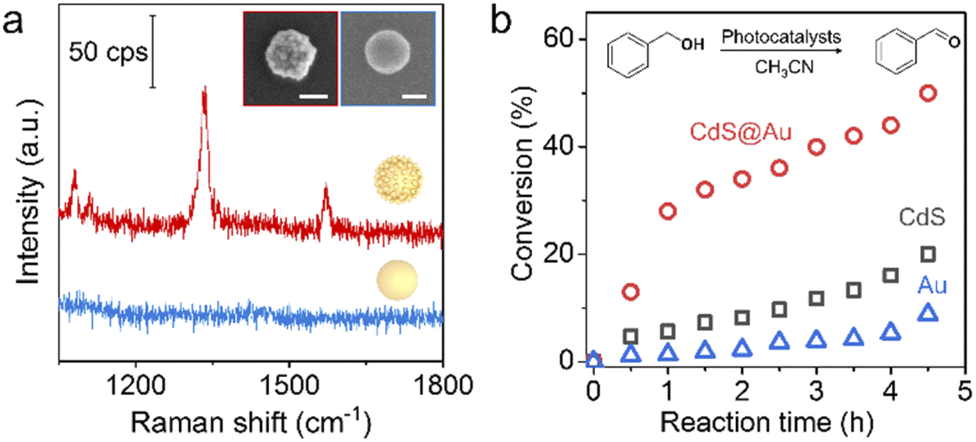 | ||
| Fig. 6 (a) SERS spectra of 4-nitrothiophenol measured on a single Au@Au core–satellite superstructure (red) and the corresponding Au core (blue). (Inset) SEM images of a single superstructure (left) and a single Au core (right). Scale bar, 50 nm. (b) Conversion rate of benzyl alcohol in a photo-oxidation reaction catalyzed by Au NPs, CdS nanowires and CdS@Au superstructures, respectively. | ||
Conclusions
To summarize, assembly of complex core–satellite superstructures is now a very simple task under our vdW force-driven assembly methodology. By weakening the repulsion between building blocks via detaching the ligand from their surfaces or neutralizing their surface charges, we trigger self-organization of small satellites on the surface of a large core. This assembly process does not involve additional molecular linkers that will be retained in the assembled superstructures. The agents used to destabilize the building blocks are nonpolar solvents or soluble inorganic salts and they can be completely removed after simple washing steps. Thus, the original nature of building blocks can be preserved after assembly. Using this facile and versatile approach, we obtained a library of distinct core–satellite superstructures. The assembly scope can be readily extended to a tremendous number of building blocks ranging from inorganic NPs to living cells, which offers great potential for application in fields such as catalysis, sensing, bioengineering, drug delivery, and others.Data availability
All data are provided in the ESI† and additional data can be available upon request.Author contributions
Wei Xie directed all aspects of the project. Wei Xie, Yanfang Hu, and Yonglong Li conceived the project. Yanfang Hu performed the superstructure assembly. Yanfang Hu, Yonglong Li, Linfeng Yu, and Yuying Zhang performed the characterization of the superstructures. Wei Zhang performed the characterization of ligands on the Au nanoparticle surface. Yanfang Hu and Yuming Lai performed the AFM measurement. Wei Xie and Yanfang Hu wrote the manuscript with input from all co-authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant 22022406), the Natural Science Foundation of Tianjin (grants 20JCJQJC00110 and 20JCYBJC00590), the Fundamental Research Funds for the Central Universities-Nankai University (000082), the 111 project (grant B12015), and the National Key R&D Program (grant 2021YFB4000600).Notes and references
- T. Udayabhaskararao, T. Altantzis, L. Houben, M. Coronado-Puchau, J. Langer, R. Popovitz-Biro, L. M. Liz-Marzán, L. Vuković, P. Král, S. Bals and R. Klajn, Science, 2017, 358, 514–518 CrossRef CAS PubMed.
- A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS PubMed.
- N. Thi, M. Fayna and A. Souad, Nanomaterials, 2018, 8, 149 CrossRef PubMed.
- M. Grzelczak, L. M. Liz-Marzán and R. Klajn, Chem. Soc. Rev., 2019, 48, 1342–1361 RSC.
- C. Yi, H. Liu, S. Zhang, Y. Yang, Y. Zhang, Z. Lu, E. Kumacheva and Z. Nie, Science, 2020, 369, 1369–1374 CrossRef CAS PubMed.
- J. Jia, G. Liu, W. Xu, X. Tian, S. Li, F. Han, Y. Feng, X. Dong and H. Chen, Angew. Chem., Int. Ed., 2020, 59, 14443–14448 CrossRef CAS PubMed.
- R. P. M. Höller, M. Dulle, S. Thomä, M. Mayer, A. M. Steiner, S. Förster, A. Fery, C. Kuttner and M. Chanana, ACS Nano, 2016, 10, 5740–5750 CrossRef PubMed.
- F. Han, S. R. C. Vivekchand, A. H. Soeriyadi, Y. Zheng and J. J. Gooding, Nanoscale, 2018, 10, 4284–4290 RSC.
- Y. Zhao, L. Du, H. Li, W. Xie and J. Chen, J. Phys. Chem. Lett., 2019, 10, 1286–1291 CrossRef CAS PubMed.
- K. Zhang, L. Yang, Y. Hu, C. Fan, Y. Zhao, L. Bai, Y. Li, F. Shi, J. Liu and W. Xie, Angew. Chem., Int. Ed., 2020, 59, 18003–18009 CrossRef CAS.
- K. Sokołowski, J. Huang, T. Földes, J. A. McCune, D. D. Xu, B. de Nijs, R. Chikkaraddy, S. M. Collins, E. Rosta, J. J. Baumberg and O. A. Scherman, Nat. Nanotechnol., 2021, 16, 1121–1129 CrossRef PubMed.
- Y. Chen, Y. Hu, J. Zhao, Y. Deng, Z. Wang, X. Cheng, D. Lei, Y. Deng and H. Duan, Adv. Funct. Mater., 2020, 30, 2000642 CrossRef CAS.
- E. A. Kwizera, E. Chaffin, X. Shen, J. Chen, Q. Zou, Z. Wu, Z. Gai, S. Bhana, R. O'Connor, L. Wang, H. Adhikari, S. R. Mishra, Y. Wang and X. Huang, J. Phys. Chem. C, 2016, 120, 10530–10546 CrossRef CAS.
- S. N. Sheikholeslami, H. Alaeian, A. L. Koh and J. A. Dionne, Nano Lett., 2013, 13, 4137–4141 CrossRef CAS PubMed.
- J. Fu, W. Zhu, Y. Chen, Z. Yin, Y. Li, J. Liu, H. Zhang, J.-J. Zhu and S. Sun, Angew. Chem., Int. Ed., 2019, 58, 14100–14103 CrossRef CAS.
- Y. Li, Y. Hu, F. Shi, H. Li, W. Xie and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 9049–9053 CrossRef CAS.
- C. Liu, C. Chen, S. Li, H. Dong, W. Dai, T. Xu, Y. Liu, F. Yang and X. Zhang, Anal. Chem., 2018, 90, 10591–10599 CrossRef CAS PubMed.
- H. S. Choi, Nat. Nanotechnol., 2014, 9, 93–94 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Zagorovsky and W. C. W. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- Z. Zhao, D. C. Pan, Q. M. Qi, J. Kim, N. Kapate, T. Sun, C. W. Shields IV, L. L.-W. Wang, D. Wu, C. J. Kwon, W. He, J. Guo and S. Mitragotri, Adv. Mater., 2020, 32, 2003492 CrossRef CAS PubMed.
- A. Huefner, W.-L. Kuan, K. H. Müller, J. N. Skepper, R. A. Barker and S. Mahajan, ACS Nano, 2016, 10, 307–316 CrossRef CAS PubMed.
- S. Lee, M. Kim and S. Yoon, Chem. Commun., 2019, 55, 1466–1469 RSC.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS PubMed.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645–2651 CrossRef CAS PubMed.
- S. Mühlig, A. Cunningham, S. Scheeler, C. Pacholski, T. Bürgi, C. Rockstuhl and F. Lederer, ACS Nano, 2011, 5, 6586–6592 CrossRef.
- N. Li, Y. Shang, Z. Han, T. Wang, Z. G. Wang and B. Ding, ACS Appl. Mater. Interfaces, 2019, 11, 13835–13852 CrossRef CAS PubMed.
- S. J. Tan, M. J. Campolongo, D. Luo and W. Cheng, Nat. Nanotechnol., 2011, 6, 268–276 CrossRef CAS PubMed.
- L.-A. Wu, W.-E. Li, D.-Z. Lin and Y.-F. Chen, Sci. Rep., 2017, 7, 13066 CrossRef PubMed.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
- X. Xu, N. L. Rosi, Y. Wang, F. Huo and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 9286–9287 CrossRef CAS.
- J. Guo, B. L. Tardy, A. J. Christofferson, Y. Dai, J. J. Richardson, W. Zhu, M. Hu, Y. Ju, J. Cui, R. R. Dagastine, I. Yarovsky and F. Caruso, Nat. Nanotechnol., 2016, 11, 1105–1111 CrossRef CAS PubMed.
- J. Guo, M. Suástegui, K. K. Sakimoto, V. M. Moody, G. Xiao, D. G. Nocera and N. S. Joshi, Science, 2018, 362, 813 CrossRef CAS PubMed.
- A. S. De Silva Indrasekara, S. J. Norton, N. K. Geitner, B. M. Crawford, M. R. Wiesner and T. Vo-Dinh, Langmuir, 2018, 34, 14617–14623 CrossRef CAS PubMed.
- H. Zhang, C. Wang, H.-L. Sun, G. Fu, S. Chen, Y.-J. Zhang, B.-H. Chen, J. R. Anema, Z.-L. Yang, J.-F. Li and Z.-Q. Tian, Nat. Commun., 2017, 8, 15447 CrossRef CAS PubMed.
- H. Zhang, X. G. Zhang, J. Wei, C. Wang, S. Chen, H. L. Sun, Y. H. Wang, B. H. Chen, Z. L. Yang, D. Y. Wu, J. F. Li and Z. Q. Tian, J. Am. Chem. Soc., 2017, 139, 10339–10346 CrossRef CAS PubMed.
- D. A. Walker, E. K. Leitsch, R. J. Nap, I. Szleifer and B. A. Grzybowski, Nat. Nanotechnol., 2013, 8, 676–681 CrossRef CAS PubMed.
- C. J. Kiely, J. Fink, M. Brust, D. Bethell and D. J. Schiffrin, Nature, 1998, 396, 444–446 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- D. V. Talapin, E. V. Shevchenko, M. I. Bodnarchuk, X. Ye, J. Chen and C. B. Murray, Nature, 2009, 461, 964–967 CrossRef CAS PubMed.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef.
- Z. Wang, Y. Zou, Y. Li and Y. Cheng, Small, 2020, 16, 1907042 CrossRef CAS.
- G. I. Guerrero-García, P. González-Mozuelos and M. Olvera de la Cruz, ACS Nano, 2013, 7, 9714–9723 CrossRef.
- Z. Zhang, S. Maji, A. B. d. F. Antunes, R. De Rycke, Q. Zhang, R. Hoogenboom and B. G. De Geest, Chem. Mater., 2013, 25, 4297–4303 CrossRef CAS.
- Z. Lee, K.-J. Jeon, A. Dato, R. Erni, T. J. Richardson, M. Frenklach and V. Radmilovic, Nano Lett., 2009, 9, 3365–3369 CrossRef CAS PubMed.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS PubMed.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J.-M. Basset, Nat. Chem., 2017, 9, 890–895 CrossRef CAS PubMed.
- B. Li, J. Yin, X. Liu, H. Wu, J. Li, X. Li and W. Guo, Nat. Nanotechnol., 2019, 14, 567–572 CrossRef CAS PubMed.
- T. Hueckel, G. M. Hocky, J. Palacci and S. Sacanna, Nature, 2020, 580, 487–490 CrossRef CAS PubMed.
- Y. Liu, X. Han, L. He and Y. Yin, Angew. Chem., Int. Ed., 2012, 51, 6373–6377 CrossRef CAS.
- J.-A. Park and S.-B. Kim, J. Contam. Hydrol., 2015, 181, 131–140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental methods and figures. See https://doi.org/10.1039/d2sc02843c |
| This journal is © The Royal Society of Chemistry 2022 |