
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplications of the unfolded state in the folding energetics of heterogeneous-backbone protein mimetics†
Jacqueline R.
Santhouse‡
,
Jeremy M. G.
Leung‡
 ,
Lillian T.
Chong
* and
W. Seth
Horne
*
,
Lillian T.
Chong
* and
W. Seth
Horne
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, PA 15211, USA. E-mail: ltchong@pitt.edu; horne@pitt.edu
First published on 20th September 2022
Abstract
Sequence-encoded folding is the foundation of protein structure and is also possible in synthetic chains of artificial chemical composition. In natural proteins, the characteristics of the unfolded state are as important as those of the folded state in determining folding energetics. While much is known about folded structures adopted by artificial protein-like chains, corresponding information about the unfolded states of these molecules is lacking. Here, we report the consequences of altered backbone composition on the structure, stability, and dynamics of the folded and unfolded states of a compact helix-rich protein. Characterization through a combination of biophysical experiments and atomistic simulation reveals effects of backbone modification that depend on both the type of artificial monomers employed and where they are applied in sequence. In general, introducing artificial connectivity in a way that reinforces characteristics of the unfolded state ensemble of the prototype natural protein minimizes the impact of chemical changes on folded stability. These findings have implications in the design of protein mimetics and provide an atomically detailed picture of the unfolded state of a natural protein and artificial analogues under non-denaturing conditions.
Introduction
The functional diversity of proteins is founded on the many architectures that can result from folding of polypeptide chains built from different combinations of the 20 canonical α-amino acids. The capacity for sequence-encoded folding is not unique to biomacromolecules, and a variety of artificial amide-based oligomers have been shown to fold into well-defined conformations.1 Historically, most studies on such entities have involved formation of secondary structure;2–5 however, recent examples show the capacity for more complex tertiary folding patterns.6 Compared to a wealth of information on folded structures possible in protein-like backbones, much less is known about the folding thermodynamics of chains artificial in chemical composition.7–10 Understanding the detailed energetic consequences of non-native backbone composition on the folding process has the potential to inform the design of more effective protein mimetics as well as yield insights into natural protein behavior.Prior work has demonstrated the value of backbone modification for elucidating fundamental aspects of protein folding and function. Substitution of a backbone amide with an ester has been applied to probe the energetic contributions of hydrogen bonding,11–13 and backbone N-methylation used to disrupt protein dimerization or aggregation.14,15 Backbone modification has also been employed to investigate influences of altered conformational freedom on folding, examining the role of turn nucleation, helix nucleation, as well as loop dynamics in different systems.16–21 Motivated by a desire to develop artificial mimetics of protein tertiary structure, we and others have explored folded structure and stability in extensively modified artificial protein-like molecules that display biological side-chain sequences.6 These efforts have shown a variety of oligomers that blend natural and artificial amino acid monomers, so-called heterogeneous backbones, can faithfully manifest the tertiary fold of a prototype protein sequence; however, backbone modification can impact folded stability in ways that are counterintuitive. As an example, comparison of variants of the B1 domain of streptococcal protein G with different backbone compositions in the α-helix showed modifications that enhance conformational freedom had a favorable effect on folding entropy, while those that increase rigidity were entropically unfavorable.7,8
To understand folding energetics in artificial protein-like chains, a vital issue to address is the impact of altered backbone composition on the unfolded state. In defining energetics associated with the folding equilibrium of a natural protein, the characteristics of the ensemble that defines the unfolded state are as important as those of the folded state.22 Furthermore, experiments conducted under strongly denaturing conditions have revealed substantial presence of residual secondary structure and sometimes tertiary structure in the unfolded state.23,24 Compared to a wealth of data on folded structures in artificial protein-like backbones, corresponding information about the unfolded states of these molecules is unknown. Given that many artificial monomers used to construct protein mimetics have fundamentally different conformational preferences than L-α-residues, it is a reasonable hypothesis that changes to backbone connectivity may have profound effects on the unfolded state. Elucidating these effects is crucial to fully understand the molecular origins of the changes to folding energetics that result from altered backbone composition. Molecular dynamics (MD) simulation has proved a powerful tool uniquely equipped to provide information about structure and dynamics of protein unfolded states in atomic detail and high temporal resolution.25–29
Here, we report a systematic examination of the consequences of altered backbone composition on structure, stability, and dynamics of the folded and unfolded states of a small helix-rich bacterial protein through a combination of experimental biophysical analysis and atomistic MD simulation. Two different artificial monomer types with opposite anticipated effects on local backbone conformational freedom are incorporated in different sequence contexts of a common host protein to produce a series of heterogeneous-backbone variants. Comparison of the folding behavior of these variants to the native protein shows effects on the unfolded state and folding energetics that depend on both the chemical composition of the backbone as well as the context for incorporation of artificial monomers. Our simulations, which employ the weighted ensemble enhanced sampling strategy,30,31 sample both unfolding events and the resulting unfolded state of a protein under native (i.e., non-denaturing) conditions with atomic detail—in contrast to prior conventional simulations of unfolding events at elevated temperatures29,32 or collapsing of fully extended chains to unfolded conformations at room temperature.26,27 Collectively, the results reported here provide fundamental insights into the behavior of natural proteins in the unfolded state as well as new considerations to guide the design of protein tertiary structure mimetics based on artificial backbones.
Results
System design and preparation of heterogeneous-backbone variants
In selecting a host protein to explore the impacts of altered backbone composition on folding, we sought the following characteristics: (1) a compact well-defined tertiary structure with multiple α-helices, (2) a folded stability that could withstand some degree of destabilization from backbone alteration, (3) a chain length accessible by total chemical synthesis, and (4) a two-state folding mechanism amenable to analysis experimentally and by atomistic MD simulations. Guided by the above considerations, we chose the B domain of protein A from Staphylococcal bacteria (BdpA) as the basis for this work. With 58-residues in its sequence, BdpA adopts an all-α tertiary fold consisting of an antiparallel three-helix bundle (Fig. 1A).33 The relatively fast, μs-timescale folding process of BdpA has been extensively studied both by experiment and simulation.29,34–42 While its three α-helices are structurally similar, they play different roles, with helix 2 thought to act as a nucleus for folding.39,41 Having multiple helices in a single compact domain provides a means to compare effects of the same backbone modification type made in different local contexts of the same protein. | ||
| Fig. 1 (A) Sequence, secondary structure map, and NMR structure (PDB 1BDC) of the B domain of Staphylococcal protein A (BdpA) alongside sequences of BdpA variants synthesized and characterized in the present work. “R” groups in β3-residues match that of the α-residue specified by the corresponding single letter code. (B) NMR structures of BdpA variants. Ensembles (10 models per protein) determined by simulated annealing with NMR-derived restraints are shown in cartoon representation for WT, β3-H2, Aib-H2, β3-H3, and Aib-H3. Artificial residues are marked with spheres colored according to the scheme in panel A. A disordered N-terminal segment consisting of residues 1–4 is omitted. | ||
A previously reported G29A point mutant of BdpA served as the prototype sequence for backbone modification.43 The structures of BdpA and this mutant are similar;44,45 however, the alanine substitution leads to three-fold faster folding kinetics along with a marginal increase in thermodynamic folded stability.37,38 We designed and prepared five proteins based on BdpAG29A: the native backbone (WT) and four heterogeneous-backbone variants in which artificial monomer type and sequence placement were systematically varied (Fig. 1A). We examined two types of artificial residues in the variants: β3-residues and the Cα-Me-α-residue aminoisobutyric acid (Aib). Both Aib and β3-residues are compatible with α-helical folds in a range of complex, protein-like tertiary architectures.6 Relative to a canonical L-α-residue, Aib has restricted backbone conformational freedom due to the geminal disubstitution on Cα. The helical propensity of Aib is comparable to that of alanine, attributed to competing factors of backbone rigidification and the unfavorable possibility of adopting a left-handed helical conformation due to its achiral nature.46 By contrast, β3-residues have enhanced conformational freedom due to the addition of a third backbone rotatable bond and are typically destabilizing to tertiary folds.10,47 In contrast to Aib, α → β3 substitution retains the side chain of the replaced α-residue.
We selected four sites in BdpA helix 2 and four sites in helix 3 to incorporate the above modifications. Artificial residues were kept distal from the hydrophobic core of the domain in all cases to minimize impact of the loss of a side chain (in the case of substitution with Aib) or subtle change to side chain orientation (in the case of substitution with β3-residues) relative to the native backbone. Each heterogeneous-backbone BdpA variant is named based on the type of artificial residue it contains and where the protein is modified (e.g., β3-H2 contains β3-residues in helix 2). Proteins were prepared by total chemical synthesis using Fmoc solid-phase methods and purified by preparative HPLC. Purity and identity of final products was confirmed by analytical HPLC and mass spectrometry (Fig. S1–S5†) before subsequent experimental analysis detailed below.
Experimental characterization of folded structure and folding thermodynamics
With BdpA and heterogeneous-backbone variants in hand, we assessed the impact of backbone alteration on tertiary folded structure using NMR spectroscopy. As global isotopic labeling is cost prohibitive in synthetic proteins of this size, our analysis exclusively relied on water-suppressed homonuclear experiments. After completing a 1H resonance assignment for each protein, a high-resolution folded structure was determined using simulated annealing with NMR-derived restraints (Fig. 1B, Tables S1–S5†). The ensemble obtained for the native-backbone protein WT is in good agreement with an NMR structure previously reported for this sequence (Fig. S6†), confirming the methods utilized here produce similar results as those involving additional heteronuclear measurements.44 Structures obtained for the four heterogeneous-backbone variants indicate the backbone alterations employed have minimal impact on the tertiary fold of the domain. The β3 and Aib residues are both well-accommodated in the local helical secondary structure (Fig. S7†), and the hydrophobic core packing is conserved across variants (Fig. S8†).Having established folded structures of the protein series, we assessed the influence of backbone alteration on the thermodynamics of the folding process. A circular dichroism (CD) spectrum of the native backbone WT featured minima at 208 nm and 220 nm consistent with its helix-rich fold (Fig. 2A), and a thermal melt showed a cooperative transition with a melting temperature (Tm) of 78.5 °C (Fig. 2B, Table 1). CD results for the heterogeneous-backbone variants revealed subtle changes in spectral features and thermal stability as a function of both artificial residue type and the context for its incorporation. The shape of the CD signatures for variants containing Aib are similar to WT, while the intensity at 220 nm is attenuated and the band ∼208 nm slightly blue shifted in the variants containing β3-residues; the latter finding is in accordance with prior CD analyses of other heterogeneous α/β-peptide backbones in a helical conformation.47,48 Variants in which the backbone of helix 2 is modified show reduced CD intensity relative to the corresponding variants where helix 3 is altered. A cooperative thermal unfolding transition is retained in all variants, with the Tm values of variants containing Aib close to WT and those with β3-residues ∼30 °C lower. Following trends seen in CD intensity, thermal stabilities of variants where the backbone of helix 2 is modified are consistently lower than the corresponding protein in which helix 3 is altered by the same residue type.
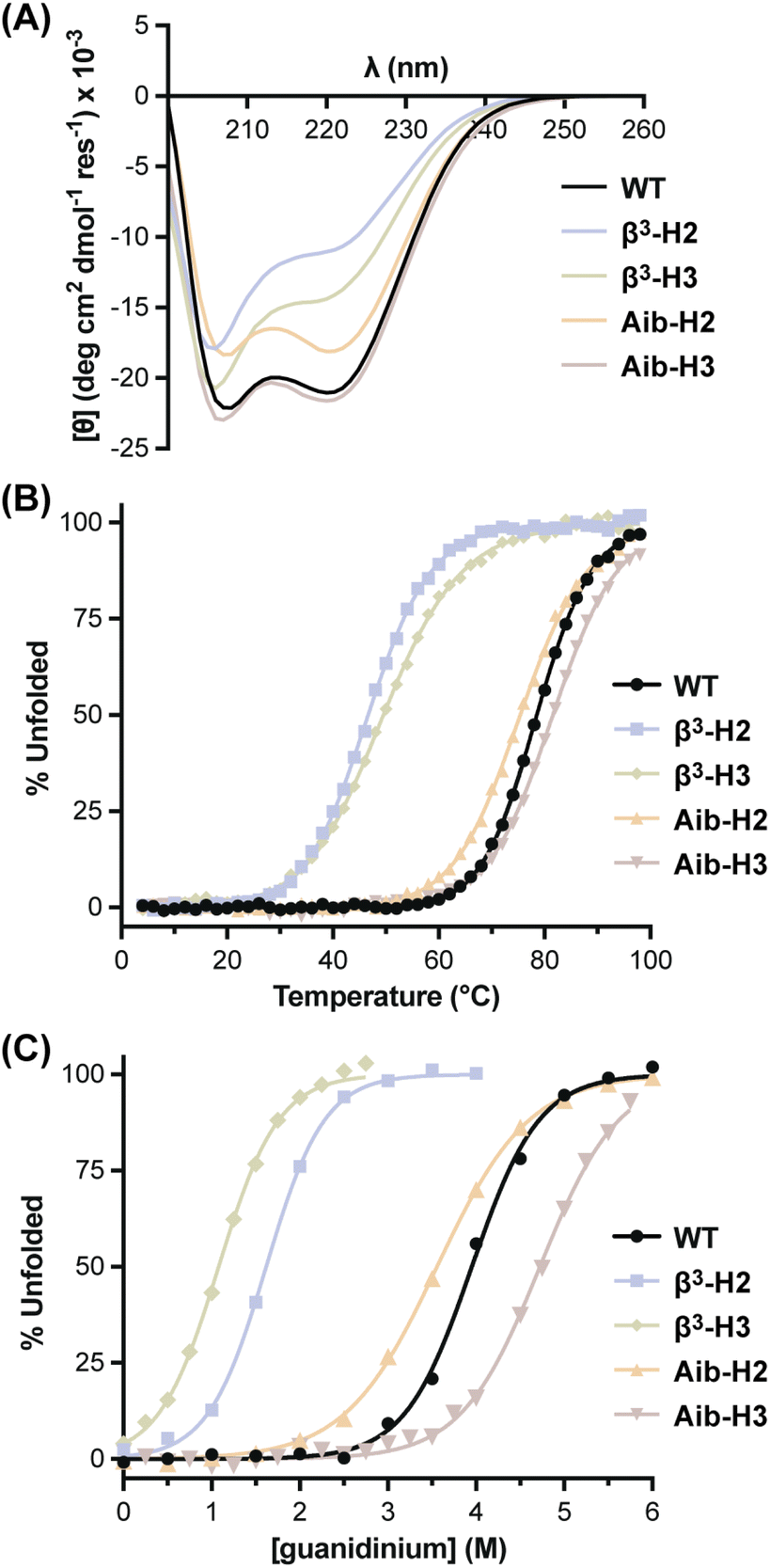 | ||
| Fig. 2 Circular dichroism (CD) analysis of the unfolding transition for BdpA variants. (A) CD scans at 20 °C. (B) Thermal melts monitored at 220 nm. (C) Chemical denaturation profiles at 4 °C. All measurements carried out on samples consisting of 50 μM protein in 10 mM phosphate buffer, pH 7. | ||
| T m (°C) | D 1/2 (M) | ΔG°d (kcal mol−1) | ΔH°d (kcal mol−1) | ΔS°d (cal. mol−1 K−1) | m gnd (kcal mol−1 M−1) | ΔCp (kcal mol−1 K−1) | |
|---|---|---|---|---|---|---|---|
| a Values obtained from fits of the unfolding transition monitored by CD as a function of temperature and concentration of guanidinium chloride denaturant in 10 mM phosphate at pH 7. Reported uncertainties are the standard error for the indicated parameter from the fit. b Midpoint of the thermal unfolding transition at 0 M denaturant. c Midpoint of the chemical denaturation transition at 4 °C. d At 25 °C. | |||||||
| WT | 78.5 ± 0.3 | 3.95 ± 0.02 | 5.2 ± 0.3 | 21.3 ± 0.3 | 54.1 ± 0.7 | 1.49 ± 0.02 | 0.57 ± 0.01 |
| β3-H2 | 46.7 ± 0.1 | 1.62 ± 0.01 | 1.9 ± 0.4 | 20.6 ± 0.3 | 63 ± 1 | 1.67 ± 0.02 | 0.62 ± 0.02 |
| Aib-H2 | 75.4 ± 0.1 | 3.56 ± 0.01 | 3.4 ± 0.2 | 14.1 ± 0.1 | 35.9 ± 0.4 | 1.08 ± 0.01 | 0.51 ± 0.01 |
| β3-H3 | 49.5 ± 0.1 | 1.08 ± 0.01 | 1.1 ± 0.4 | 12.5 ± 0.3 | 38.2 ± 0.8 | 1.52 ± 0.03 | 0.43 ± 0.02 |
| Aib-H3 | 81.6 ± 0.1 | 4.72 ± 0.01 | 5.1 ± 0.3 | 20.8 ± 0.2 | 52.6 ± 0.5 | 1.22 ± 0.01 | 0.51 ± 0.01 |
To gain insights into the thermodynamic basis for observed thermal stability differences seen as a function of altered backbone composition, we subjected each protein to chemical denaturation with guanidinium chloride (Fig. 2C) and monitored the unfolding transition as a function of temperature in parallel samples with differing concentrations of denaturant (Fig. S9†).49,50 A global fit of the resulting combined thermal/chemical denaturation data sets to a two-state folding model yielded the thermodynamic parameters for the folding equilibrium of each BdpA variant (Table 1). The unfolding free energy observed for WT at 25 °C (5.2 ± 0.2 kcal mol−1) agrees well with that reported previously for the closely related sequence BdpAG29A,F13W by the same method (5.1 ± 0.5 kcal mol−1).37
The tertiary fold of native-backbone WT was the most thermodynamically stable among the proteins examined. Comparison of folding free energy, entropy, and enthalpy for each heterogeneous-backbone variant relative to WT illustrates effects on folding energetics that depend on both the type of artificial residues present as well as where they are located. Variants containing Aib were closest to WT in overall stability, with Aib-H3 identical within uncertainty and Aib-H2 only modestly destabilized. Enthalpy and entropy parameters for Aib-H3 are also close to those of WT, while the small change to folding free energy in the case of Aib-H2 conceals large compensating unfavorable enthalpic and favorable entropic components. Context dependent effects of β3-residue incorporation on folding free energy showed the opposite trend as Aib, with incorporation of β3-residues in helix 2 more destabilizing than the same substitutions in helix 3. The destabilization for β3-H2 relative to WT is primarily entropic, while that for β3-H3 is entirely enthalpic and compensated for by a favorable entropic effect on the folding process. In general, Aib-containing variants were less susceptible to chaotropic agents than the native backbone, reflected by lower linear dependence of ΔG° on the concentration of guanidinium (m). By contrast, m values for β3-residue containing variants were somewhat elevated relative to WT, particularly for β3-H2. These differences suggest altered solvation effects associated with the folding process,7 a hypothesis explored further in the simulations discussed below.
Characterization of folded state and unfolded state ensembles by simulation
To examine the effects of altered backbone composition on the structure and dynamics of the folded and unfolded states of BdpA, we carried out atomistic MD simulations to generate conformational ensembles of both states for WT and each variant. We employed the weighted ensemble (WE) approach (as implemented in WESTPA57) to extensively sample both states under native conditions. The starting point for each simulation was the folded structure of the corresponding protein determined by NMR above, parameterized using the AMBER ff15ipq-m force field recently developed and validated for application to heterogeneous-backbone protein mimetics.51 Simulations were run in two stages and reweighted using a history-augmented Markov State model (haMSM) analysis procedure52 to estimate state populations under equilibrium conditions. In the first stage, simulations were run at 25 °C to sample the folded state as well as unfolding transitions to generate representative unfolded conformations. In the second stage, simulations were initiated from the unfolded conformations to extensively sample the unfolded state.The folded and unfolded state ensembles of the native-backbone protein WT can be defined as well-separated, highly populated regions in a two-dimensional probability distribution (Fig. 3A) as a function of fraction of native contacts and radius of gyration (Rg); the same is the case for the heterogeneous-backbone variants (Fig. S10, Table S6†). On average, the folded state ensembles among the series exhibit 80–95% native contacts (i.e., contacts present in a reference model from the NMR structure) with 52–67% native contacts between the helices, while the corresponding unfolded state ensembles exhibit 67–80% native contacts with only 2–13% native contacts between the helices (Fig. 3B, Table S7†). While only 52–67% of the interhelical native contacts are maintained in the folded states, the majority of native contacts in the hydrophobic cores (79–80%) are present in these states. Similar results were obtained for the WT folded state using conventional simulations with two other force fields and water models (Table S8†). The unfolded state ensembles exhibit larger most probable Rg values and a wider distribution relative to the corresponding folded state ensembles; however, the characteristics of the distributions vary considerably as a function of backbone composition (Fig. 3C).
 | ||
| Fig. 3 Probability distributions of various features that describe the unfolded state and folded state ensembles of BdpA variants obtained from simulation. (A) State definitions of folded and unfolded states of native-backbone protein WT based on two-dimensional probability distributions as a function of fraction of native contacts and radius of gyration (Rg). States are defined as regions with values of −ln(P) < 4, where P is the statistical weight (probability). (B–D) One-dimensional probability distributions for the folded state (top) and unfolded state (bottom) of each BdpA variant as a function of (B) fraction of native contacts, (C) Rg, and (D) all-atom solvent accessible surface area (SASA). | ||
The enhanced backbone conformational flexibility resulting from β3-residue substitution results in more expanded unfolded state ensembles relative to WT, indicated by a ∼15% increase in the most probable Rg value (12.7 Å for WTvs. 14.5 and 14.8 Å for β3-H2 and β3-H3, respectively). The impact of backbone rigidification from Aib substitution on the compactness of the unfolded state varies with the context for incorporation of the artificial residue. The Rg distribution for the unfolded state ensemble of Aib-H3 resembles that of WT, while that for Aib-H2 exhibits an additional peak (∼11.5 Å). Hierarchical clustering of the corresponding unfolded state ensemble using the Rg as a structure similarity metric reveals that this additional peak corresponds to a more compact misfolded sub-population (Fig. S11†). In contrast to differences seen in the unfolded states of the proteins, Rg probability distributions for the folded state ensembles are similar among WT and the heterogeneous-backbone variants (most probable values within 1 Å).
As experimental observations on the folding energetics of the BdpA variants suggested possible roles for altered solvent effects as a function of backbone composition, we mined the simulation results for insights related to this issue. We calculated probability distributions for the solvent accessible surface area (SASA) of the folded state ensemble and the unfolded state ensemble of each BdpA variant. In addition to all-atom SASA (Fig. 3D), SASA values were calculated using three atom subsets (Fig. S12†): hydrocarbons, backbone amides, and all amides. The SASA probability distributions reveal that the Aib variants have a lower total solvent accessible surface area than their β3 counterparts in both folded state and unfolded state ensembles. While the Aib variants also exhibit smaller all-atom SASA values than WT, the corresponding hydrocarbon SASA values and backbone amide SASA remain similar to WT. This suggests that apparent differences in solvation of the Aib variants compared to the native backbone primarily result from side chain functional groups removed by backbone alteration.
To gain atomic-level insights into the effects of altered backbone composition in BdpA on the conformational diversity of the unfolded state ensembles, we applied hierarchical clustering to the entire set of unfolded conformations obtained from simulation of WT and each variant using the pairwise “best-fit” Cα RMSD of the three helices. The conformational diversity of the unfolded state ensemble, as quantified by the number of clusters needed to describe it, follows the trend Aib variants < WT < β3 variants (Fig. S13†). This result shows that the altered backbone conformational freedom expected for each monomer type from first principles has a corresponding measurable impact on the structural diversity of the unfolded state—rigidification leads to a less disordered unfolded state ensemble and flexibility enhancement leads to a more disordered unfolded state ensemble.
To illustrate the diversity of the unfolded state ensemble for each BdpA variant, we selected one representative conformation from each of the clusters that collectively account for 85% of the unfolded state ensemble of that variant (Fig. 4A). Inspection of these conformations shows substantial residual helical content in the unfolded state of each protein—consistent with previous NMR studies, which revealed the preorganization of helices in the unfolded state as a determinant for the fast-folding kinetics of BdpA.53 A detailed helix population analysis of the entire ensemble bears this out; however, no clear correlation is seen between the extent or location of this residual structure with the position or type of residue substitution (Table S7†). Although the unfolded state ensembles exhibit a large extent of native secondary structure (i.e., relatively preformed helices), the extent of native tertiary structure is relatively low, ranging from 0–20% interhelical native contacts (Table S7†).
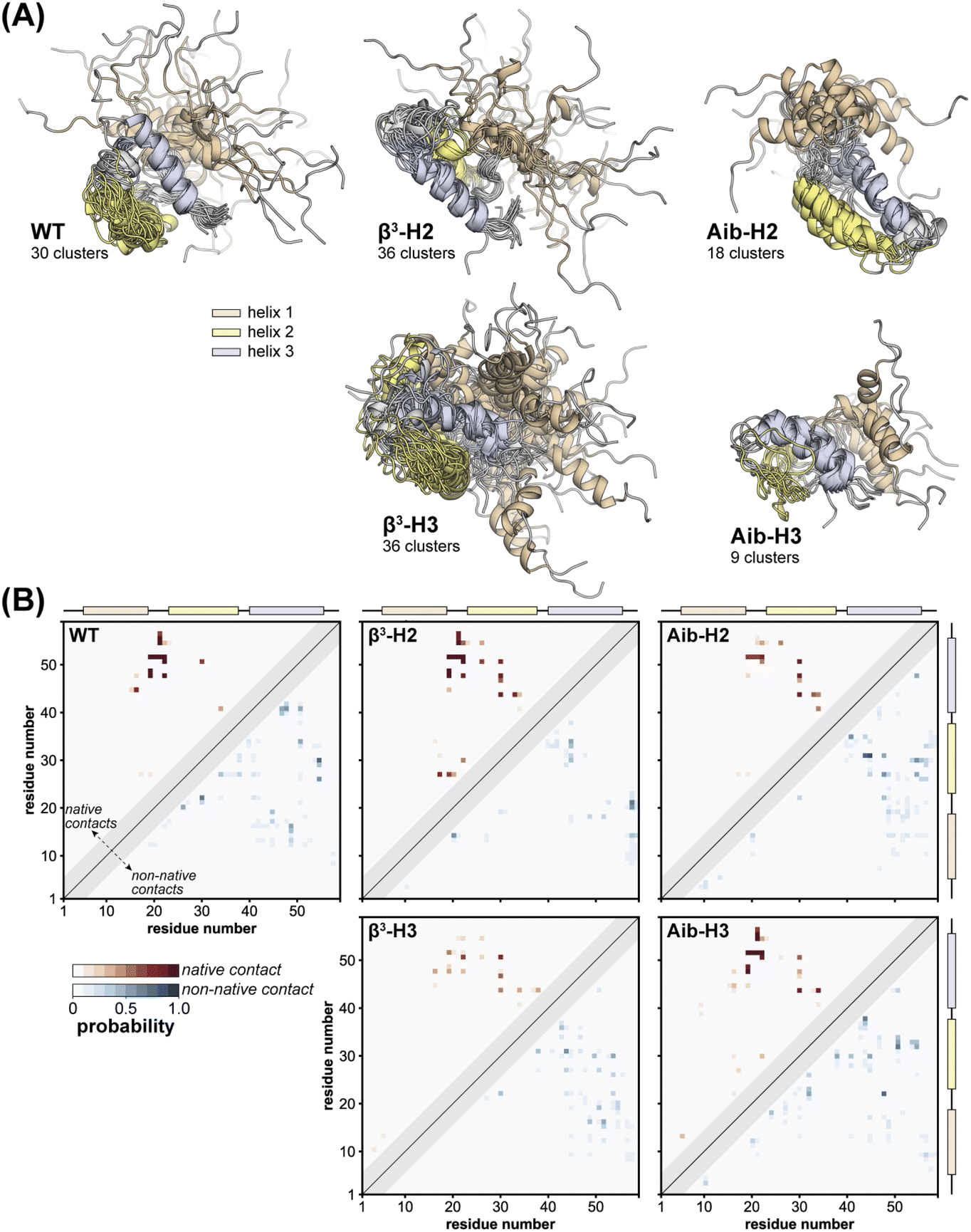 | ||
| Fig. 4 Characterization of unfolded state ensembles of BdpA variants obtained from simulation. (A) Representative structures that are closest to the average structure of each cluster obtained from agglomerative hierarchical clustering on pairwise “best-fit” Cα RMSD of the three helices. Clusters shown account for 85% of the unfolded state ensemble and all models are aligned on helix 3 of the respective structure of the most populated cluster. (B) Probability maps of residue-level tertiary contacts (|i − j| ≥ 6) for each unfolded state ensemble. The region above and left of the diagonal shows probabilities (red) for contacts present in the reference folded structure (i.e., “native”), while the region below and to the right of the diagonal shows probabilities (blue) for contacts absent in the reference structure (i.e., “non-native”). Residues are considered in contact when the residue pair contains heavy atoms within 5 Å. | ||
In an effort to obtain more quantitative insights into the structural similarities and differences among the unfolded states, we calculated a probability map for pairwise tertiary contacts (|i − j| ≥ 6) in each protein, categorizing each contact as “native” or “non-native” based on whether it was also present in the corresponding NMR reference structure of the folded state (Fig. 4B). All five proteins retain significant native tertiary contacts in the unfolded state ensemble involving residues in the vicinity of helix 3, with high probabilities surrounding the folded state hydrophobic core contacts between Leu22 and Leu51. Further, regardless of backbone composition, a substantial loss in tertiary native contacts is seen between helix 1 and helix 2, and between helix 2 and helix 3 (Fig. S14 and S15†). Some characteristics of the tertiary contacts present in the unfolded state vary in a systematic way with backbone composition. For example, in both WT and the Aib containing variants, we observe extensive medium-range non-native contacts (6 ≤ |i − j| ≤ 8) that are less probable in the unfolded state ensembles of the β3 variants, possibly due to the increased flexibility of the backbone. These contacts, along with non-native contacts in the vicinity of helix 3, are the most probable in the unfolded state ensembles.
Discussion
Despite the structural similarity of the heterogeneous-backbone BdpA variants in their folded states, our experiments reveal that the corresponding folding energetics varies considerably as a function of the type of artificial residue incorporated (Aib vs. β3) and position of the substitution (helix 2 vs. helix 3 of the domain). Aib substitution is better accommodated in helix 3 than helix 2, while the opposite is seen for β3-residue incorporation. Interpreting the molecular basis for effects of backbone alteration on folding energetics from folded structure alone is difficult; however, the picture becomes clearer when characteristics of the unfolded states, as probed by atomistic MD simulation, are considered. In some respects, Aib and β3-residues have effects on the unfolded state that follow from their fundamental chemical characteristics. Incorporation of Aib, which is more conformationally restricted than an L-α-residue, leads to an unfolded state ensemble that is more compact and less heterogeneous than the native backbone. In contrast, increasing backbone conformational freedom through incorporation of β3-residues leads to an unfolded state ensemble that is both more expansive and more heterogeneous.A comparative analysis of unfolded states also reveals insights into the context dependence of thermodynamic impacts of backbone alteration (Fig. 5). Our simulation results suggest that the majority of residual helical structure present in the unfolded state of native BdpA is found in helix 3. Backbone rigidification in this region (i.e., Aib-H3) leads to an unfolded state ensemble with increased helix 3 content as well as a pattern of long-range contacts that most closely resembles that of the natural backbone among the analogues examined. Similarities between the unfolded states of Aib-H3 and WT correlate with similarities in folding energetics observed by experiment, where Aib-H3 was found to be closest to the prototype natural protein in folding free energy. Helix 2 is less populated than helix 3 in the unfolded state of native BdpA. Bolstering rigidity in helix 2 through Aib incorporation (i.e., Aib-H2) leads to an unfolded state that less resembles that of the natural backbone, with greater secondary structure content in helix 2 and a lower probability of tertiary contacts between helix 2 and 3 in the unfolded state. Correspondingly, Aib incorporation in helix 2 is destabilizing to the fold and leads to a significant change in the balance of enthalpic/entropic contributions. Enhancing backbone conformational freedom in helix 3 of BpdA (i.e., β3-H3) leads to an unfolded state with decreased helicity in this region and altered long-range contacts relative to the natural backbone. These changes in unfolded state characteristics are accompanied by a significant destabilization of the fold. The same flexibility enhancing modifications made in helix 2 (β3-H2) exert a smaller thermodynamic penalty and lead to an unfolded state with a network of long-range contacts more like that seen for the natural backbone.
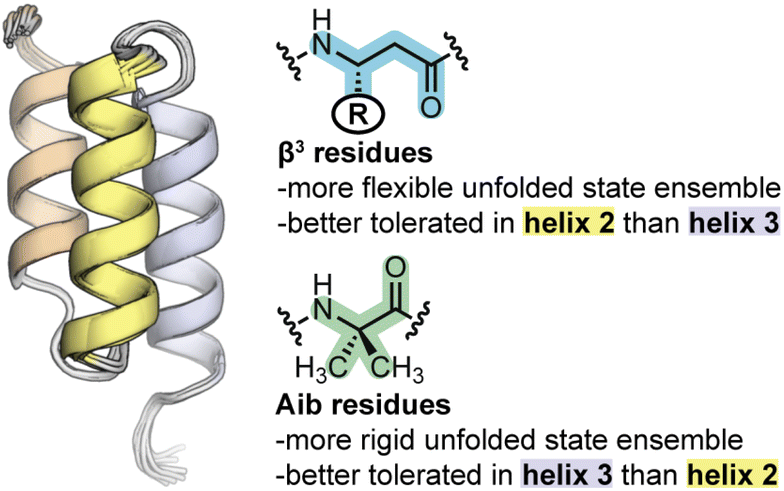 | ||
| Fig. 5 Summary of the effects of backbone alteration on the unfolded state ensemble and folded stability observed in the BdpA system. | ||
Conclusions
Collectively, the findings reported here suggest a previously unappreciated consideration important to the design of heterogeneous-backbone protein mimetics of high folded stability—backbone modification should be guided by the unfolded state of a prototype protein as well as its folded tertiary structure. Incorporation of artificial monomers in way that is compatible with structural features of the folded state of the prototype and reinforces underlying characteristics of the unfolded state ensemble minimizes the impact of altered backbone composition on folding energetics. Beyond implications related to protein mimetic design, our results provide new insights into natural protein behavior in the form of an atomically detailed picture of the unfolded state of the well-studied protein BdpA under non-denaturing conditions. In contrast to collapsing an extended chain, this unfolded state was obtained directly from unfolding the protein. Thus, it has value in being guaranteed to be on-path for the folding/unfolding process. Not normally possible using conventional simulations without biasing potentials or other changes to conditions (e.g., elevated temperature), we obtain room temperature unfolding events by employing the WE enhanced sampling strategy. While the power of the WE strategy for generating rare events and kinetic observables is now well established for diverse processes (e.g., protein folding,54 protein–protein binding,55 and the large-scale opening of the coronavirus spike protein56), the present results highlight its effectiveness to also provide enhanced conformational sampling of stable states of interest and their corresponding equilibrium observables. The unfolded state ensembles obtained here for BdpA and variants have potential future utility in studying the impacts of backbone alteration on folding kinetics and mechanism. Efforts to this end are ongoing in our laboratories.Data availability
Additional data supporting the findings of this study can be found in the ESI† for the article. Coordinates and associated experimental data supporting the NMR structures reported are deposited in the PDB (7TIO, 7TIP, 7TIQ, 7TIR, 7TIS) and BMRB (30980, 30981, 30982, 30983, 30984). Other data are available from the corresponding authors upon request.Author contributions
J. R. S., J. M. G. L., L. T. C. and W. S. H. designed research; J. R. S. and J. M. G. L. performed research; J. R. S., J. M. G. L., L. T. C. and W. S. H. analyzed data; J. R. S., J. M. G. L., L. T. C. and W. S. H. wrote the paper.Conflicts of interest
L. T. C. is a member of the Scientific Advisory Board of OpenEye Scientific, Cadence Molecular Sciences; and an Open Science Fellow with Psivant Therapeutics.Acknowledgements
Funding for this work was provided by the National Science Foundation (CHE-1807301 to L. T. C and W. S. H.). Computational resources were provided through NSF XSEDE Award No. MCB-100109 for the use of PSC's Bridges-258 and by the University of Pittsburgh Center for Research Computing for use of their shared cluster. We are also grateful for helpful discussions with Daniel Zuckerman and John Russo on the use of their haMSM plugin for the WESTPA 2.0 software package.References
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed.
- A. D. Bautista, C. J. Craig, E. A. Harker and A. Schepartz, Curr. Opin. Chem. Biol., 2007, 11, 685–692 CrossRef CAS PubMed.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed.
- Z. E. Reinert and W. S. Horne, Chem. Sci., 2014, 5, 3325–3330 RSC.
- N. A. Tavenor, Z. E. Reinert, G. A. Lengyel, B. D. Griffith and W. S. Horne, Chem. Commun., 2016, 52, 3789–3792 RSC.
- K. L. George and W. S. Horne, J. Am. Chem. Soc., 2017, 139, 7931–7938 CrossRef CAS PubMed.
- B. F. Fisher, S. H. Hong and S. H. Gellman, J. Am. Chem. Soc., 2018, 140, 9396–9399 CrossRef CAS.
- W. Lu, M. A. Qasim, M. Laskowski and S. B. H. Kent, Biochemistry, 1997, 36, 673–679 CrossRef CAS.
- E. Chapman, J. S. Thorson and P. G. Schultz, J. Am. Chem. Soc., 1997, 119, 7151–7152 CrossRef CAS.
- S. Deechongkit, H. Nguyen, E. T. Powers, P. E. Dawson, M. Gruebele and J. W. Kelly, Nature, 2004, 430, 101–105 CrossRef CAS PubMed.
- K. Rajarathnam, B. D. Sykes, C. M. Kay, B. Dewald, T. Geiser, M. Baggiolini and I. Clark-Lewis, Science, 1994, 264, 90–92 CrossRef CAS PubMed.
- A. G. Kreutzer and J. S. Nowick, Acc. Chem. Res., 2018, 51, 706–718 CrossRef CAS PubMed.
- F. I. Valiyaveetil, M. Sekedat, R. MacKinnon and T. W. Muir, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 17045–17049 CrossRef CAS PubMed.
- D. Bang, A. V. Gribenko, V. Tereshko, A. A. Kossiakoff, S. B. Kent and G. I. Makhatadze, Nat. Chem. Biol., 2006, 2, 139–143 CrossRef CAS.
- F. Liu, D. Du, A. A. Fuller, J. E. Davoren, P. Wipf, J. W. Kelly and M. Gruebele, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 2369–2374 CrossRef CAS PubMed.
- A. A. Fuller, D. Du, F. Liu, J. E. Davoren, G. Bhabha, G. Kroon, D. A. Case, H. J. Dyson, E. T. Powers, P. Wipf, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 11067–11072 CrossRef CAS PubMed.
- V. Y. Torbeev, H. Raghuraman, D. Hamelberg, M. Tonelli, W. M. Westler, E. Perozo and S. B. H. Kent, Proc. Natl. Acad. Sci. U.S.A., 2011, 108, 20982–20987 CrossRef CAS PubMed.
- V. Bauer, B. Schmidtgall, G. Gógl, J. Dolenc, J. Osz, Y. Nominé, C. Kostmann, A. Cousido-Siah, A. Mitschler, N. Rochel, G. Travé, B. Kieffer and V. Torbeev, Chem. Sci., 2021, 12, 1080–1089 RSC.
- D. Shortle, FASEB J., 1996, 10, 27–34 CrossRef CAS.
- D. Neri, M. Billeter, G. Wider and K. Wuthrich, Science, 1992, 257, 1559–1563 CrossRef CAS PubMed.
- F. J. Blanco, L. Serrano and J. D. Forman-Kay, J. Mol. Biol., 1998, 284, 1153–1164 CrossRef CAS PubMed.
- B. Zagrovic, C. D. Snow, S. Khaliq, M. R. Shirts and V. S. Pande, J. Mol. Biol., 2002, 323, 153–164 CrossRef CAS.
- B. Zagrovic and V. S. Pande, Nat. Struct. Biol., 2003, 10, 955–961 CrossRef CAS.
- S. Chowdhury, H. Lei and Y. Duan, J. Phys. Chem. B, 2005, 109, 9073–9081 CrossRef CAS.
- M. E. McCully, D. A. Beck and V. Daggett, Biochemistry, 2008, 47, 7079–7089 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, R. O. Dror and D. E. Shaw, Science, 2011, 334, 517–520 CrossRef CAS.
- G. A. Huber and S. Kim, Biophys. J., 1996, 70, 97–110 CrossRef CAS PubMed.
- D. M. Zuckerman and L. T. Chong, Annu. Rev. Biophys., 2017, 46, 43–57 CrossRef CAS PubMed.
- D. O. Alonso and V. Daggett, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 133–138 CrossRef CAS PubMed.
- H. Gouda, H. Torigoe, A. Saito, M. Sato, Y. Arata and I. Shimada, Biochemistry, 1992, 31, 9665–9672 CrossRef CAS PubMed.
- Z. Guo, C. L. Brooks III and E. M. Boczko, Proc. Natl. Acad. Sci. U.S.A., 1997, 94, 10161–10166 CrossRef CAS.
- A. E. Garcia and J. N. Onuchic, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 13898–13903 CrossRef CAS PubMed.
- S. Jang, E. Kim, S. Shin and Y. Pak, J. Am. Chem. Soc., 2003, 125, 14841–14846 CrossRef CAS PubMed.
- G. Dimitriadis, A. Drysdale, J. K. Myers, P. Arora, S. E. Radford, T. G. Oas and D. A. Smith, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 3809–3814 CrossRef CAS PubMed.
- P. Arora, T. G. Oas and J. K. Myers, Protein Sci., 2004, 13, 847–853 CrossRef CAS.
- S. Sato, T. L. Religa, V. Daggett and A. R. Fersht, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 6952–6956 CrossRef CAS PubMed.
- S. Cheng, Y. Yang, W. Wang and H. Liu, J. Phys. Chem. B, 2005, 109, 23645–23654 CrossRef CAS PubMed.
- S. Sato, T. L. Religa and A. R. Fersht, J. Mol. Biol., 2006, 360, 850–864 CrossRef CAS PubMed.
- H. Lei, C. Wu, Z.-X. Wang, Y. Zhou and Y. Duan, J. Chem. Phys., 2008, 128, 235105 CrossRef PubMed.
- B. Nilsson, T. Moks, B. Jansson, L. Abrahmsén, A. Elmblad, E. Holmgren, C. Henrichson, T. A. Jones and M. Uhlén, Protein Eng., Des. Sel., 1987, 1, 107–113 CrossRef CAS.
- M. Tashiro, R. Tejero, D. E. Zimmerman, B. Celda, B. Nilsson and G. T. Montelione, J. Mol. Biol., 1997, 272, 573–590 CrossRef CAS PubMed.
- D. Zheng, J. M. Aramini and G. T. Montelione, Protein Sci., 2004, 13, 549–554 CrossRef CAS.
- K. T. O'Neil and W. F. DeGrado, Science, 1990, 250, 646–651 CrossRef PubMed.
- Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS PubMed.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 9151–9156 CrossRef CAS.
- B. Kuhlman and D. P. Raleigh, Protein Sci., 1998, 7, 2405–2412 CrossRef CAS.
- J. R. Santhouse, S. R. Rao and W. S. Horne, Methods Enzymol., 2021, 656, 93–122 CAS.
- A. T. Bogetti, H. E. Piston, J. M. G. Leung, C. C. Cabalteja, D. T. Yang, A. J. DeGrave, K. T. Debiec, D. S. Cerutti, D. A. Case, W. S. Horne and L. T. Chong, J. Chem. Phys., 2020, 153, 064101 CrossRef CAS PubMed.
- J. Copperman and D. M. Zuckerman, J. Chem. Theory Comput., 2020, 16, 6763–6775 CrossRef CAS PubMed.
- J. K. Myers and T. G. Oas, Nat. Struct. Biol., 2001, 8, 552–558 CrossRef CAS PubMed.
- U. Adhikari, B. Mostofian, J. Copperman, S. R. Subramanian, A. A. Petersen and D. M. Zuckerman, J. Am. Chem. Soc., 2019, 141, 6519–6526 CrossRef CAS PubMed.
- A. S. Saglam and L. T. Chong, Chem. Sci., 2019, 10, 2360–2372 RSC.
- T. Sztain, S. H. Ahn, A. T. Bogetti, L. Casalino, J. A. Goldsmith, E. Seitz, R. S. McCool, F. L. Kearns, F. Acosta-Reyes, S. Maji, G. Mashayekhi, J. A. McCammon, A. Ourmazd, J. Frank, J. S. McLellan, L. T. Chong and R. E. Amaro, Nat. Chem., 2021, 13, 963–968 CrossRef CAS PubMed.
- J. D. Russo, S. Zhang, J. M. G. Leung, A. T. Bogetti, J. P. Thompson, A. J. DeGrave, P. A. Torrillo, A. J. Pratt, K. F. Wong, J. Xia, J. Copperman, J. L. Adelman, M. C. Zwier, D. N. LeBard, D. M. Zuckerman and L. T. Chong, J. Chem. Theory Comput., 2022, 18, 638–649 CrossRef CAS PubMed.
- S. T. Brown, P. Buitrago, E. Hanna, S. Sanielevici, R. Scibek and N. A. Nystrom, Practice and Experience in Advanced Research Computing, Association for Computing Machinery, New York, NY, USA, 2021, pp. 1–4 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S16, Tables S1–S7, materials and methods. See https://doi.org/10.1039/d2sc04427g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |