
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-promoted cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction: efficient access to δ-carboline derivatives†
Xi-Shang
Sun‡
a,
Xin-Yu
Diao‡
a,
Xiu-Qin
Dong
 *a and
Chun-Jiang
Wang
*ab
*a and
Chun-Jiang
Wang
*ab
aCollege of Chemistry and Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: xiuqindong@whu.edu.cn; cjwang@whu.edu.cn
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin, 300071, P. R. China
First published on 15th August 2022
Abstract
A serendipitous and highly efficient approach for the construction of a variety of δ-carboline derivatives was developed through base-promoted cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction of N-2,2,2-trifluoroethylisatin ketoimine esters with alkynes in good to high yields with excellent regio-/chemoselectivity control. Moreover, a reasonable reaction pathway was proposed, which was in accordance with the prepared reaction intermediate and control experiment results. The δ-carboline product could be easily converted into a new chiral Py-box-type ligand through simple synthetic transformations. This salient strategy featured the advantages of metal-free conditions, excellent regio-/chemoselectivity, good to high yields, and outstanding substrate tolerance. Importantly, the potential application of these fascinating δ-carboline derivative products is well demonstrated in the recognition of ferric ions.
Introduction
Carbolines, usually known as pyrido[x,y-b]indoles, have emerged as important N-heterocycles with a broad spectrum of biological activities, which could be divided into α-, β-, γ- and δ-carbolines according to the position of the N atom.1,2 Among these various types of carbolines, δ-carbolines are identified as privileged units in plenty of natural alkaloids, pharmaceuticals, and functional materials (Scheme 1a).2 For example, CzBPDCb was used as an electron transport unit bipolar host material for blue phosphorescent organic light emitting didoes,2aN5-ω-cyclohexylpentyl-N1-methyl-δ-carbolinium displayed antibacterial and antifungal activities,2b PIQ could serve as a potential structure-selective G-quadruplex binding molecule,2c natural alkaloid Jusbetonin was isolated from Justicia betonica,2d and SYUIQ-5 is a potential anticancer therapeutic.2e,f Great efforts have been devoted to the development of the synthesis of these promising synthetic target δ-carboline derivatives, but limited efficient synthetic protocols have been explored until now.3 Yao and coworkers disclosed a metal-free protocol to construct δ-carboline derivatives through the I2-promoted cycloaddition of indolylchalcone oxime esters.3c Tang's group3e and Chang's group3b respectively developed different [2 + 2 + 2] cycloadditions as attractive approaches for the synthesis of δ-carbolines. Reddy and coworkers developed Yb(OTf)3-catalyzed decarboxylative condensation of 2-aminoindole-3-carboxylates with ynals, followed by uncommon [3 + 2]-spirocycloaddition and 2,3-aza migration to afford dihydrochromeno-fused δ-carbolines in moderate yields.3f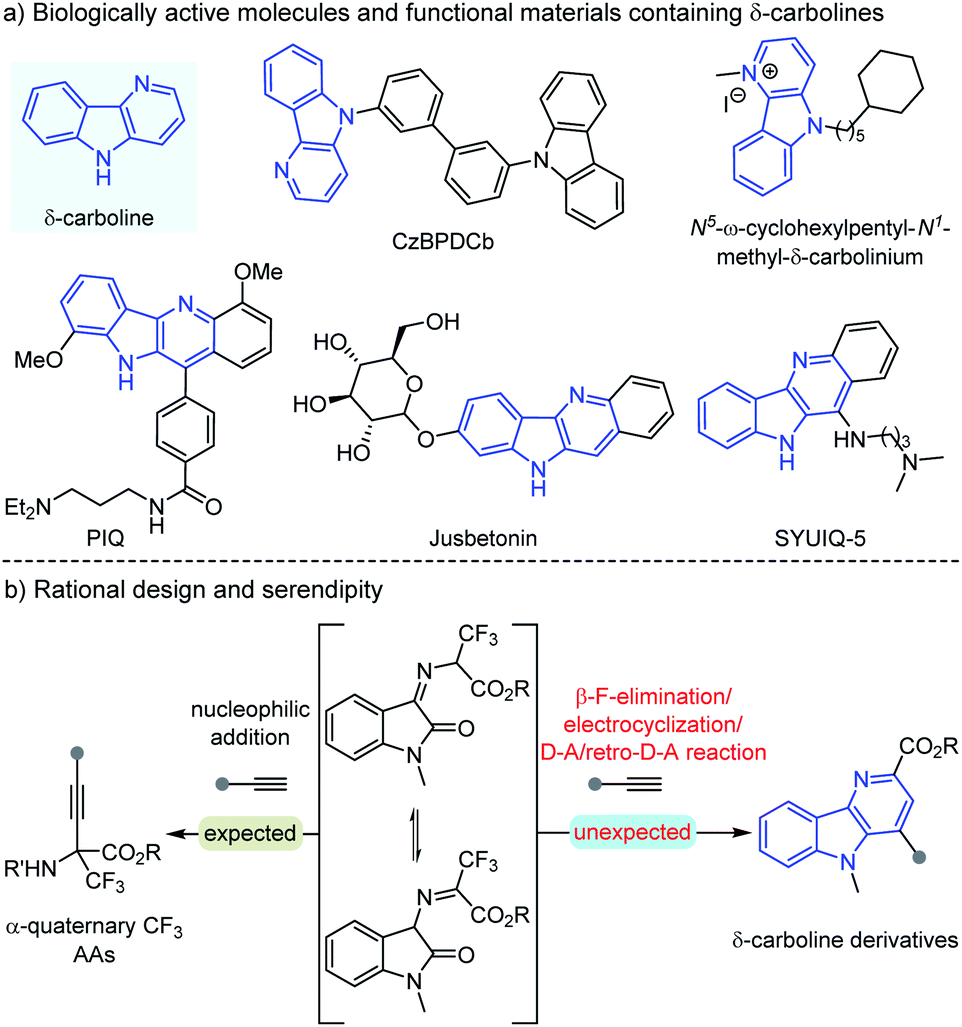 | ||
| Scheme 1 Examples of biologically active molecules and functional materials containing δ-carboline motifs and synthesis of δ-carboline derivatives. | ||
On the other hand, quaternary α-trifluoromethyl α-amino acids (α-Tfm-AAs) and derivatives as important subunits have been extensively distributed in many bioactive molecules.4 And we are interested in the synthesis of these important molecules using the easily available arylethynylenes as the nucleophilic reagents, which would suffer from the challenge of umpolung functionalization of α-trifluoromethyl ketoimine esters5 (Scheme 1b left). However, the expected α-Tfm-AA derivative product was not found in the initial investigation. To our surprise, an unexpected compound was obtained, whose structure was clearly assigned as a δ-carboline derivative. This biologically important product δ-carboline derivative is intriguing, and we speculated that this transformation may undergo an unprecedented cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction (Scheme 1b right). Considering these current scarce synthetic methods, the intriguing structures and the biological relevance of δ-carboline derivatives, we decided to investigate this transformation in detail and establish another facile and efficient synthetic protocol.
Results and discussion
In order to optimize the reaction conditions, the reactions between trifluoroethylisatin ketimine ester 1a and phenylacetylene 2a as model substrates were carried out in the presence of different Lewis acids and NEt3. A series of Lewis acids, such as Zn(OTf)2, Cu(OTf)2, InCl3 and Sc(OTf)3, were then applied in this transformation. Although the expected α-quaternary trifluoromethyl amino acid derivative 3a was not observed, structurally important δ-carboline derivative 4a could be obtained in poor yields and exclusive regioselectivity, and another δ-carboline derivative 4a′ was not observed (8–15% yields, Table 1, entries 1–4). In order to improve the reactivity, chemoselectivity and regioselectivity, other bases like Cs2CO3 and DBU were then investigated, but no better result was afforded (Table 1, entries 5 and 6). When this transformation was performed in toluene, higher reactivity was provided with excellent selectivity (Table 1, entry 7). To our delight, when the reaction temperature was increased to 100 °C, the reactivity was greatly improved, the δ-carboline derivative 4a could be obtained with high yield and exclusive regioselectivity (82% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr, Table 1, entry 8). In addition, it was found that very poor reactivity was furnished in the absence of a base (Table 1, entry 9). Remarkably, this transformation could be performed smoothly just promoted by NEt3, leading to the product 4a in good yield with exclusive regioselectivity (Table 1, entry 10). These results demonstrated that a Lewis acid was not essential in this cascade reaction. And we speculated that ketoimine ester 1a and phenylacetylene 2a may go through the cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction to produce δ-carboline derivative 4a.
:1 rr, Table 1, entry 8). In addition, it was found that very poor reactivity was furnished in the absence of a base (Table 1, entry 9). Remarkably, this transformation could be performed smoothly just promoted by NEt3, leading to the product 4a in good yield with exclusive regioselectivity (Table 1, entry 10). These results demonstrated that a Lewis acid was not essential in this cascade reaction. And we speculated that ketoimine ester 1a and phenylacetylene 2a may go through the cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction to produce δ-carboline derivative 4a.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Lewis acid | Base | Solvent | T (°C) | 3a/4a/4a′b | Yieldc (%) |
| a All reactions were carried out with 0.20 mmol ketoimine ester 1a, 0.40 mmol phenylacetylene 2a, 20 mol% Lewis acid, and 0.4 mmol base in 2 mL solvent. b The selectivity was determined by using a 1H NMR analysis. c Isolated yield. | ||||||
| 1 | Zn(OTf)2 | NEt3 | DCM | 25 | 0/100/0 | 15 |
| 2 | Cu(OTf)2 | NEt3 | DCM | 25 | 0/100/0 | 8 |
| 3 | InCl3 | NEt3 | DCM | 25 | 0/100/0 | 8 |
| 4 | Sc(OTf)3 | NEt3 | DCM | 25 | 0/100/0 | 9 |
| 5 | Zn(OTf)2 | Cs2CO3 | DCM | 25 | 0/100/0 | 11 |
| 6 | Zn(OTf)2 | DBU | DCM | 25 | — | — |
| 7 | Zn(OTf)2 | NEt3 | Toluene | 25 | 0/100/0 | 25 |
| 8 | Zn(OTf)2 | NEt3 | Toluene | 100 | 0/100/0 | 82 |
| 9 | Zn(OTf)2 | — | Toluene | 100 | — | — |
| 10 | — | NEt3 | Toluene | 100 | 0/100/0 | 81 |
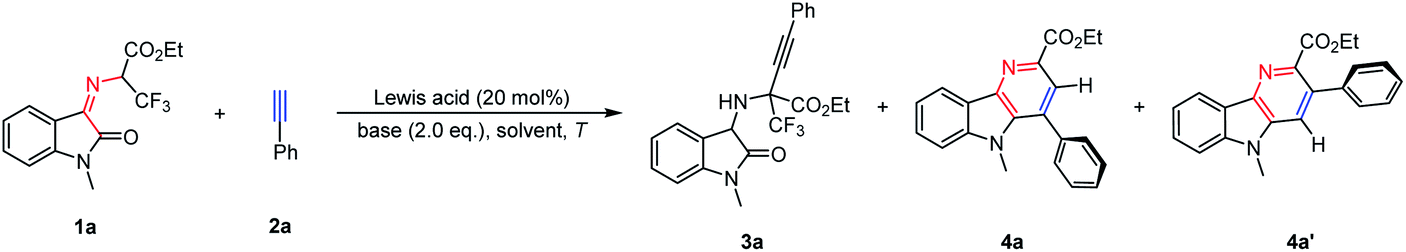
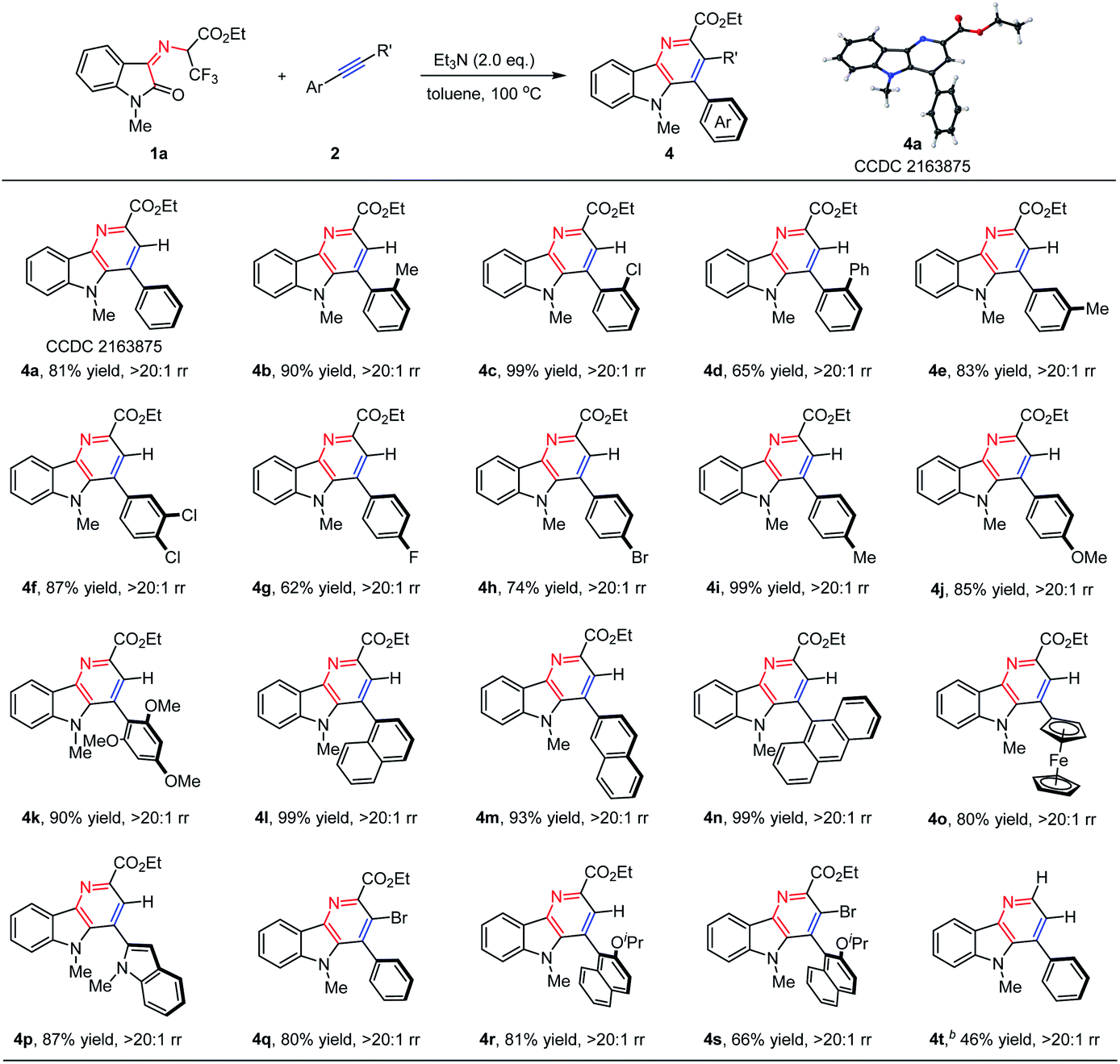
With the optimal reaction conditions in hand, the substrate generality of this protocol was then assessed. As shown in Table 2, N-2,2,2-Trifluoroethylisatin ketoimine ester 1a was treated with a wide range of aromatic alkynes, and a variety of desired δ-carboline derivatives 4 were furnished with excellent results. When aryl alkynes were applied as reaction partners, the electronic properties of the substituted groups on the phenyl ring have little influence on the reactivities and regioselectivities. We found that the electron-neutral, electron-donating and electron-withdrawing groups could be well tolerated, resulting in the corresponding δ-carboline products 4a–4k in moderate to high yields with exclusive regioselectivity (62–99% yields, >20:1 rr). The structure of product 4a was determined by X-ray analysis (CCDC 2163875†). It is worth noting that other polycyclic aromatic substituted alkynes bearing steric hindrance, such as 1-naphthyl (2l), 2-naphthyl (2m), 9-anthracyl (2n) and 2-indolyl (2p) groups, underwent cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction smoothly to give the desired products 4l–4n and 4p in 87–99% yields. In addition, the ferrocenyl substituted alkyne (2o) was also surveyed, and the corresponding product (4o) could be obtained in 80% yield. Remarkably, compared with an aryl-terminated alkyne, disubstituted (bromoethynyl)benzene 2q was also amenable under the standard reaction conditions, giving the desired product 4q in 80% yield. Notably, bulky 1-ethynyl-2-isopropoxynaphthalene 2r and 1-(bromoethynyl)-2-isopropoxy-naphthalene 2s also performed well to deliver the desired products 4r and 4s with satisfying results (81% yield and 66% yield, respectively). In addition, ethyl-(Z)-2-((1-methyl-2-oxoindolin-3-ylidene)amino)acetate 1b could be tolerated in this cascade reaction, delivering the desired product 4t in 46% yield. When ethyl-(Z)-2-((1-methyl-2-oxoindolin-3-ylidene)amino)-2-phenylacetate was then examined in this reaction system, no expected δ-carboline derivative was obtained with 3,3-difluoro-5-methyl-2-phenyl-3,5-dihydro-[1,4]oxazino[2,3-b]indole III′ being separated in 80% yield as the intermediate (vide infra in Scheme 2b).
| a All reactions were carried out with 0.20 mmol ketoimine ester 1a, 0.40 mmol arylalkyne 2, and 0.4 mmol NEt3 in 2 mL toluene at 100 °C. The regioselectivity was determined by 1H NMR analysis. The yield is isolated yield. b (Z)-1-Methyl-2-methylene-N-(2,2,2-trifluoroethyl)indolin-3-imine 1b was used as the substrate. |
|---|
|
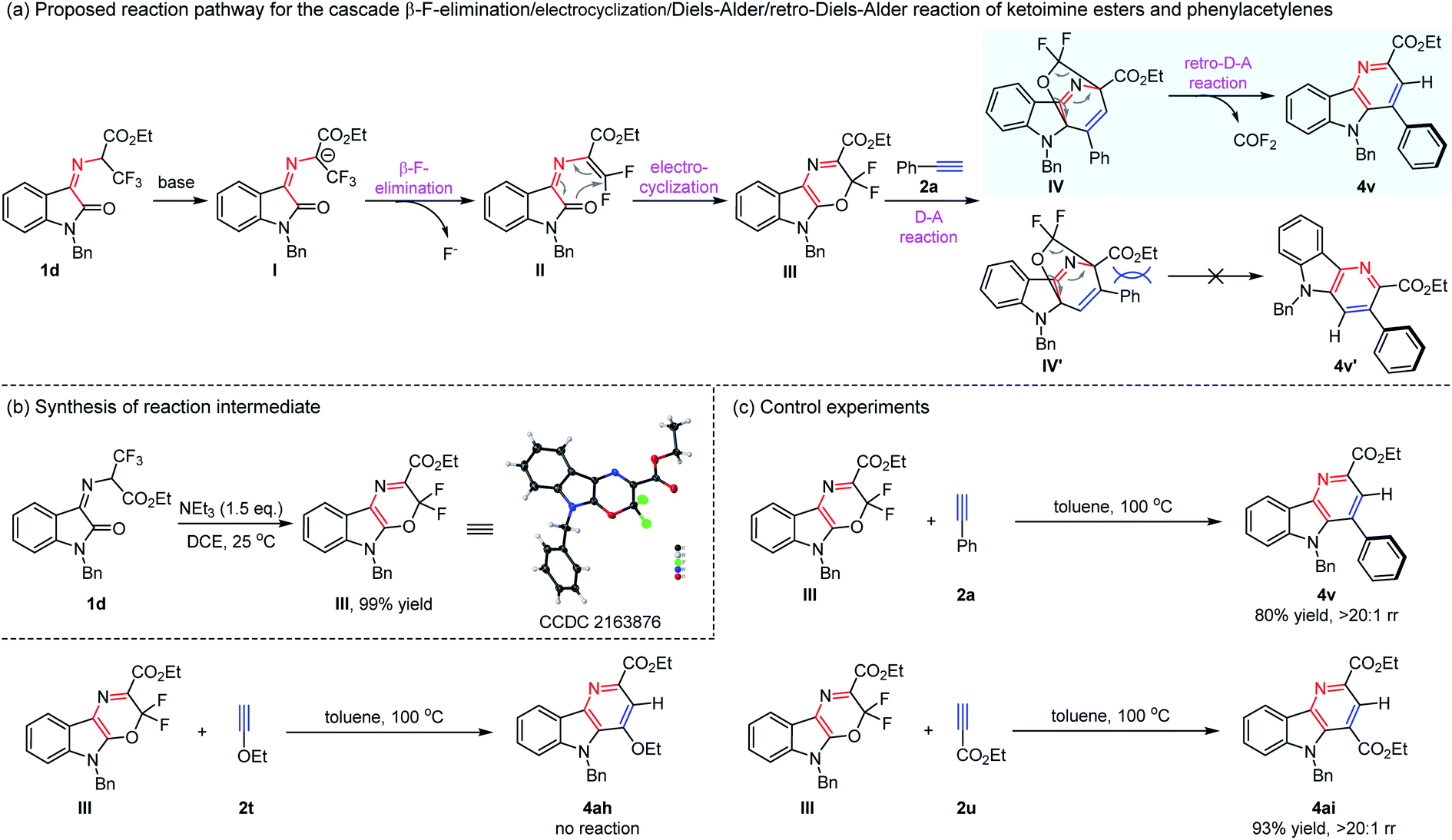 | ||
| Scheme 2 Proposed reaction pathway, synthesis of the reaction intermediate and the control experiment. | ||
Next, we turned our attention to extending the above synthetic methodology for N-2,2,2-trifluoroethylisatin ketoimine esters. As shown in Table 3, the influence of protecting groups on the N atom of isatin moieties, such as allyl (1c), benzyl (1d), and n-butyl (1e) substituted groups, was first evaluated. They worked as compatible partners with phenylacetylene (2a) and steric 1-ethynyl-2-isopropoxynaphthalene (2r) smoothly, generating the corresponding products 4u–4y in moderate to high yields with exclusive regioselectivity (67–98% yields, >20:1 rr). These reaction results demonstrated that the N-substituent of the N-2,2,2-trifluoroethylisatin ketoimine esters has little influence on the reactivity and selectivity. In addition, the effect of substitution at the benzene ring of the isatin motif was further explored. It was found that the substituted groups, such as halogen, methyl, and methoxy groups at different positions could be well tolerated. They reacted with steric 1-ethynyl-2-isopropoxynaphthalene (2r) efficiently, and excellent results were generally achieved for a wide range of N-2,2,2-trifluoroethylisatin ketoimine esters. A series of desired products (4z–4ag) were obtained with good to high yields (80–99% yields).
| a All reactions were carried out with 0.20 mmol ketoimine ester 1, 0.40 mmol arylalkyne 2, and 0.4 mmol NEt3 in 2 mL toluene at 100 °C. The regioselectivity was determined by 1H NMR analysis. The yield is isolated yield. |
|---|
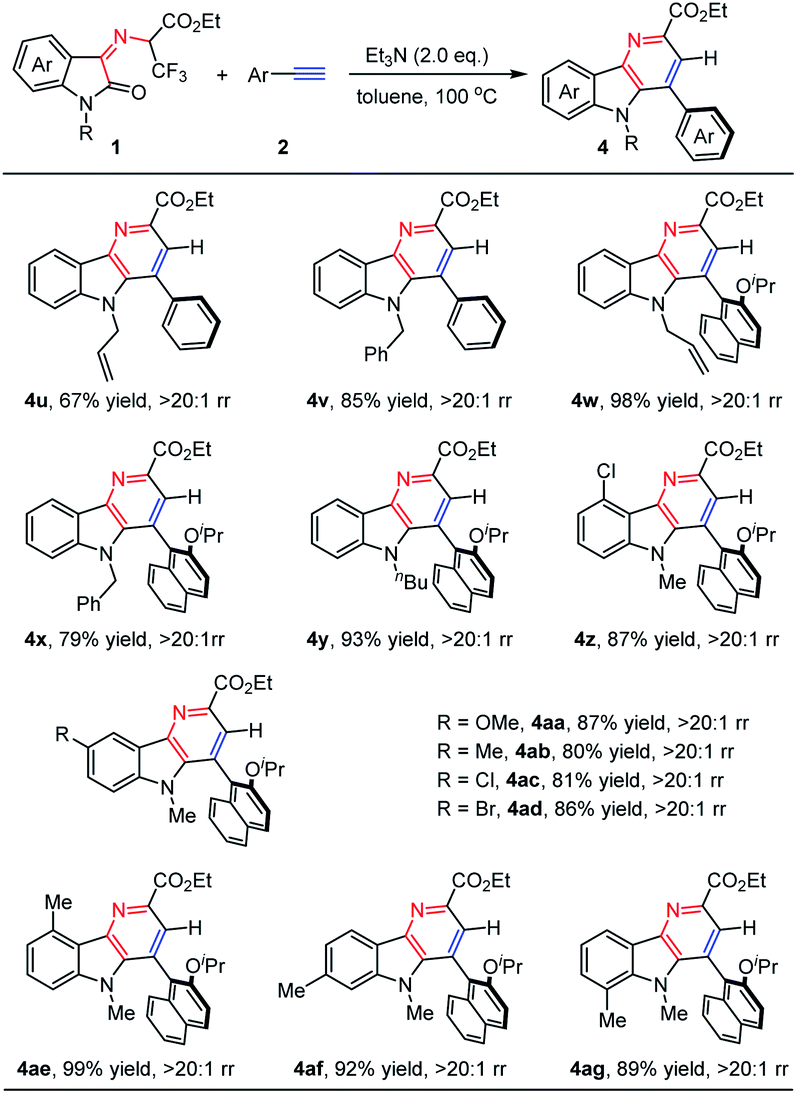
|
Having explored the new cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction, we were intrigued by the reaction mechanism. As depicted in Scheme 2a, a possible pathway for the synthesis of δ-carboline derivatives was proposed. Owing to the strong electron-withdrawing property of the trifluoromethyl group and ester group, the cascade transformation could be easily initiated by the loss of a proton in the presence of a base to form carbon anion I. Subsequently, it was converted to intermediate IIvia the β-F-elimination reaction, which underwent an intramolecular 6π-electrocyclic reaction to afford compound III.6 The following Diels–Alder reaction with phenylacetylene 2a gave the cyclization products IV and IV′. The product IV′ should be more thermodynamically disfavored due to the steric repulsion, and the more preferential IV proceeded retro-Diels–Alder reaction smoothly, delivering the desired δ-carboline compound 4v with high regioselectivity via extrusion of carbonyl fluoride. To further explore and confirm the reaction pathway, control experiments were conducted as shown in Scheme 2b. N-2,2,2-Trifluoroethylisatin ketoimine ester 1d was treated with NEt3, and it was found that 3,3-difluoro-3,5-dihydro-[1,4]oxazino[2,3-b]indole-2-carboxylate ester III is the sole product in nearly quantitative yield, and its structure was confirmed by X-ray diffraction analysis (CCDC 2163876,†Scheme 2b).7 Moreover, the intermediate III could react with phenylacetylene 2a easily to give the desired product 4v in 80% yield (Scheme 2c). These results demonstrated that this transformation should go through intermediate III, which is consistent with the above-proposed pathway through the β-F-elimination/electrocyclization/Diels–Alder reaction. When electron-rich ethoxyacetylene 2t was allowed to react with intermediate III, no reaction was observed. We found that electron-deficient ethyl propiolate 2u reacted smoothly to furnish the corresponding δ-carboline compound 4ai in 93% yield. Therefore, it is possible that the normal Diels–Alder reaction between the intermediate diene III with alkyne molecules proceeded in this reaction system.
To demonstrate the potential practicality of this cascade methodology, a scale-up experiment on 3 mmol was carried out smoothly, and the desired product δ-carboline derivative 4a could be obtained in 79% yield (Scheme 3a). In addition, a further synthetic transformation of the δ-carboline derivative product was explored. The development of chiral ligands with a new skeleton is an important research topic in the field of asymmetric catalysis. A chiral oxazoline backbone has been regarded as a privileged structure for chiral ligands in the field of asymmetric catalytic synthesis.8 As shown in Scheme 3b, a structurally novel chiral Py-box-type ligand could be easily prepared through simple transformations.9 The ester group of a δ-carboline derivative was easily hydrolyzed to give carboxylic acid 5 under basic conditions, which underwent condensation with the commercially available chiral (S)-tert-leucinol and the subsequent intramolecular cyclization to give a new functionalized chiral Py-box-type ligand 6 smoothly with 53% yield in two steps. To our delight, chiral ligand 6 could be well utilized in the Ni-catalyzed asymmetric α-fluorination reaction of 2-tert-butoxycarbonyl-1-indanone 7 with N-fluorobenzenesulfon-imide (NFSI) 8, and preliminary results could be obtained to give the desired product 9 in good yield and moderate enantioselectivity. Ligand 6 was found to be more superior than the commonly used Py-box ligand 10, which displayed poor enantioselective control in this asymmetric transformation (Scheme 3c).
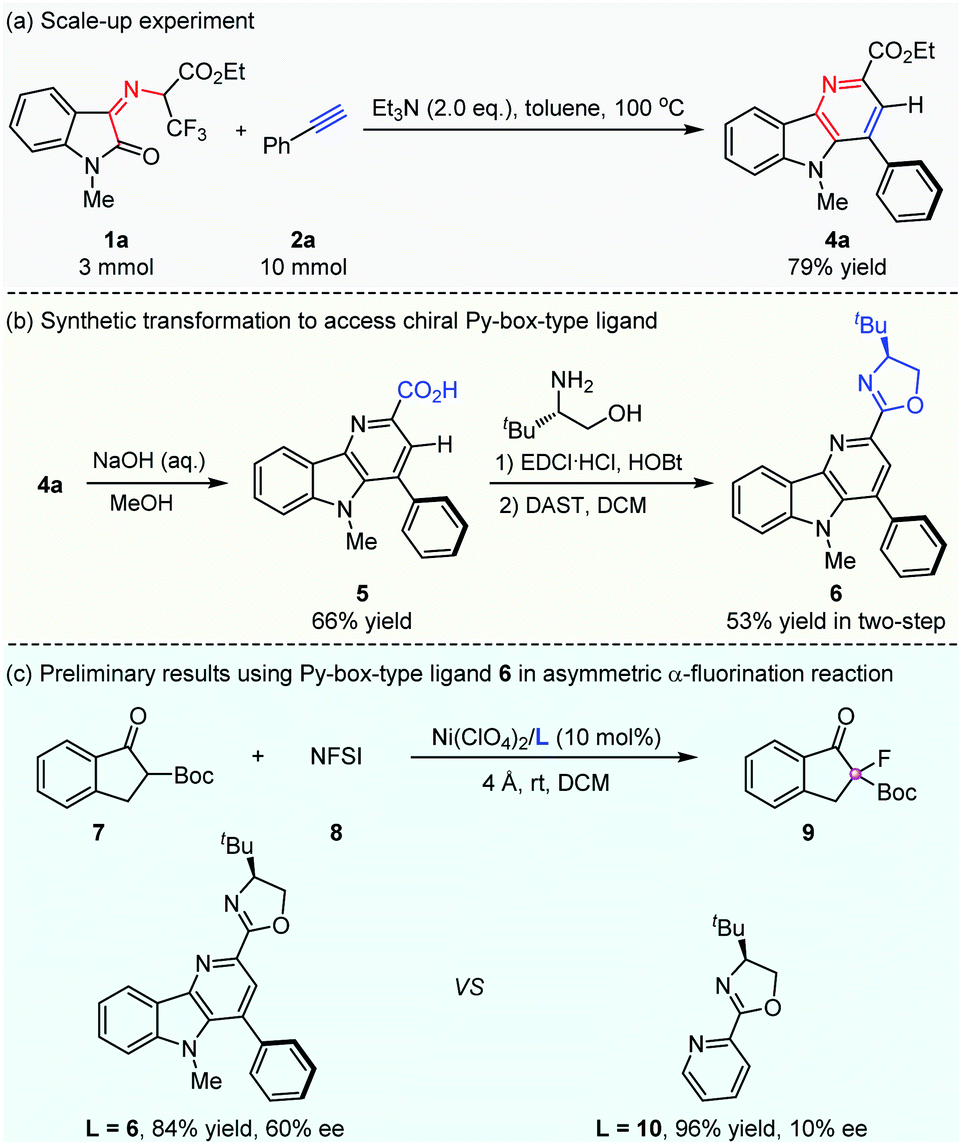 | ||
| Scheme 3 Scale-up experiment and application in asymmetric synthesis. | ||
In view of such a δ-carboline derivative containing π-conjugated system, we then turned our attention to investigate their spectral properties and potential application. The UV-visible absorption of a series of δ-carboline derivatives, including 4b, 4p, 4r, 4z, 4aa, 4ab, 4ae, and 4ag, has shown the adsorption ranges up to the visible light region within 460 nm in anhydrous dichloromethane (DCM) solution (Fig. 1a).10 The fluorescence spectra of these molecules showed that the emission band maxima fell in the range between 395 (4z) and 460 nm (4aa) with a slight red-shift (Fig. 1b and c). As shown in Fig. 1d, the δ-carboline derivatives 4p, 4z and 4aa exhibited different fluorescence emissions and colors under irradiation with UV light (λex = 365 nm) in DCM. All these useful findings would offer a promising opportunity for these δ-carboline derivatives as potential fluorescent materials, metal ion recognition reagents, and biosensors.11 Therefore, considering the possibility of the recognition ability of δ-carboline derivatives towards metal ions, we then focused on the investigation of the metal ion recognition effect of product 4a in acetonitrile. As shown in Fig. 1e, the addition of Fe(OTf)3 to the solution of 4a led to a slight redshift but a great reduction of fluorescence intensity, while the addition of Fe(OTf)2 or Cu(OTf)2 only resulted in a slight redshift of the fluorescence. In addition, we found that the wavelength led to a redshift effect in the range of 409–448 nm in the process of gradual increase of the Fe(OTf)3 concentration. These results demonstrated that ferric ions (Fe3+) could be selectively recognized by δ-carboline derivative 4a.12
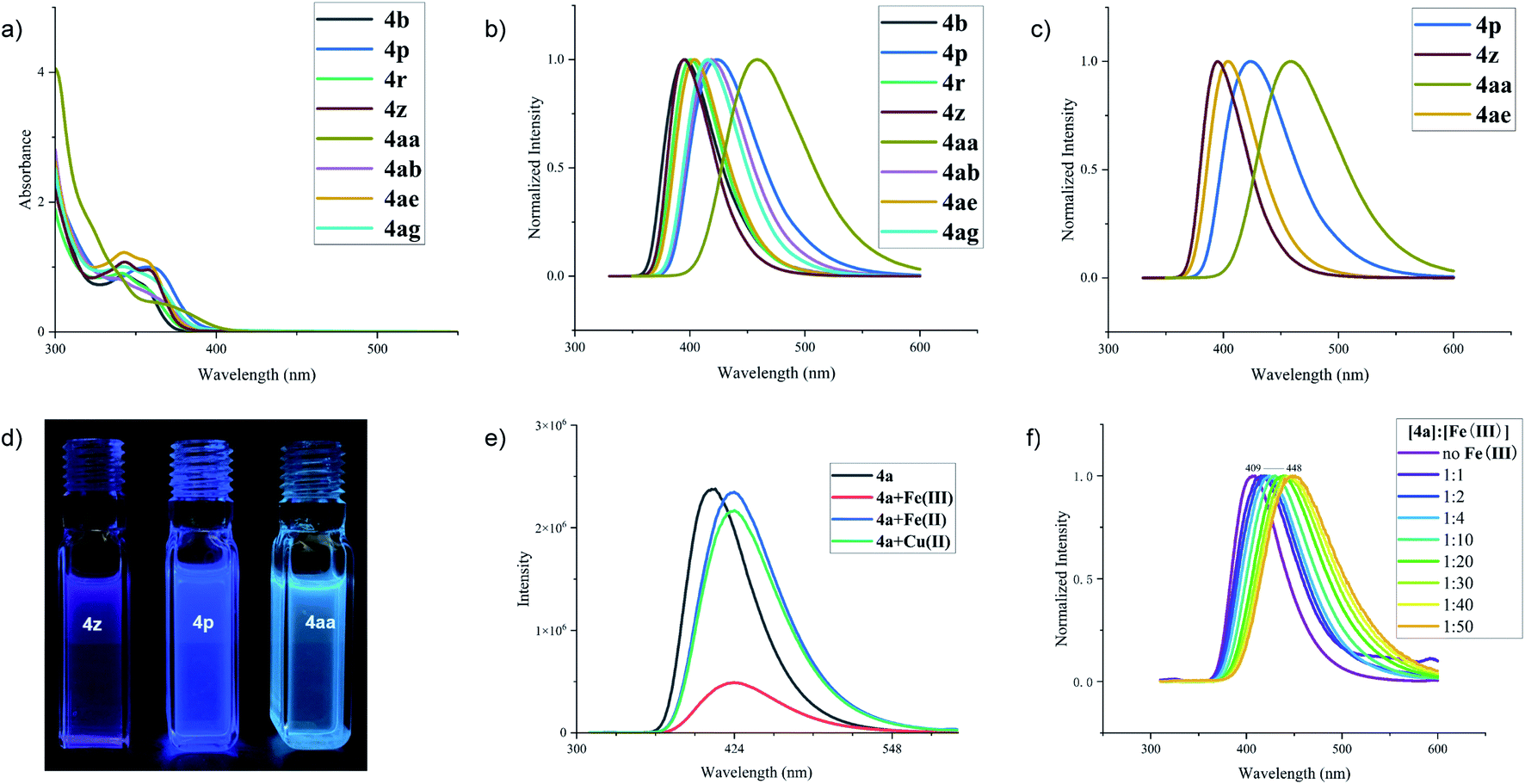 | ||
| Fig. 1 (a) UV-vis absorption spectra. (b and c) Fluorescence spectra of δ-carboline derivatives. (d) Photographs of δ-carboline derivatives under irradiation with UV light (λex = 365 nm) in DCM. (e) Fluorescence spectra of 4a at 5 × 10−5 M with or without metal precursors (2 × 10−4 M) in CH3CN. (f) Fluorescence spectra of 4a with gradual addition of different amounts of Fe3+. | ||
Conclusions
In summary, we developed a new and highly efficient base-promoted cascade β-F-elimination/electrocyclization/Diels–Alder/retro-Diels–Alder reaction of N-2,2,2-trifluoroethylisatin ketoimine esters with alkynes, which provided a facile and efficient approach to access a wide range of nitrogen-containing heterocyclic δ-carboline derivatives in good to high yields. This synthetic strategy had the advantages of metal-free conditions, excellent regio-/chemoselectivity, good to high yields, wide substrate generality, and various synthetic transformations. Moreover, a possible reaction pathway was proposed for this cascade protocol, which was confirmed by the reaction intermediate and control experiment results. Importantly, the δ-carboline derivative product had good recognition ability of ferric ions through fluorescence spectrum analysis.Data availability
The ESI† contains method description, product characterization data, and NMR spectra.Author contributions
CJW conceptualized the project. CJW and XQD supervised the investigation. XSS and XYD performed the research. CJW and XQD co-wrote the paper. All authors analyzed the data, discussed the results, and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22071186 and 22071187), Natural Science Foundation of Hubei Province (2020CFA036 and 2021CFA069), Natural Science Foundation of Jiangsu Province (BK20190213), and Fundamental Research Funds for the Central Universities (2042022kf1180).Notes and references
- (a) A. Kamboj, B. Sihag, D. S. Brar, A. Kaur and D. B. Salunke, Eur. J. Med. Chem., 2021, 221, 113536–113556 CrossRef CAS PubMed; (b) Y. Yao, M. Alami, A. Hamze and O. Provot, Org. Biomol. Chem., 2021, 19, 3509–3526 RSC; (c) S. Aaghaz, K. Sharma, R. Jain and A. Kamal, Eur. J. Med. Chem., 2021, 216, 113321–113346 CrossRef CAS PubMed; (d) F. Faheem, K. B. Kumar, G. C. K. V. Sekhar, S. Kunjiappan, J. Jamalis, R. Balaña-Fouce and M. Sankaranarayanan, Mini-Rev. Med. Chem., 2021, 21, 398–425 CrossRef PubMed; (e) U. Tewari, D. Sharma, S. Srivastava, B. K. Kumar, F. Faheem and S. Murugesan, ChemistrySelect, 2021, 6, 2428–2445 CrossRef CAS; (f) H. Zhang, R. H. Zhang, L. X. Wang, Y. J. Li, S. G. Liao and M. Zhou, Asian J. Org. Chem., 2021, 10, 429–452 CrossRef CAS; (g) A. Gupta, B. Kamble, N. Moola Joghee and C. Moola Joghee Nanjan, Curr. Org. Synth., 2012, 9, 377–396 CrossRef CAS; (h) R. S. Alekseyev, A. V. Kurkin and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2012, 47, 1469–1501 CrossRef CAS; (i) O. B. Smirnova, T. V. Golovko and V. G. Granik, Pharm. Chem. J., 2011, 45, 389–400 CrossRef CAS; (j) A. A. Semenov and V. V. Tolstikhina, Chem. Heterocycl. Compd., 1984, 20, 345–356 CrossRef; (k) M. L. Trudell, A. S. Basile, H. E. Shannon, P. Skolnick and J. M. Cook, J. Med. Chem., 1987, 30, 456–458 CrossRef CAS PubMed; (l) M. L. Trudell, S. L. Lifer, Y. C. Tan, M. J. Martin, L. Deng, P. Skolnick and J. M. Cook, J. Med. Chem., 1990, 33, 2412–2420 CrossRef CAS PubMed; (m) X. Chang, C. Che, Z.-F. Wang and C.-J. Wang, CCS Chem, 2022, 4, 1414–1428 CrossRef CAS; (n) R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479–500 CrossRef CAS PubMed; (o) K. Sako, H. Aoyama, S. Sato, Y. Hashimoto and M. Baba, Bioorg. Med. Chem., 2008, 16, 3780–3790 CrossRef CAS PubMed; (p) R. S. Alekseyev, A. V. Kurkin and M. A. Yurovskaya, Chem. Heterocycl. Compd., 2009, 45, 889 CrossRef CAS; (q) T. Takeuchi, S. Oishi, T. Watanabe, H. Ohno, J.-i. Sawada, K. Matsuno, A. Asai, N. Asada, K. Kitaura and N. Fujii, J. Med. Chem., 2011, 54, 4839–4846 CrossRef CAS PubMed; (r) A. Le Gresley, V. Gudivaka, S. Carrington, A. Sinclair and J. E. Brown, Org. Biomol. Chem., 2016, 14, 3069–3079 RSC.
- (a) Z. Zhang, J. Xie, Z. Wang, B. Shen, H. Wang, M. Li, J. Zhang and J. Cao, J. Mater. Chem. C, 2016, 4, 4226–4235 RSC; (b) T. K. Mazu, J. R. Etukala, M. R. Jacob, S. I. Khan, L. A. Walker and S. Y. Ablordeppey, Eur. J. Med. Chem., 2011, 46, 2378–2385 CrossRef CAS PubMed; (c) A. Funke, J. Dickerhoff and K. Weisz, Chem.–Eur. J., 2016, 22, 3170–3181 CrossRef CAS PubMed; (d) G. V. Subbaraju, J. Kavitha, D. Rajasekhar and J. I. Jimenez, J. Nat. Prod., 2004, 67, 461–462 CrossRef CAS PubMed; (e) J. M. Zhou, X. F. Zhu, Y. J. Lu, R. Deng, Z. S. Huang, Y. P. Mei, Y. Wang, W. L. Huang, Z. C. Liu, L. Q. Gu and Y. X. Zeng, Oncogene, 2006, 25, 503–511 CrossRef CAS PubMed; (f) J. N. Liu, R. Deng, J. F. Guo, J. M. Zhou, G. K. Feng, Z. S. Huang, L. Q. Gu, Y. X. Zeng and X. F. Zhu, Leukemia, 2007, 21, 1300–1302 CrossRef CAS PubMed; (g) F. Riechert-Krause, K. Autenrieth, A. Eick and K. Weisz, Biochemistry, 2013, 52, 41–52 CrossRef CAS PubMed; (h) R. Jiang, Y. Wang and Z. Zhou, Tetrahedron, 2016, 72, 6444–6449 CrossRef CAS; (i) E. Arzel, P. Rocca, P. Grellier, M. Labaeïd, F. Frappier, F. Guéritte, C. Gaspard, F. Marsais, A. Godard and G. Quéguiner, J. Med. Chem., 2001, 44, 949–960 CrossRef CAS PubMed.
- (a) K. Selvaraj and K. C. K. Swamy, J. Org. Chem., 2018, 83, 15043–15056 CrossRef CAS PubMed; (b) J. Zhang, M. Guo, Y. Chen, S. Zhang, X.-N. Wang and J. Chang, Org. Lett., 2019, 21, 1331–1336 CrossRef CAS PubMed; (c) T. H. Yang, C. W. Kuo, V. Kavala, A. Konala, C. Y. Huang and C.-F. Yao, Chem. Commun., 2017, 53, 1676–1679 RSC; (d) Z. Z. Chen, S. Q. Li, Y. J. Zhang, D. Y. Tang, J. P. Meng, J. Lei, H. Y. Li and Z. G. Xu, Org. Lett., 2018, 20, 7811–7815 CrossRef CAS PubMed; (e) H. Wen, W. Cao, Y. Liu, L. Wang, P. Chen and Y. Tang, J. Org. Chem., 2018, 83, 13308–13324 CrossRef CAS PubMed; (f) M. Rajesh, R. Kumar, S. Puri, J. B. Nanubolu and M. S. Reddy, Org. Lett., 2020, 22, 1117–1123 CrossRef CAS PubMed.
- J. T. Welch, A. Gyenes and M. J. Jung, in General Features of Biological Activity of Fluorinated Amino Acids: Design, Pharmacology and Biochemistry, V. P. Kuhhar and V. A. Soloshonok, ed. John Wiley & Sons, Chichester, 1995, pp. 311–331 Search PubMed.
- (a) P. Chen, Z. Yue, J. Zhang, X. Lv, L. Wang and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 13316–13320 CrossRef CAS PubMed; (b) L. M. Shi, X. S. Sun, C. Shen, Z. F. Wang, H. Y. Tao and C.-J. Wang, Org. Lett., 2019, 21, 4842–4848 CrossRef CAS PubMed; (c) X. S. Sun, X. H. Wang, H. Y. Tao, L. Wei and C. J. Wang, Chem. Sci., 2020, 11, 10984–10990 RSC; (d) W. R. Zhu, K. Liu, J. Weng, W. H. Huang, W. J. Huang, Q. Chen, N. Lin and G. Lu, Org. Lett., 2020, 22, 5014–5019 CrossRef CAS PubMed; (e) W. C. Jang, M. Jung and H. M. Ko, Org. Lett., 2021, 23, 1510–1515 CrossRef CAS PubMed; (f) M. Zhang, P. Zhao, H. Yuan, A. Zhang, W. Zhang, L. Zheng, D. Wu and L. Liu, Chin. J. Org. Chem., 2021, 41, 669 CrossRef.
- M. B. Smith and J. March, March's Advanced Organic Chemistry-Reaction, Mechanism, and Structure, John Wiely & Sons, 5th edn, 2001 Search PubMed.
- CCDC 2163876 (III) contains the ESI†.
- G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
- (a) H. Brunner and U. Obermann, Chem. Ber., 1989, 122, 499–507 CrossRef CAS; (b) H. Shimizu, J. C. Holder and B. M. Stoltz, Beilstein J. Org. Chem., 2013, 9, 1637–1642 CrossRef PubMed; (c) J. Lai, W. Li, S. Wei and S. Li, Org. Chem. Front., 2020, 7, 2263–2268 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- (a) J. Hwang, C. Lee, J.-E. Jeong, C. Y. Kim, H. Y. Woo, S. Park, M. J. Cho and D. H. Choi, ACS Appl. Mater. Interfaces, 2020, 12, 8485–8494 CrossRef CAS PubMed; (b) J. S. Moon, D. H. Ahn, S. W. Kim, S. Y. Lee, J. Y. Lee and J. H. Kwon, RSC Adv., 2018, 8, 17025–17033 RSC; (c) M. Singh, R. Vaishali, R. Kumar and V. Singh, ChemistrySelect, 2020, 5, 5172–5179 CrossRef CAS; (d) L.-J. Fan, Y. Zhang, C. B. Murphy, S. E. Angell, M. F. L. Parker, B. R. Flynn and W. E. Jones Jr, Coord. Chem. Rev., 2009, 253, 410–422 CrossRef CAS; (e) M.-C. Radulescu, M.-P. Bucur, B. Bucur and G. L. Radu, Talanta, 2015, 137, 94–99 CrossRef CAS PubMed; (f) A. Doménech-Carbóa, G. Cebrián-Torrejón, L. de Miguel, V. Tordera, D. Rodrigues-Furtadoe, S. Assad-Kahnf, A. Fournet, B. Figadère, R. P. Vázquez-Manrique and E. Poupon, Electrochim. Acta, 2014, 115, 546–552 CrossRef.
- W. Xiao, Y. Mo, J. Guo, Z. Su, S. Dong and X. Feng, Chem. Sci., 2021, 12, 2940–2947 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2163875 and 2163876. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03166c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |