
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structure of the medium-pore zeolite PST-35 with two interconnected cages of unusual orthorhombic shape†
Kingsley Christian
Kemp
a,
Wanuk
Choi
a,
Donghui
Jo
b,
Sung Hwan
Park
 a and
Suk Bong
Hong
*a
a and
Suk Bong
Hong
*a
aCenter for Ordered Nanoporous Materials Synthesis, Division of Environmental Science and Engineering, POSTECH, Pohang 3763, Korea. E-mail: sbhong@postech.ac.kr
bPetrochemical Catalyst Research Center, Korea Research Institute of Chemical Technology, Daejon 34114, Korea
First published on 15th August 2022
Abstract
The search for new zeolite structures and compositions remains important in synthetic materials science due to the high impact on developing new chemical technologies, as well as on improving existing ones. Herein we present the synthesis and structure of PST-35, a novel medium-pore germanosilicate (Si/Ge = 2.1–6.6) zeolite, achieved by combining the excess fluoride approach and the unique structure directing ability of Ge in the presence of 1,2,3-triethylimidazolium ions as an organic structure-directing agent. PST-35 contains a zig-zag 10-ring (4.6 × 6.7 Å) channel system constructed of strictly alternating large 28-hedral ([48·58·68·82·102]) and smaller 18-hedral ([46·54·64·82·102]) cages of anomalous orthorhombic shape. The PST-35 structure is built from the connection of pst-35 layers consisting of small 8-hedral ([43·54·6]) cages, previously unobserved zeolite building layers, through single 4-rings.
Introduction
Zeolites are a class of crystalline materials with uniform micropores, and their commercial application in adsorption and catalysis touches our daily lives.1–3 Yet there is still a need for new zeolites to develop more efficient and greener chemical technologies.4,5 However, although the structural and compositional regimes of this important class of microporous materials have been dramatically expanded by the use of organic structure-directing agents (OSDAs),6–8 only about 260 framework type codes (FTCs), which is four orders of magnitude smaller than the predicted number of chemically feasible hypothetical structures,9,10 have been recognised by the Structure Commission of the International Zeolite Association.11Zeolites can be classified into several categories, depending on the number of tetrahedral atoms (T-atoms) in the largest pore aperture. While there are 52 medium-pore structures with 10-ring pore openings among the framework types known to date, only seven of them are cage-based and are characterised by one type of accessible cage.11 In fact, the presence of two accessible interconnected cages even in other classes of zeolites is rather rare. Furthermore, most of such structures are small-pore zeolites. As a consequence, phosphate-based molecular sieves UCSB-8 (FTC SBE) with 14-hedral atn ([48·64·82]) and 26-hedral sbe ([412·810·124]) cages and cloverite (CLO) with 18-hedral clo ([48·68·82]) and 26-hedral lta ([412·68·86]) cages are the only examples of large- and extra-large-pore structures, respectively.12,13
The combined use of fluoride (F−) and OSDA under highly concentrated conditions (typically H2O/SiO2 ratio lower than 10) is one of the most successful synthetic strategies in the search for new silica-based zeolite structures.7 On the other hand, since F− encapsulation within many different small cages of the crystallised zeolites competes with Al substitution for Si, we considered the possibility that F− concentration in the aluminosilicate synthesis mixture containing already known OSDA cations could act as a phase selectivity factor in zeolite crystallisation. This simple hypothesis has enabled us to synthesise three novel aluminosilicate (Si/Al ∼ 10) structures, i.e., PST-21 (PWO, HF/OSDA = 2.0), PST-22 (PWW, HF/OSDA = 2.0), and PST-30 (PTY, HF/OSDA = 4.0), where all OSDAs are diazolium-based cations.8,14,15 We also found that PST-21 with two intersecting 9-ring channels is much more active and selective in 1-butene skeletal isomerization than ferrierite (FER), known as the best catalyst for this reaction.14,16
Another new but unexpected material discovered via the so-called excess fluoride approach is PST-24, a pure-silica medium-pore intergrowth zeolite, which has been synthesised at HF/OSDA = 3.0, using pentamethylimidazolium (PMI+) ions as an OSDA.17 Because HPM-1 (STW) is the phase formed using the same OSDA but in normal fluoride media (HF/OSDA = 1.0), we have reconsidered the F− concentration effect on the phase selectivity of the crystallisation and speculated whether interactions at the nucleation stage between the OSDA and the silicate species that subsequently constitute the zeolite framework can change with the increase of F− concentration in the synthesis mixture. Therefore, although the nature and extent of such interactions can also be influenced by the competition of F− encapsulation and Al substitution, we considered the other possibility that combining the excess fluoride concept with the high tendency of Ge to form double 4-ring (d4r, [46]) cages8 would allow us to find unprecedented zeolite structures. Up to now, a total of 33 FTCs could be obtained as the germanosilicate composition. However, the majority (21) of them are large-pore or extra-large-pore structures,11 so that the known germanosilicate medium-pore structures are rather rare.18–20
In this contribution, we report on the synthesis of PST-35, a new germanosilicate medium-pore zeolite, based on the hypothesis described above. Synchrotron single-crystal X-ray diffraction (SXRD) analysis shows that the PST-35 pore system is one-dimensional (1D) in nature and consists of zig-zag 10-ring (4.6 × 6.7 Å) channels built from orthorhombic 28-hedral ([48·58·68·82·102]) and 18-hedral ([46·54·64·82·102]) cages. These two interconnected cages are formed when the pst-35 layers, a new type of zeolite building layer consisting of 8-hedral ([435461]) cages, are connected through single 4-rings (s4rs) along the b axis. The small ([435461]) cage has not been assigned as a composite building unit yet, but was first found in SSZ-23 (STT) and subsequently in ITQ-52 (IFW).21,22 Thus, we will tentatively refer to this cage as stt.
Experimental section
Synthesis
Seven different imidazolium-based OSDAs were prepared following the procedures given in the ESI† and converted to the hydroxide form by anion exchange in aqueous solution using Amberlite IRN-78 anion-exchange resin (Alfa). The resulting solution was then concentrated by rotary evaporation at 80 °C, and the final hydroxide concentration was determined by titration using 0.1 M HCl and phenolphthalein as an indicator. The other reagents employed in zeolite synthesis included tetraethylorthosilicate (TEOS, 98%, Aldrich), germanium dioxide (GeO2, 99.99%, Samchun), aluminum isopropoxide (Al[OCH(CH3)2]3, 98%, Aldrich), hydrofluoric acid (HF, 48%, J.T. Baker) and deionised water. The composition of the final synthesis mixtures was 0.5ROH·(1 − x)SiO2·xGeO2·yAl2O3·zHF·5H2O, where R is the OSDA prepared here and x, y and z are varied between 0.125 ≤ x ≤ 0.333, 0 ≤ y ≤ 0.167 and 0.5 ≤ z ≤ 2.0, respectively. The final synthesis mixture was transferred into a 23 mL Teflon-lined autoclave and heated under rotation (60 rpm) at 160 °C for 3–14 days. The solid product was recovered using filtration, washed repeatedly with distilled water and dried overnight at room temperature. The as-synthesised zeolites were calcined in air at 550 °C for 8 h to remove the occluded OSDA.Analytical methods
Powder X-ray diffraction (PXRD) patterns were recorded on a PANalytical X'Pert diffractometer (Cu Kα radiation) with an X'Celerator detector. Elemental analysis for Si, Ge, and Al was carried out using a Shimadzu ICPE-9000 inductively coupled plasma spectrometer. The C, H and N contents of the samples were analyzed using a Vario EL III elemental organic analyzer. Thermogravimetric analyses (TGA) were performed in air on an SII EXSTAR 6000 thermal analyzer, where the weight losses related to the combustion of OSDAs were further confirmed by differential thermal analyses (DTA) using the same analyzer. The crystal morphology and average size were determined using a Hitachi S-3400N scanning electron microscope (SEM). N2 sorption measurements were carried out at −196 °C on a Mirae SI nanoPorosity-XG analyzer.1H and 13C liquid nuclear magnetic resonance (NMR) measurements on OSDAs were performed in 5 mm quartz tubes using a Bruker AVANCE III 300 spectrometer. 1H NMR spectra were measured at an 1H frequency of 300.13 MHz with a π/2 rad pulse length of 11 ms and a recycle delay of 2 s. 13C NMR spectra were recorded at a 13C frequency of 75.475 MHz with a π/2 rad pulse length of 10.2 ms and a recycle delay of 1.5 s. Solid-state multinuclear magic angle spinning (MAS) NMR measurements were carried out using a Bruker AVANCE III 500 HD spectrometer. 27Al and 29Si MAS spectra at a spinning speed of 21.0 kHz were recorded at 27Al and 29Si frequencies of 130.351 and 99.357 MHz, π/6 and π/2 rad pulse lengths of 1.0 and 4.0 μs, recycle delays of 2.0 and 20 s, and an acquisition of ca. 100 and 500 pulse transients, respectively. The 27Al and 29Si chemical shifts are reported relative to Al(H2O)63+ solution and TMS, respectively. 1H–13C cross polarization (CP) MAS NMR spectra at a spinning speed of 5.0 kHz were recorded at a 13C frequency of 125.795 MHz with a π/2 rad pulse length of 4.8 μs, a recycle delay of 3.0 s and an acquisition of about 2000 pulse transients. The 13C chemical shift is reported relative to TMS. 19F MAS NMR spectra at a spinning speed of 7.0, 10.0 and 15.0 kHz were recorded at a 19F frequency of 470.527 MHz, π/4 rad pulse length of 4.0 μs, a recycle delay of 5.0 s and an acquisition of about 6000 pulse transients. The 19F chemical shifts are referenced relative to CFCl3.
SXRD study
Synchrotron SXRD data of an as-synthesised, hydrated PST-35(3.9) crystal were collected at 100 K using monochromated X-rays (λ = 0.95116 Å) with a Rayonix MX225-HS CCD detector in the 6D beamline of the Pohang Accelerator Laboratory (PAL), Korea. The data were processed and scaled using the HKL2000 software package, and a structure solution was obtained by direct methods and refined using SHELXL2018.23,24 A summary of experimental and crystallographic data is presented in Table S1.†The positions of F− ions, disordered OSDAs, and partially occupied water molecules were derived from the Fourier difference map. Four tetrahedral sites (T-sites) in the double four-ring (d4r) with average T–O bond distances of 1.664, 1.668, 1.664 and 1.652 Å were assigned to Ge sites, and the other six T-sites with T–O bond distances of 1.606, 1.602, 1.605, 1.595, 1.606 and 1.594 Å to Si sites, following the conventional tetrahedral Ge–O and Si–O bond distances of 1.74 and 1.61 Å, respectively. On the other hand, it was found that while the F− ions reside in the d4r, disordered OSDAs and water molecules are partially occupied within the orthorhombic 28-hedral ([48586882102]) and 18-hedral ([46546482102]) cages, denoted ptf-1 and ptf-2 cages, respectively. The final atomic positions and occupancy factors are listed in Table S2† and the anisotropic displacement parameters in Table S3.† The selected bond lengths and angles are listed in Table S4.†
Stabilisation energy calculations
The stabilization energy of OSDAs within the PST-35 framework was calculated using the method developed by Deem and co-workers.25 The Dreiding force field was used in the Forcite module in Materials Studio 7.0 to calculate the stabilization energies.26,27 The experimentally determined number of OSDAs was first located in the unit cell and then energetically minimised using three molecular dynamics cycles which were carried out at 70 °C for 30 ps. The initial location of the OSDAs was done manually by placement of the OSDAs in the crystallographically determined positions within the framework before minimization. The stabilization energy (SE) is defined as follows:| SE = Ecomplex − Ezeolite − nEOSDA, |
Generation of hypothetical structures
The hypothetical periodic models (P1 symmetry) related to PST-35 were built as a silica polymorph using Materials Studio 7.0.27 These models were optimised using the Sanders–Leslie–Catlow (SLC) potential in the GULP program.28,29 The highest symmetries of the models were identified by the ‘Find Symmetry’ tool implemented in Materials Studio 7.0. The symmetrised structures were standardised in the VESTA program.30 To evaluate the feasibility of hypothetical models with P1 symmetry, their framework energies relative to α-quartz were calculated using the SLC potential in the GULP program. In addition, the local interatomic distance (LID) criteria were applied.31,32Results and discussion
Table 1 lists the representative products from zeolite synthesis at Si/Ge = 4.0 and HF/OSDA = 1.0–4.0 using seven alkyl-substituted imidazolium cations as OSDAs carried out under rotation (60 rpm) at 160 °C for 7 days. These organocations with C/N ratios of 3.0–5.0 were selected here because all of them have already been used as OSDAs in F−-mediated zeolite synthesis.33–35 No significant changes in the product when increasing the HF/OSDA ratio from 1.0 to 3.0 in the presence of each OSDA were observed. In the case of 1-ethyl-2,3-dimethylimidazolium (1E23DMI) with C/N = 3.5 and 2-ethyl-1,3,4-trimethylimidazolium (2E134TMI) with C/N = 4.0, for example, we always obtained ITQ-12 (ITW) and HPM-1 (STW), regardless of the HF/OSDA ratio, respectively. However, we noticed the first sign of the formation of a new zeolite phase (i.e., PST-35) from synthesis using 1,3-diethyl-2-methylimidazolium (13DE2MI), the C/N ratio (4.0) of which is the same as that of 2E134TMI, as an OSDA, at HF/OSDA = 2.0. Eventually, we were able to crystallise its pure phase under the same conditions, but using 1,2,3-triethylimidazolium (123TEI) with C/N = 4.5, slightly larger than 13DE2MI.| HF/OSDA |
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
| Productb | |||||||
| a The composition of the synthesis mixture is 0.5ROH·0.8SiO2·0.2GeO2·xHF·5.0H2O, R is OSDA prepared here and x is varied in the range 0.5 ≤ x ≤ 2.0. All syntheses were performed under rotation (60 rpm) at 160 °C for 7 days. OSDA abbreviation: 1E3MI, 1-ethyl-3-methylimidazolium; 13DEI, 1,3-diethylimidazolium; 1E23DMI, 1-ethyl-2,3-dimethylimidazolium; 2E134TMI, 2-ethyl-1,3,4-trimethylimidazolium; 13DE2MI, 1,3-diethyl-2-methylimidazolium; 123TEI, 1,2,3-triethylimidazolium; 123TE4MI, 1,2,3-triethyl-4-methylimidazolium. b The product appearing first is the major phase, and the product obtained in a trace amount is given in parentheses. A and D indicate amorphous and dense phases, respectively. | |||||||
| 1.0 | MFI + STW | TON + MFI | ITW | ITW | STW | PST-35 + DON | PST-35 + *STO |
| 2.0 | MFI + STW | TON + MFI | ITW | ITW + (PST-35) | STW | PST-35 | PST-35 + *STO |
| 3.0 | STW | TON | ITW | ITW + A | STW | PST-35 + (STW) | PST-35 + *STO |
| 4.0 | D | D | D + ITW | D | D | D | D |
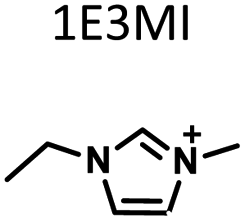
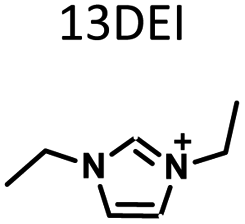
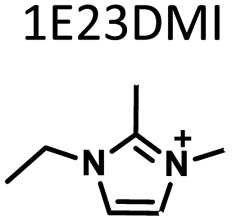
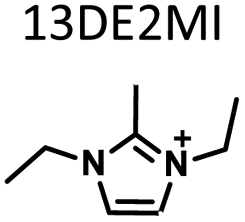
Table 1 also shows that the 123TEI-mediated synthesis in normal fluoride media (HF/OSDA = 1.0) gave a mixture of PST-35 and UTD-1F (DON), a 1D extra-large-pore zeolite.36 This suggests that F− concentration in the synthesis mixture containing 123TEI is crucial for obtaining pure PST-35, as evidenced by the synthesis results at HF/OSDA ratios (3.0–4.0). The molecular size and shape of the OSDA are another important factor governing the phase selectivity of the crystallisation, because the use of 1,2,3-triethyl-4-methylimidazolium (123TE4MI) with C/N = 5.0, again slightly larger than 123TEI, always yielded SSZ-31 (STO*), a 1D large-pore intergrowth zeolite,37 together with PST-35. On the other hand, this new zeolite was found to crystallise in its pure form using 123TEI as an OSDA and synthesis mixtures with Si/Ge ratios ranging from 2.0 to 7.0 (Fig. S1†). We also note no noticeable differences in the bulk Si/Ge ratio between the resulting product and its synthesis mixture. From now on, we will refer to PST-35 zeolites synthesised in this study as PST-5(n), where n is the bulk Si/Ge ratio determined by elemental analysis.
1H–13C CP MAS NMR spectroscopy shows that the 123TEI cations remain intact during PST-35 formation (Fig. S2†). In addition, although the as-synthesised PST-35(3.9) typically appears as prism-like crystals ca. 10 μm in length (Fig. S3†), we were able to collect the SXRD data of its hydrated form at the 6D beamline of the Pohang Acceleration Laboratory (PAL; Pohang, Korea) using synchrotron X-rays (λ = 0.95116 Å) and to solve the structure by direct methods: monoclinic, C2/m, a = 21.573(4) Å, b = 22.578(5) Å, c = 12.562(3) Å and β = 124.63(3)° (Table S1†). We next determined the occupancies of extraframework species in the as-synthesised, hydrated PST-35(3.9) from its unit cell composition |(C9H17N2)7.2F7.2(H2O)3.8|[Si63.7Ge16.3O160], determined by elemental and thermal analyses (Fig. S4†) and anisotropically refined all the framework and extraframework atoms, with final R1 and wR2 values of 0.0671 and 0.2217, respectively. Furthermore, a good match between the experimental and simulated PXRD patterns is observed (Fig. S5†). The final atomic positions and selected bond lengths and angles for the as-synthesised PST-35(3.9) can be found in Tables S2–S4.†
The structure of PST-35 has 10 crystallographically distinct T-sites, and its framework is built up of stt cages as a key composite building unit (CBU) (Fig. 1a). Two stt cages related by an inversion symmetry, which have an s4r in both the bottom and top sides, respectively, are connected to each other by sharing a 4-ring to form a larger 15-hedral ([45·58·62]) CBU (Fig. 1a and b). The pst-35 building chain is created by edge-sharing 15-hedral cages in a two-fold rotation symmetry arrangement (Fig. 1b and c). These pst-35 building chains are then linked in a nonjointed manner to generate the pst-35 building layer where 4- and 6-rings are formed between adjacent chains (Fig. 1d). The pst-35 layers are stacked through s4rs along the b axis in sequence AAmAAm…, where layer Am is mirrored from layer A along the same axis. Once this stacking completes, the entire PST-35 structure forms and has d4r cages as pillars (Fig. 1e). The PST-35 was calculated to have a framework density of 15.8 T-atoms per 1000 Å3, even lower than the value (16.5) of MCM-22 (MWW) with large cylindrical ([512·614·106]) supercages.11
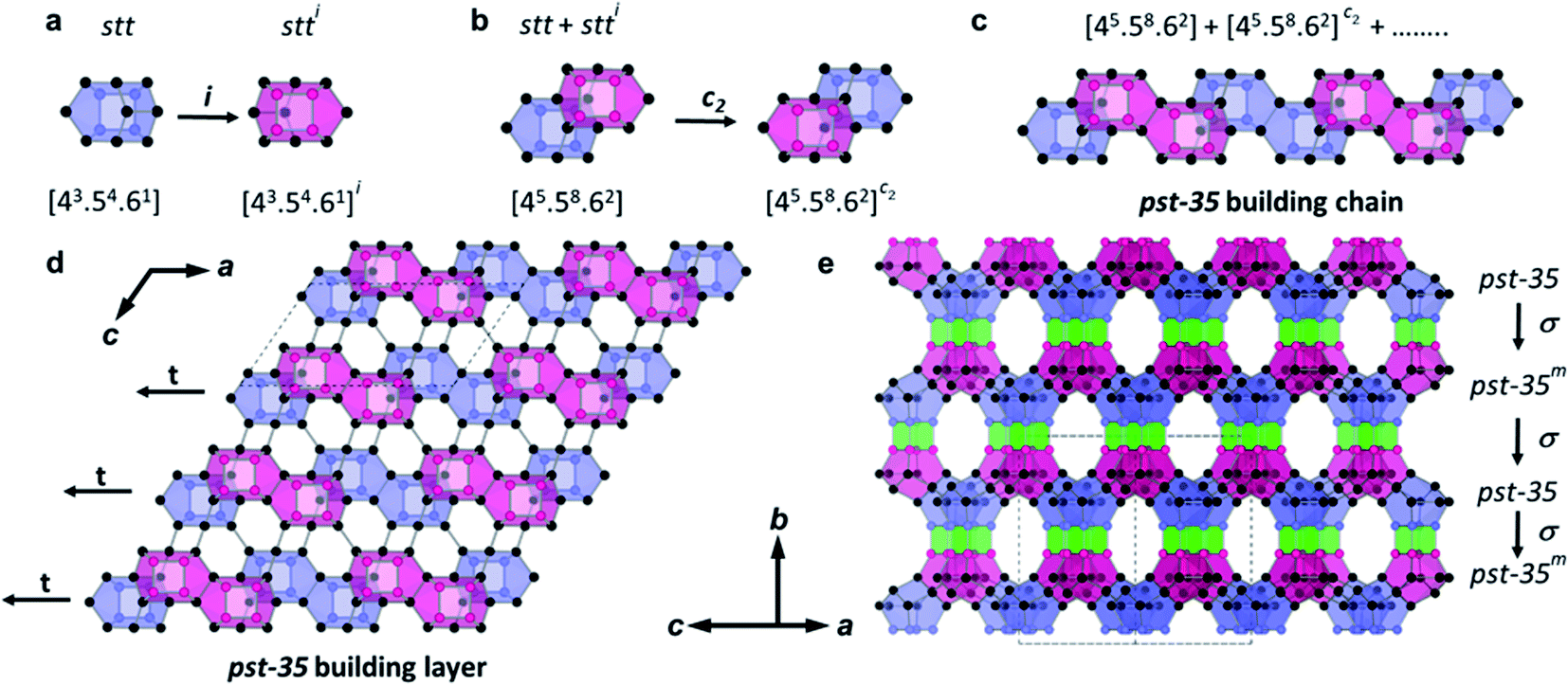 | ||
| Fig. 1 (a) Two 8-hedral stt ([43·54·6]) cages related by an inversion symmetry. While the T-atoms of s4r units in their bottom and top sides are indicated by blue and pink balls, respectively, the rest of them are shown as black balls. All faces in the stt cage with bottom and top s4r units are colored transparent blue and pink, respectively. Bridging O atoms have been omitted for clarity. (b) Two 15-hedral ([45·58·62]) cages, each of which is formed by face-sharing two stt cages related by a 2-fold rotation symmetry. (c) PST-35 building chain generated by edge-sharing 15-hedral cages. (d) PST-35 building layer viewed along the b axis. Two adjacent PST-35 building chains are nonjointly connected to form the 4- and 6-rings. (e) PST-35 structure viewed so as to show the 10-ring pore windows. The building layers are stacked in sequence AAmAAm… along the b axis, where the superscript m indicates a mirror symmetry along the b axis. Faces of the d4r cages formed by connection of adjacent layers are colored transparent green. | ||
Interconnection of pst-35 layers yields a 2D pore system, which consists of mutually intersecting 8- and 10-ring channels (Fig. 2). The zig-zag 10-ring (4.6 × 6.7 Å) channel is made up of two novel orthorhombic-shaped cages, labelled ptf-1 ([48·58·68·82·102]; 13.7 × 15.0 × 10.1 Å) and ptf-2 ([46·54·64·82·102]; 11.0 × 13.9 × 10.2 Å), consisting of 52 and 36 T-atoms, respectively, which are arranged in a strictly alternating fashion. On the other hand, the two types of 8-ring channels present in the ptf-1 and ptf-2 cages are characterised by very narrow pore dimensions (6.3 × 2.3 and 6.3 × 1.3 Å, respectively; Fig. 2 and S6†). Therefore, the sorption dimensionality (molecular cross section > 3.4 Å) of PST-35 is one.11 It is worth noting that the “modified” natural tiling structure of PST-35, where the tiles can be split at non-strong rings with negative curvature, is constructed from two t-cub ([46]; equivalent to the d4r), four t-ifw-1 ([43·54·6]; identical to the stt cage in Fig. 1a), two t-kdk ([4·52·82]), one t-ptf-1 ([48·58·68·82·102]) and one t-ptf-2 ([46·54·64·82·102]) tiles, as shown in Fig. 2. Here, the 18 T-atom ([425482]) cavity is formed by a merger of two t-kdk tiles.38 Also, if the rules for natural tiling were strictly applied, four t-ifw-1 tiles (with a non-strong 6-ring) and one ptf-1 tile would then be merged to give a significantly larger ([4205246482102]) tile.
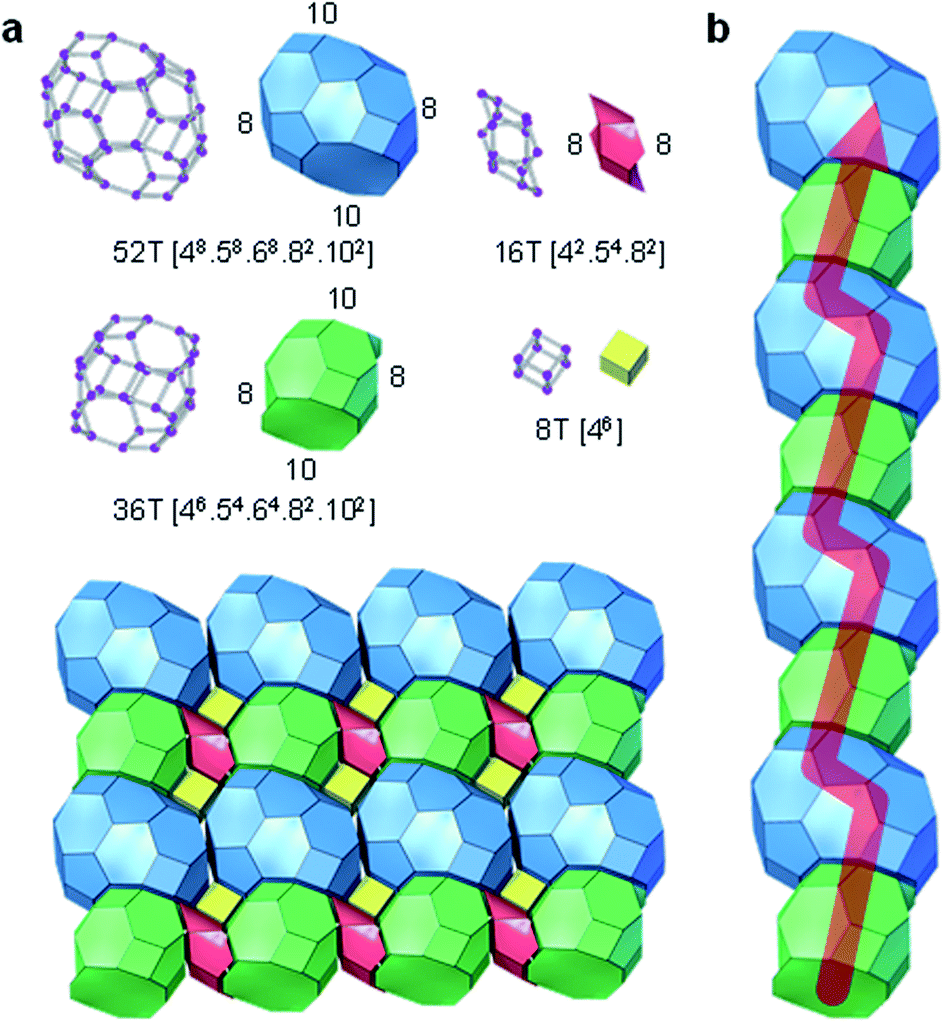 | ||
| Fig. 2 (a) The tiling presentation of the 2D pore structure of PST-35 and (b) its zig-zag 10-ring channel consisting of strictly alternating 52 T-atom (ptf-1 ([48586882102])) and 36 T-atom (ptf-2 ([46546482102])) cages. | ||
The SXRD refined structure of the as-synthesised PST-35(3.9) reveals the presence of two and one 123TEI ions within the ptf-1 and ptf-2 cages, respectively (Fig. S7†). Framework stabilization energy calculations based on these locations gave a value of −11.3 kJ molSi−1. However, the replacement of 123TEI with slightly smaller and larger OSDAs (i.e., 13DE2MI and 123TE4MI, respectively) showed comparably higher stabilization energies of −10.6 and −5.3 kJ molSi−1, respectively (Fig. S8†). This suggests that 123TEI is a better OSDA for the formation of PST-35 than the latter two organocations, in agreement with the synthesis results in Table 1.
The 19F MAS NMR spectra of three as-synthesised PST-35 zeolites with different Ge contents are compared in Fig. S9.† The spectrum of PST-35(2.2) with the largest Ge content shows one prominent resonance at −5 ppm due to the F− ions occluded in the small 4Si4Ge-d4r cages.39–41 However, while the spectrum of PST-35(3.9) is characterised by another two but weaker resonances at 16 and 34 ppm, assignable to the F− ions in the 6Si2Ge- and 8Si-d4rs, respectively, that of PST-35(6.6) with the smallest Ge content exhibits another sharp resonance at −125 ppm, attributable to the free F− ions encapsulated within the considerably larger ptf-1 and/or ptf-2 cages. It should be noted here that alternative assignments of F− encapsulation within d4rs have also been proposed.42,43 Therefore, the preference of Ge substitution into the d4r cages of PST-35 does not appear to be high compared to the so-called ADORable germanosilicate zeolites such as IM-12 (UTL).44 This can be further supported by the fact that all the 29Si MAS NMR spectra of the as-synthesised PST-35(2.2), PST-35(3.9) and PST-35(6.6) show a featureless line shape (Fig. S10†).
The as-calcined PST-35(3.9) has a micropore volume of 0.17 cm3 g−1. However, its exposure to ambient moisture led to a structural collapse (Fig. S11†), mainly due to the cleavage of Ge–O bonds, as previously reported,45 and this was also observed for PST-35(6.6) with a lower Ge content (Fig. S12†). In addition, although we were able to introduce a small of amount of Al into the framework of PST-35, the calcined form of the resulting material was not stable under ambient conditions (Fig. S13 and S14†). Furthermore, none of our attempts to synthesise new zeolite structures by applying the assembly–disassembly–organization–reassembly (ADOR) concept46,47 to PST-35 were successful, probably owing to the more random nature of Ge distribution in the d4r cages of PST-35. Indeed, the average T–O bond length (1.65 Å) of d4rs in the as-synthesised PST-35(3.9) is far shorter than the lengths (1.73 and 1.71 Å, respectively) in ADORable zeolites IM-12 and IM-17 (UOV).46,47 On the other hand, the PST-35 structure can be theoretically manipulated to create two hypothetical zeolites by removing s4rs and d4rs in the ac plane, denoted PST-35h1 and PST-35h2, respectively (Table S5 and Fig. S15–S17†). Both structures with pure-silica composition were calculated to have framework energies relative to α-quartz lower than 30 kJ molSi−1 and to meet all local interatomic distance criteria (Table S6†),32 like in the PST-35 case, revealing their high synthetic feasibility. It is interesting to note that PST-35h2 is isostructural with the 12-layer ABC-6 structure with stacking sequence ABABCACABCBC and thus belongs to the clathrasil family.9,10
Conclusions
In summary, a new cage-based medium-pore zeolite, denoted PST-35, has been synthesised by combining the excess fluoride approach and the strong directing effect of Ge on d4r formation. The structure of PST-35, solved using synchrotron single-crystal X-ray diffraction data of its as-synthesised form, can be described as a zig-zag 10-ring channel system consisting of one large and one small orthorhombic cage in a strictly alternating manner. Thus, to our knowledge, PTS-35 is the first medium-pore zeolite with two accessible interconnected cages. We have also generated two chemically synthesisable hypothetical structures by removing the s4rs and d4rs in PST-35 along the ac plane, respectively. We anticipate that the excess fluoride approach will be useful for finding novel industrially relevant zeolite structures.Data availability
Crystallographic data for PST-35 structure has been deposited at the CCDC under 2173296 and included as part of the ESI.†Author contributions
Kingsley Christian Kemp: conceptualization, formal analysis, investigation, methodology. Wanuk Choi: formal analysis, investigation. Donghui Jo: formal analysis, methodology. Sung Hwan Park: formal analysis, investigation. Suk Bong Hong: conceptualization, methodology, project administration, funding acquisition. All authors co-wrote the paper, which involved writing the original draft as well as review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Creative Research Initiative Program (2012R1A3A-2048833) through the National Research Foundation of Korea. We acknowledge PAL for beam time at beamlines 5A (H. H. Lee), 6D (T. J. Shin), and 9B (D. Ahn). PAL is supported by MSIP and POSTECH. We also thank I.-C. Hwang (POSTECH) for helpful discussion.Notes and references
- W. Vermeiren and J.-P. Gilson, Top. Catal., 2009, 52, 1131 CrossRef CAS.
- Q. Zhang, J. Yu and A. Corma, Adv. Mater., 2020, 32, 2002927 CrossRef PubMed.
- K. C. Kemp, J. G. Min, H. J. Choi and S. B. Hong, in New Developments in Adsorption/Separation of Small Molecules by Zeolites, Structure and Bonding, ed. S. Valencia and F. Rey, Springer, Cham, 1st edn, 2020, ch. 1, vol. 184, pp. 1–30 Search PubMed.
- M. Moliner, C. Martínez and A. Corma, Chem. Mater., 2014, 26, 246 CrossRef CAS.
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156 CrossRef CAS.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880 CrossRef CAS PubMed.
- M. Moliner, C. Martínez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 3560 CrossRef CAS PubMed.
- J. Shin, D. Jo and S. B. Hong, Acc. Chem. Res., 2019, 52, 1419 CrossRef CAS PubMed.
- A Database of Hypothetical Zeolite Structures, accessed June 29, 2022, http://www.hypotheticalzeolites.net/.
- Predicted Crystallography Open Database, accessed June 29, 2022, http://www.crystallography.net/pcod/.
- Database of Zeolite Structures, accessed June 29, 2022, http://www.iza-structure.org/databases/.
- X. Bu, P. Feng and G. D. Stucky, Science, 1997, 278, 2080 CrossRef CAS PubMed.
- M. Estermann, L. B. McCusker, C. Baerlocher, A. Merrouche and H. A. Kessler, Nature, 1991, 352, 320 CrossRef CAS.
- D. Jo, G. T. Park, J. Shin and S. B. Hong, Angew. Chem., Int. Ed., 2018, 57, 2199 CrossRef CAS PubMed.
- D. Jo and S. B. Hong, Angew. Chem., Int. Ed., 2019, 58, 13845 CrossRef CAS PubMed.
- S. van Donk, J. H. Bitter and K. P. de Jong, Appl. Catal., A, 2001, 212, 97 CrossRef CAS.
- D. Jo, J. Zhao, J. Cho, J. H. Lee, Y. Liu, C.-J. Liu, X. Zou and S. B. Hong, Angew. Chem., Int. Ed., 2020, 59, 17691 CrossRef CAS PubMed.
- A. Corma, M. J. Díaz-Cabañas, J. L. Jorda, F. Rey, G. Sastre and K. G. Strohmaier, J. Am. Chem. Soc., 2008, 130, 16482 CrossRef CAS PubMed.
- M. O. Cichocka, Y. Lorgouilloux, S. Smeets, J. Su, W. Wan, P. Caullet, N. Bats, L. B. McCusker, J. L. Paillaud and X. D. Zou, Cryst. Growth Des., 2018, 18, 2441 CrossRef CAS.
- X. Liu, W. Mao, J. Jiang, X. Lu, M. Peng, H. Xu, L. Han, S. Che and P. Wu, Chem.–Eur. J., 2019, 25, 4520 CrossRef CAS PubMed.
- M. A. Camblor, M.-J. Díaz-Cabanas, J. Perez-Pariente, S. J. Teat, W. Clegg, I. J. Shannon, P. Lightfoot, P. A. Wright and R. E. Morris, Angew. Chem., Int. Ed., 1998, 37, 2122 CrossRef CAS PubMed.
- R. Simancas, J. L. Jorda, F. Rey, A. Corma, A. Cantin, I. Peral and C. A. Popescu, J. Am. Chem. Soc., 2014, 136, 3342 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Molecular Crystallography Part A, ed. C. W. Carter Jr, Academic Press, 1st edn, 1997, ch. 20, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- R. Pophale, F. Daeyaert and M. W. Deem, J. Mater. Chem. A, 2013, 1, 6750 RSC.
- S. L. Mayo, B. D. Olafson and W. Goddard, J. Phys. Chem., 1990, 94, 8897 CrossRef CAS.
- Materials Studio 7.0, Accelrys Inc., San Diego, CA, 2013 Search PubMed.
- K.-P. Schröder, J. Sauer, M. Leslie, C. R. A. Catlow and J. M. Thomas, Chem. Phys. Lett., 1992, 188, 320 CrossRef.
- J. D. Gale and A. L. Rohl, Mol. Simul., 2003, 29, 291 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- M. D. Foster, A. Simperler, R. G. Bell, O. D. Friedrichs, F. A. A. Paz and J. Klinowski, Nat. Mater., 2004, 3, 234 CrossRef CAS PubMed.
- Y. Li, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2013, 52, 1673 CrossRef CAS PubMed.
- R. H. Archer, S. I. Zones and M. E. Davis, Microporous Mesoporous Mater., 2010, 130, 255 CrossRef CAS.
- A. Rojas and M. A. Camblor, Dalton Trans., 2014, 43, 10760 RSC.
- Y. M. Variani, A. Rojas, L. Gómez-Hortigüela and S. B. C. Pergher, New J. Chem., 2016, 40, 7968 RSC.
- T. Wessels, C. Baerlocher, L. B. McCusker and E. J. Creyghton, J. Am. Chem. Soc., 1999, 121, 6242 CrossRef CAS.
- R. F. Lobo, M. Tsapatsis, C. C. Freyhardt, I. Chan, C. Y. Chen, S. I. Zones and M. E. Davis, J. Am. Chem. Soc., 1997, 119, 3732 CrossRef CAS.
- V. A. Blatov, O. Delgado-Friedrichs, M. O'Keeffe and D. M. Proserpio, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 63, 418 CrossRef CAS PubMed.
- T. Blasco, A. Corma, M. J. Diaz-Cabanas, F. Rey, J. A. Vidal-Moya and C. M. Zicovich-Wilson, J. Phys. Chem. B, 2002, 106, 2634 CrossRef CAS.
- A. Pulido, G. Sastre and A. Corma, ChemPhysChem, 2006, 7, 1092 CrossRef CAS PubMed.
- X. Liu, U. Ravon, F. Bosselet, G. Bergeret and A. Tuel, Chem. Mater., 2012, 24, 3016 CrossRef CAS.
- N. Kasian, A. Tuel, E. Verheyen, C. E. A. Kirschhock, F. Taulelle and J. A. Martens, Chem. Mater., 2014, 26, 5556 CrossRef CAS.
- S. P. Gramatikov, P. St. Petkov and G. N. Vayssilov, Inorg. Chem. Front., 2022, 9, 3747 RSC.
- J.-L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990 CrossRef CAS PubMed.
- L. A. Villaescusa and M. A. Camblor, Chem. Mater., 2016, 28, 7544 CrossRef CAS.
- W. J. Roth, P. Nachtigall, R. E. Morris, P. S. Wheatley, V. R. Seymour, S. E. Ashbrook, P. Chlubná, L. Grajciar, M. Položij, A. Zukal, O. Shvets and J. A. Čejka, Nat. Chem., 2013, 5, 628 CrossRef CAS PubMed.
- V. Kasneryk, M. Shamzhy, M. Opanasenko, P. S. Wheatley, S. A. Morris, S. E. Russell, A. Mayoral, M. Trachta, J. Čejka and R. E. Morris, Angew. Chem., Int. Ed., 2017, 56, 4324 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: OSDA syntheses, structural characterization data of PST-35 and hypothetical structures from PST-35. CCDC 2173296. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03628b |
| This journal is © The Royal Society of Chemistry 2022 |