
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed intramolecular Heck dearomative gem-difluorovinylation of indoles†
Gang
Wang
,
Wenqi
Li
,
Tianxiang
Liu
,
Yonghong
Zhang
 ,
Bin
Wang
,
Fei
Xue
,
Weiwei
Jin
,
Caiyan
Ma
,
Yu
Xia
* and
Chenjiang
Liu
*
,
Bin
Wang
,
Fei
Xue
,
Weiwei
Jin
,
Caiyan
Ma
,
Yu
Xia
* and
Chenjiang
Liu
*
Urumqi Key Laboratory of Green Catalysis and Synthesis Technology, Key Laboratory of Oil and Gas Fine Chemicals, Ministry of Education & Xinjiang Uygur Autonomous Region, State Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, College of Chemistry, Xinjiang University, Urumqi 830017, P. R. China. E-mail: 18509487642@163.com; pxylcj@126.com
First published on 13th September 2022
Abstract
A palladium-catalyzed dearomative reaction of indoles has been developed through a domino Heck/gem-difluorovinylation sequence. By taking advantage of a difluorocarbene precursor (ClCF2COONa), the palladium difluorocarbene ([Pd]![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) species was formed smoothly. Then, a migratory insertion/β-H elimination process enabled access to polycyclic indolines containing 1,1-difluoroethylene units in acceptable yields with a broad substrate scope, which also showed dearomative gem-difluorovinylation for the first time. Remarkably, the superb diversified transformations allowed the product to install various functional groups.
CF2) species was formed smoothly. Then, a migratory insertion/β-H elimination process enabled access to polycyclic indolines containing 1,1-difluoroethylene units in acceptable yields with a broad substrate scope, which also showed dearomative gem-difluorovinylation for the first time. Remarkably, the superb diversified transformations allowed the product to install various functional groups.
Introduction
gem-Difluoroalkenes are widely used in the fields of pharmaceuticals, agrochemicals, materials and fine chemicals (Fig. 1A).1–4 Moreover, a number of valuable compounds can be converted by the transformations of gem-difluoroalkenes with radicals, carbon-metal species, and nucleophiles.5–7 So far, a range of methods have been developed to prepare gem-difluoroalkene derivatives.7–11 Traditionally, the well-known Julia-Kocienski12–14 and Wittig15–19 reactions are confined to aldehyde and ketone substrates. In recent years, the defluorinative functionalization of trifluoromethyl olefins has been explored in detail,20–22 but the synthesis of trifluoromethyl olefins is cumbersome.23 Besides, gem-difluoroalkenes can also be acquired from diazo compounds,24–27 which have potential safety hazards. Consequently, it is necessary to develop a new strategy to get gem-difluoroalkenes.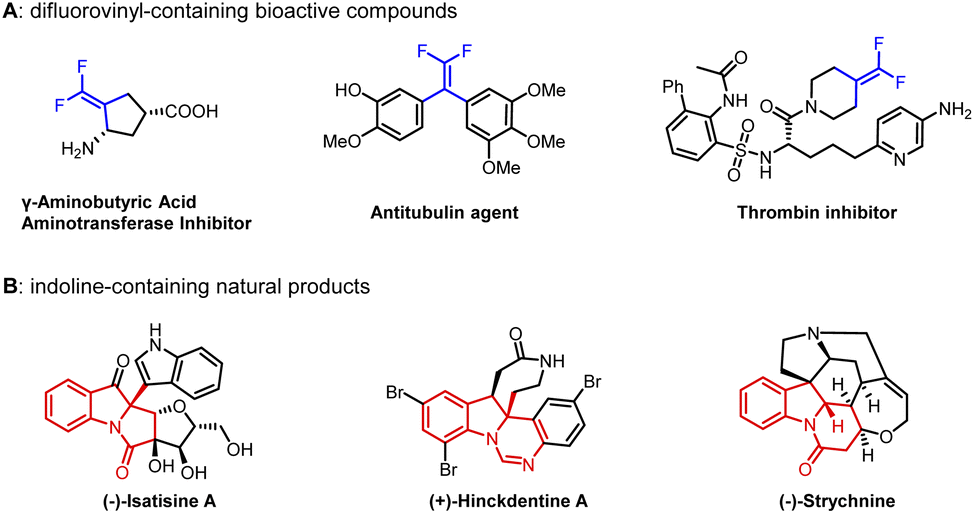 | ||
| Fig. 1 Molecules containing difluorovinyl and indoline units. | ||
Transition-metal-catalyzed cross-coupling reactions with carbenes have been extensively studied.28 CC double bonds can be constructed smoothly through general procedures of carbene migratory insertion and β-H elimination.29 Studies mainly focus on nonfluorinated carbenes, and β-H elimination involving difluorocarbene has not been reported. Even the conversion of metal difluorocarbene ([M]CF2) is a massive challenge due to limited reaction types.30 In 2015, Zhang and coworkers reported the first metal difluorocarbene coupling (MeDiC) reaction for synthesizing organofluorine compounds via palladium catalysis.31 Thereafter, several successful transformations of [Pd]CF2 were reported,31–37 but most of the coupling reagents are limited to nucleophilic arylboronic acids and esters (Scheme 1a).31–35 Furthermore, in the only research involving the coupling of electrophilic aryl halides, the hydrolysis of [Pd]CF2 is inevitable (Scheme 1b).38 On this foundation, we expect to inhibit the hydrolysis by the β-H elimination process to prepare gem-difluoroalkenes and expand the coupling of aryl halides with difluorocarbene ulteriorly. Meanwhile, considering the operability as a crystalline solid and low cost, ClCF2COONa is chosen as the difluorocarbene precursor.39
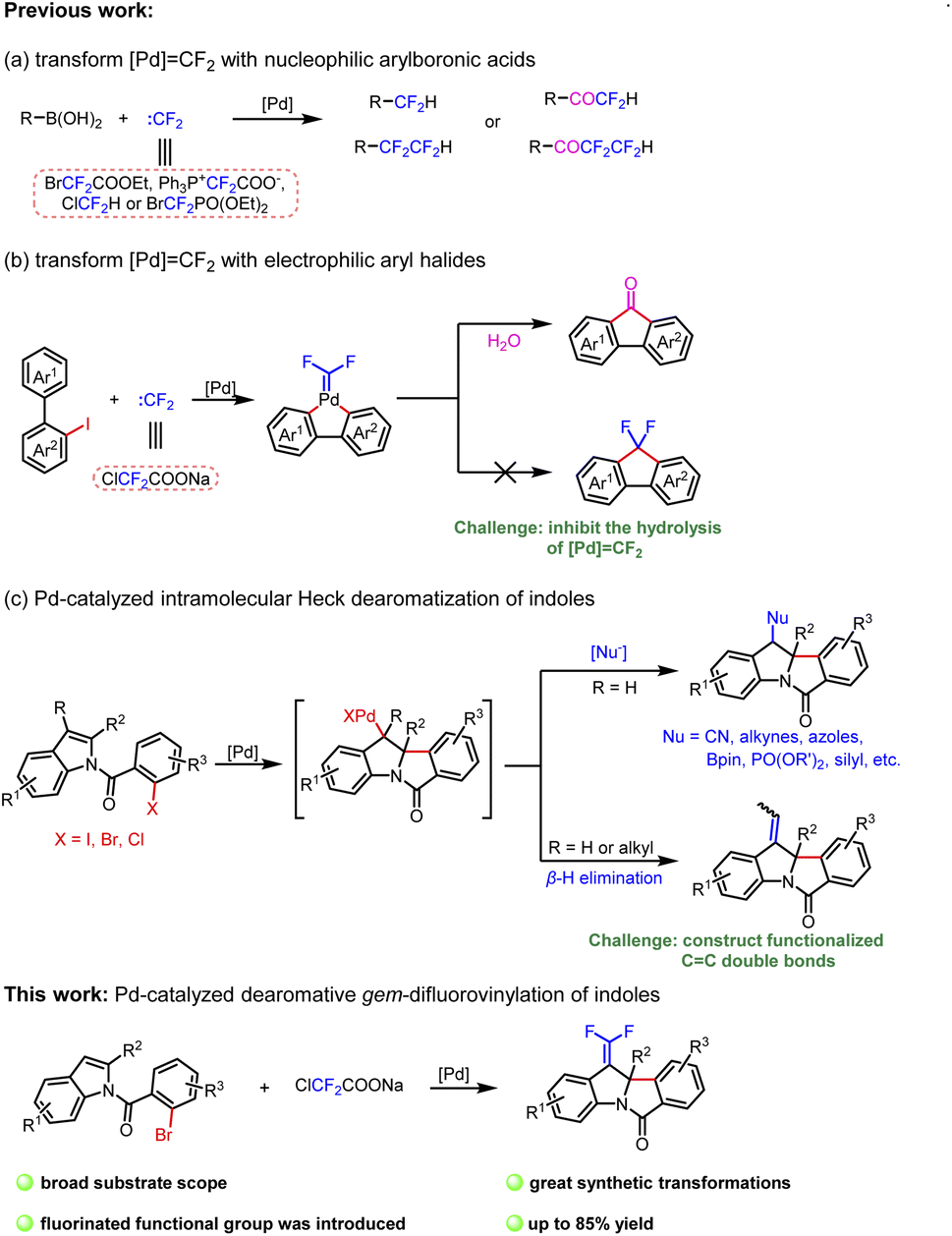 | ||
| Scheme 1 Pd-catalyzed transformations of :CF2 and dearomatization of indoles. | ||
The assembly of polycyclic indoline derivatives remains one of the most interesting subjects in organic synthesis40–45 due to the fact that many natural products contain constitutional units (Fig. 1B).46–49 At present, the production of polycyclic indoline scaffolds has been established by palladium-catalyzed intramolecular Heck dearomatization of indoles (Scheme 1c).50–68 On the one hand, 1,2-difunctionalization of indoles is achieved when the resulting benzyl-Pd species are captured with diverse nucleophiles that can effectively form various C–X (H, C, N, O, P, B, and Si) bonds. On the other hand, the reactions can be terminated by β-H elimination to construct CC double bonds.48,60,64–66 However, limited precursors restrict the installation of functionalized CC double bonds. Inspired by our first attempt at dearomatization of indoles,67 we present herein an innovative idea for synthesizing gem-difluoroalkenes from N-(2-bromobenzoyl)indoles and ClCF2COONa. Remarkably, it is the first report of dearomative gem-difluorovinylation.
Results and discussion
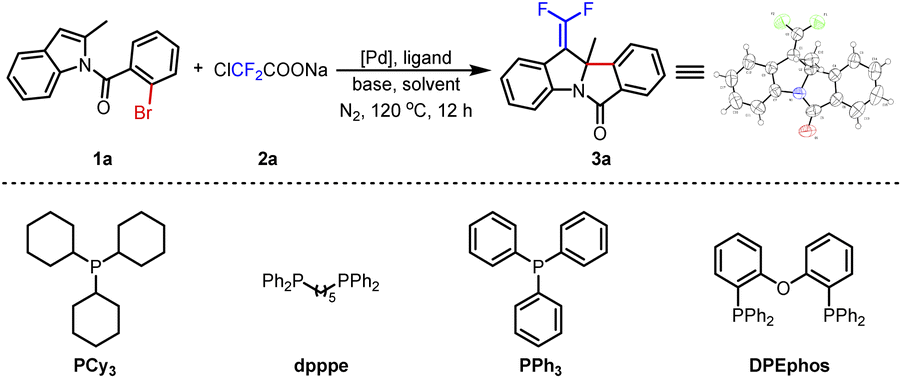
With this thinking in mind, N-acyl indole 1a and ClCF2COONa were selected as model substrates in the palladium/ligand catalytic system to explore the optimal conditions (Table 1). As expected, when Pd(OAc)2/PCy3 was used as a catalyst with K2CO3 in 1,2-dichloroethane (DCE) at 120 °C for 12 h, the process of dearomative gem-difluorovinylation was carried out smoothly to afford the expectant product 3a in 45% yield (Table 1, entry 1). The structure of 3a was confirmed by X-ray crystallographic analysis (see the ESI†). Then, K3PO4 and NEt3 as bases were tested, but they were unfavorable for the reaction compared to K2CO3 (Table 1, entries 2 and 3). And several ligands such as dpppe, PPh3, and DPEphos were studied, and the results showed that DPEphos was a suitable ligand to get 3a in 60% yield (Table 1, entries 4–6). Substituting Pd(TFA)2 for Pd(OAc)2, the yield of the target product decreased to 42% (Table 1, entry 7). When PdCl2 was used as the catalyst, 3a was not generated (Table 1, entry 8). Only a trace amount of 3a was obtained by changing the solvent to 1,4-dioxane or toluene (Table 1, entries 9 and 10). We reduced the amount of ligand to 12 mol%, and the yield of the final product was 63% (Table 1, entry 11). To our delight, when 4 Å molecular sieves (4 Å MS) were added to the reaction, the yield of the desired product improved to 73% (Table 1, entry 12). On this foundation, the amount of DCE was raised to 3 mL, and the yield of product 3a further increased to 79% (Table 1, entry 13). Afterwards, difluorocarbene precursors such as BrCF2TMS and ClCF2COOEt were investigated with unsatisfactory results (Table 1, entries 14 and 15). The result was poor when the amount of Pd(OAc)2 was lowered to 5 mol% (Table 1, entry 16). Finally, we determined that the best reaction conditions were to use Pd(OAc)2 (10 mol%), DPEphos (12 mol%), K2CO3 (3.0 equiv.), and 4 Å MS (100 mg) in DCE (3 mL) under N2 at 120 °C for 12 h.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | Ligand | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (2.0 equiv.), [Pd] (10 mol%), ligand (20 mol%), base (3.0 equiv.) and solvent (2.0 mL) at 120 °C for 12 h under a N2 atmosphere. b Isolated yield. c DPEphos (12 mol%). d 4 Å MS (100 mg) was added. e DCE (3.0 mL). f 2a changed by BrCF2TMS. g 2a changed by ClCF2COOEt. h Pd(OAc)2 (5 mol%). NR = no reaction. | |||||
| 1 | Pd(OAc)2 | PCy3 | K2CO3 | DCE | 45 |
| 2 | Pd(OAc)2 | PCy3 | K3PO4 | DCE | 20 |
| 3 | Pd(OAc)2 | PCy3 | NEt3 | DCE | Trace |
| 4 | Pd(OAc)2 | dpppe | K2CO3 | DCE | 39 |
| 5 | Pd(OAc)2 | PPh3 | K2CO3 | DCE | 26 |
| 6 | Pd(OAc)2 | DPEphos | K2CO3 | DCE | 60 |
| 7 | Pd(TFA)2 | DPEphos | K2CO3 | DCE | 42 |
| 8 | PdCl2 | DPEphos | K2CO3 | DCE | NR |
| 9 | Pd(OAc)2 | DPEphos | K2CO3 | 1,4-Dioxane | Trace |
| 10 | Pd(OAc)2 | DPEphos | K2CO3 | Toluene | Trace |
| 11c | Pd(OAc)2 | DPEphos | K2CO3 | DCE | 63 |
| 12c,d | Pd(OAc)2 | DPEphos | K2CO3 | DCE | 73 |
| 13 , , | Pd(OAc) 2 | DPEphos | K 2 CO 3 | DCE | 79 |
| 14c,d,e,f | Pd(OAc)2 | DPEphos | K2CO3 | DCE | NR |
| 15c,d,e,g | Pd(OAc)2 | DPEphos | K2CO3 | DCE | 20 |
| 16c,d,e,h | Pd(OAc)2 | DPEphos | K2CO3 | DCE | Trace |
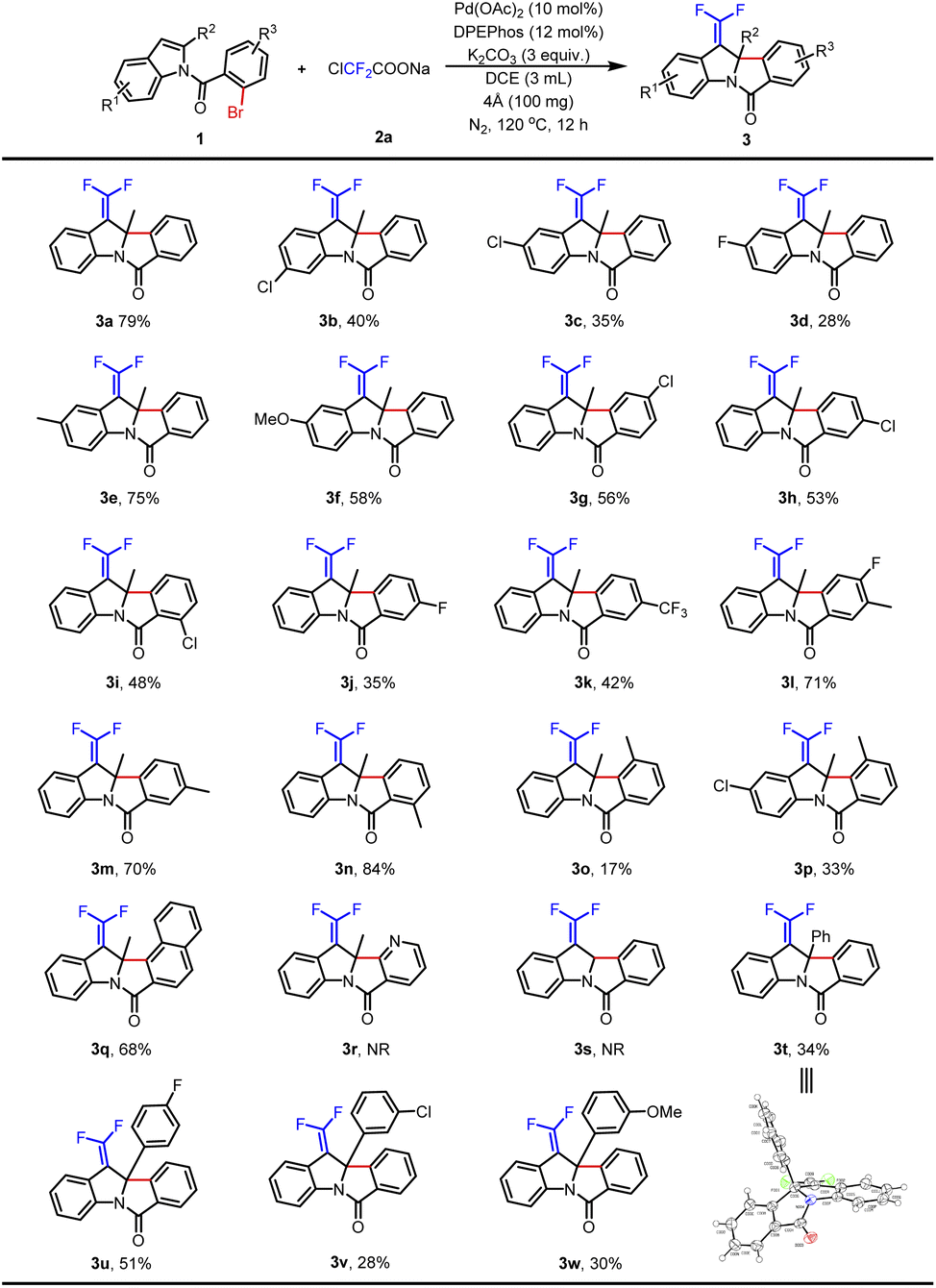
The optimal reaction conditions for the synthesis of 3a were evaluated by using indole derivatives containing different substituents and ClCF2COONa. As shown in Table 2, electron-donating and electron-withdrawing groups on indoles' C5 and C6 positions could afford the desired products in 28–79% yields (3a–3f), and electron-donating groups helped to get better results (3e–3f). Compared with the electron-withdrawing groups that provided related products in 35–56% yields (3g–3k), the electron-donating group on N-benzoyl was more conducive to the reaction in 70–84% yields (3l–3n). However, due to the influence of steric hindrance, the substrate with a methyl group at the C3 position of N-benzoyl provided the corresponding product 3o in only 17% yield. When both indole and N-benzoyl were substituted, the desired product 3p was obtained in 33% yield. A substrate containing a naphthalene ring was tried and led to 3q in 68% yield, but heteroaromatic bromine (pyridine-containing) and non-substituted indole (R2 = H) failed (3r–3s). In the cases of phenyl and substituted phenyl (4-fluorophenyl, 3-chlorophenyl, and 3-methoxyphenyl) attached at the C2 position of indoles, the protocol went smoothly with 28–51% yields (3t–3w). The structure of 3t was confirmed by X-ray crystallographic analysis (see the ESI†).
| a Reaction conditions: 1 (0.2 mmol), 2a (2.0 equiv.), Pd(OAc)2 (10 mol%), DPEPhos (12 mol%), K2CO3 (3.0 equiv.), 4 Å MS (100 mg) and DCE (3.0 mL) at 120 °C for 12 h under a N2 atmosphere. Isolated yield. NR = no reaction. |
|---|
|
Furthermore, the substrate scope was extended with a methoxyl group substituent at the N-benzoyl position (Table 3). Indole without a substituent on the benzene ring produced 3x in 81% yield. With electron-donating groups (Me- and MeO-) at the C5 position of indoles, the desired products 3y–3z were obtained in 69–85% yields. In contrast, the corresponding products were obtained in lower yields when halogen groups (F- and Cl-) were assembled at the same position (3aa–3ab). And then, 7-azaindole was tested but failed to obtain product 3ac. Next, the desired product 3ad was smoothly produced in 72% yield by changing the methoxy group at the C4 position of N-benzoyl. In addition, a dimethoxy group substituted substrate was also tolerated with this procedure and provided the product 3ae in 64% yield. Finally, C2-substituted indoles were studied. Although 2-ethyl indole and 2-phenyl indole could afford the anticipated products 3af–3ag in 58–74% yields, cyano and ester groups were the failed choices (3ah–3ai). In short, installing MeO- on the N-benzoyl was an excellent choice to improve the yields.
| a Reaction conditions: 1 (0.2 mmol), 2a (2.0 equiv.), Pd(OAc)2 (10 mol%), DPEPhos (12 mol%), K2CO3 (3.0 equiv.), 4 Å MS (100 mg) and DCE (3.0 mL) at 120 °C for 12 h under a N2 atmosphere. Isolated yield. NR = no reaction. |
|---|
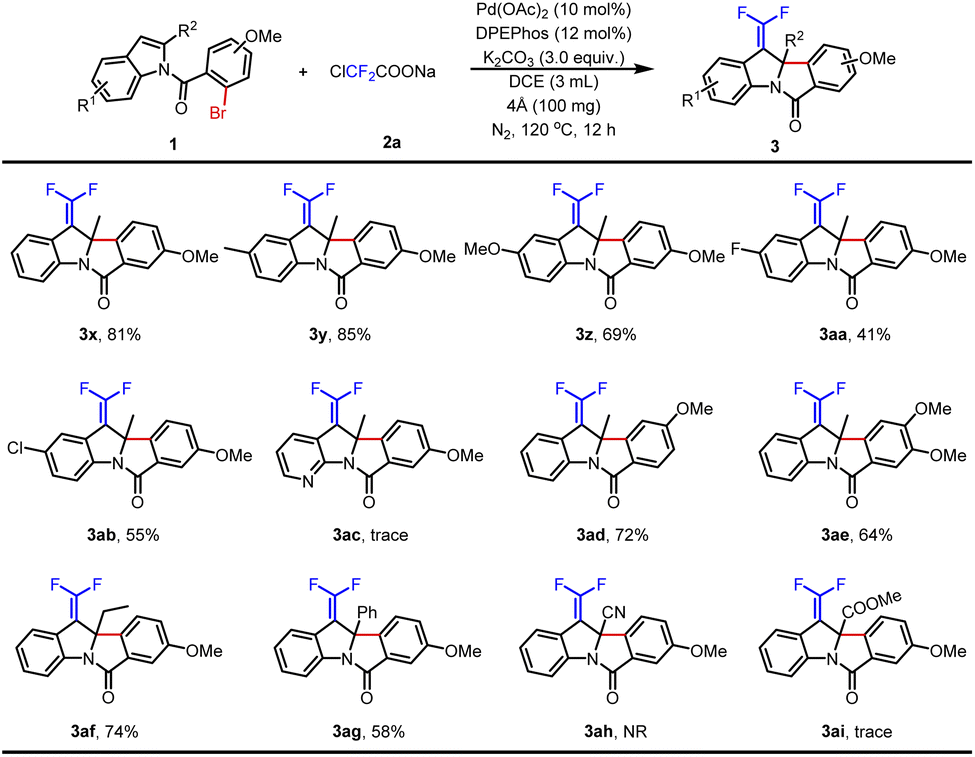
|
The synthetic transformations of product 3a indicated the practicability of the method (Scheme 2). gem-Difluorovinyl was successfully transformed into eight useful functional groups. Firstly, α-difluoro(thio)methylated alcohol 4a was obtained in 66% yield by a three-component reaction between compound 3a, p-toluenethiol and oxygen under mild conditions. And the coupling of 3a with p-toluenethiol resulted in α,α-difluoroalkylthioether 4b in 85% yield in dry DCE at 80 °C for 15 min. When 3a was treated with trimethylsilyl cyanide and imidazole, the nucleophilic vinylic substitution reaction (SNV) worked well and afforded the corresponding products 4c and 4d in good yields. Moreover, the cyclization of 3a with benzoyl hydrazine was explored, thus accessing unsymmetrical 2,5-disubstituted 1,3,4-oxadiazole in 79% yield with the assistance of Cs2CO3 (4e). Besides, the fluoro-functionalization reactions of 3a with selectfluor and O-nucleophiles were conducted smoothly in acetonitrile. Therefore, α-CF3 derivatives 4f and 4g could be synthesized in ideal yields. Remarkably, in the presence of potassium tert-butoxide and water, 3a has successfully worked with dibutylamine to provide arylacetamide 4h in 43% yield. And we confirmed the structure of 4h by X-ray crystallographic analysis (see the ESI†).
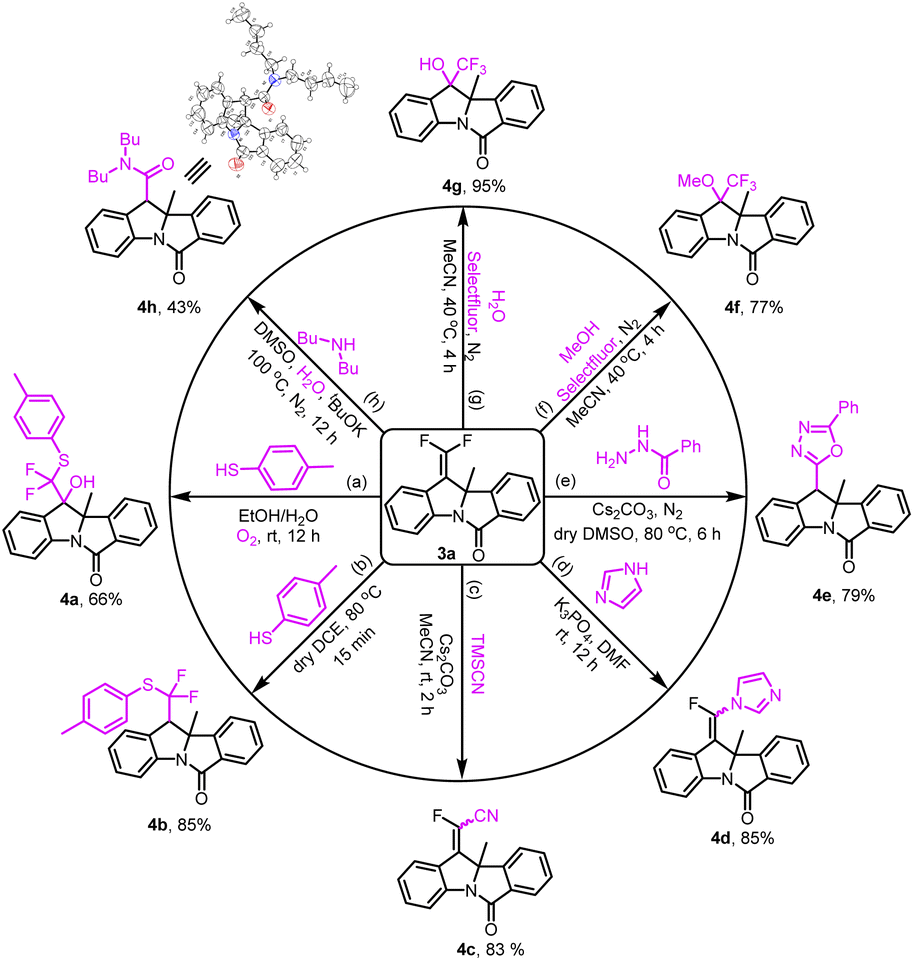 | ||
Scheme 2 Synthetic transformations of 3a: (a) 3a (0.2 mmol), 4-methylbenzenethiol (0.4 mmol), and EtOH/H2O (2.0 mL, v/v = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at rt for 12 h under an O2 atmosphere; (b) 3a (0.2 mmol), 4-methylbenzenethiol (0.24 mmol), and dry DCE (40 μL) at 80 °C for 15 min; (c) 3a (0.3 mmol), Cs2CO3 (0.03 mmol), TMSCN (0.9 mmol), and dry MeCN (1.0 mL) at rt for 2 h; (d) 3a (0.6 mmol), K3PO4 (1 mmol), imidazole (0.5 mmol), and DMF (1 mL) at rt for 12 h; (e) 3a (0.2 mmol), benzoyl hydrazide (0.24 mmol), Cs2CO3 (0.4 mmol), and dry DMSO (1 mL) at 80 °C for 6 h under a N2 atmosphere; (f) 3a (0.2 mmol), selectfluor (0.3 mmol), dry MeOH (1 mmol), and dry CH3CN (0.8 mL) at 40 °C for 4 h under N2 atmosphere; (g) 3a (0.2 mmol), selectfluor (0.3 mmol), H2O (1.6 mmol), and dry CH3CN (0.8 mL) at 40 °C for 4 h under a N2 atmosphere; (h) 3a (0.2 mmol), tBuOK (0.6 mmol) and dibutylamine (0.4 mmol), H2O (0.02 mL),and DMSO (1.2 mL) at 100 °C for 12 h under a N2 atmosphere. Isolated yield. :1) at rt for 12 h under an O2 atmosphere; (b) 3a (0.2 mmol), 4-methylbenzenethiol (0.24 mmol), and dry DCE (40 μL) at 80 °C for 15 min; (c) 3a (0.3 mmol), Cs2CO3 (0.03 mmol), TMSCN (0.9 mmol), and dry MeCN (1.0 mL) at rt for 2 h; (d) 3a (0.6 mmol), K3PO4 (1 mmol), imidazole (0.5 mmol), and DMF (1 mL) at rt for 12 h; (e) 3a (0.2 mmol), benzoyl hydrazide (0.24 mmol), Cs2CO3 (0.4 mmol), and dry DMSO (1 mL) at 80 °C for 6 h under a N2 atmosphere; (f) 3a (0.2 mmol), selectfluor (0.3 mmol), dry MeOH (1 mmol), and dry CH3CN (0.8 mL) at 40 °C for 4 h under N2 atmosphere; (g) 3a (0.2 mmol), selectfluor (0.3 mmol), H2O (1.6 mmol), and dry CH3CN (0.8 mL) at 40 °C for 4 h under a N2 atmosphere; (h) 3a (0.2 mmol), tBuOK (0.6 mmol) and dibutylamine (0.4 mmol), H2O (0.02 mL),and DMSO (1.2 mL) at 100 °C for 12 h under a N2 atmosphere. Isolated yield. | ||
Based on previous research and our understanding on palladium-catalyzed Heck dearomatization of indoles, a possible reaction process is shown in Scheme 3. The Pd(0) species was formed under the action of the ligand, and the reaction was initiated by the oxidative addition of Pd(0) to aryl bromide to afford the Pd(II) species A. The benzyl-Pd(II) intermediate B was generated by the intramolecular coordination and migratory insertion of the Pd(II) species into indole. Difluorocarbene, which formed from ClCF2COONa in situ in the presence of bases, was captured by intermediate B to provide the Pd(II)=CF2 species C. Following carbene migratory insertion,34 the σ-alkyl-Pd(II) species D was obtained. Then, the β-hydride elimination of the species D resulted in the final product 3a and Pd(II) species E. Finally, with the assistance of a base, Pd(0) was regenerated for the next catalytic cycle.
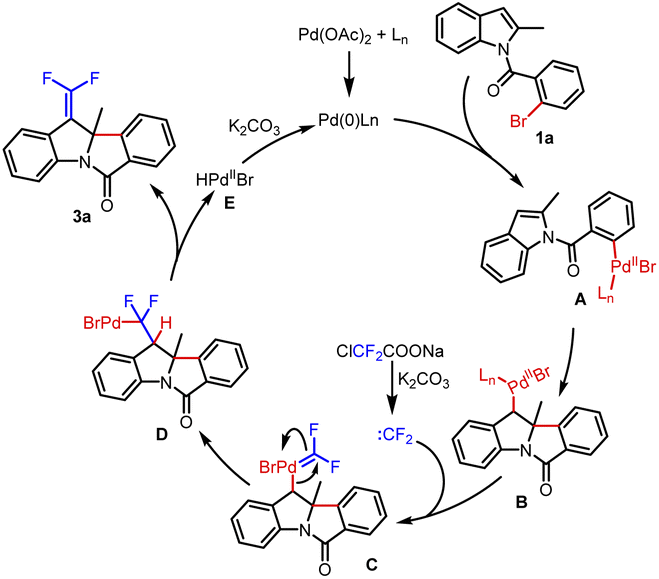 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, polycyclic indoline derivatives containing 1,1-difluoroethylene units are prepared by palladium-catalyzed intramolecular Heck dearomatization of indoles with ClCF2COONa. β-H elimination involving [Pd]CF2 provides a new synthetic course for gem-difluoroalkenes. In addition, it is the first report of dearomative gem-difluorovinylation, which has a broad substrates scope and acceptable yields. Further diversified transformations of the product show the practicability of this methodology.
Data availability
All data associated with this study are available in the article and ESI.†Author contributions
G. Wang, W. Q. Li, T. X. Liu, B. Wang, and C. Y. Ma carried out the methodology, synthesis, characterization, and analysis. Y. Xia and G. Wang prepared the manuscript. W. W. Jin, Y. H. Zhang, and F. Xue revised the manuscript. C. J. Liu and Y. Xia directed the project and supervised the whole experiment. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2020D01C077), the National Natural Science Foundation of China (Grant No. 21961037, 21861036, 21702175, and 21572195), the Program for Tianshan Innovative Research Team of Xinjiang Uygur Autonomous Region (2021D14011), the Key Program of Natural Science Foundation of Xinjiang Uygur Autonomous Region (2022D01D06) and the Program of Tianchi Doctor (tcbs201909) for the support of this research.Notes and references
- Y. Pan, J. Qiu and R. B. Silverman, J. Med. Chem., 2003, 46, 5292–5293 CrossRef CAS PubMed.
- S. Messaoudi, B. Tréguier, A. Hamze, O. Provot, J.-F. Peyrat, J. R. De Losada, J.-M. Liu, J. Bignon, J. Wdzieczak-Bakala, S. Thoret, J. Dubois, J.-D. Brion and M. Alami, J. Med. Chem., 2009, 52, 4538–4542 CrossRef CAS PubMed.
- J.-M. Altenburger, G. Y. Lassalle, M. Matrougui, D. Galtier, J.-C. Jetha, Z. Bocskei, C. N. Berry, C. Lunven, J. Lorrain, J.-P. Herault, P. Schaeffer, S. E. O'Connor and J.-M. Herbert, Bioorg. Med. Chem., 2004, 12, 1713–1730 CrossRef CAS PubMed.
- M. Bobek, I. Kavai and E. De Clercq, J. Med. Chem., 1987, 30, 1494–1497 CrossRef CAS PubMed.
- G. Landelle, M. Bergeron, M.-O. Turcotte-Savard and J.-F. Paquin, Chem. Soc. Rev., 2011, 40, 2867–2908 RSC.
- C. Liu, H. Zeng, C. Zhu and H. Jiang, Chem. Commun., 2020, 56, 10442–10452 RSC.
- X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS.
- X.-J. Zhang, Y.-M. Cheng, X.-W. Zhao, Z.-Y. Cao, X. Xiao and Y. Xu, Org. Chem. Front., 2021, 8, 2315–2327 RSC.
- G. Chelucci, Chem. Rev., 2012, 112, 1344–1462 CrossRef CAS PubMed.
- M. Decostanzi, J. M. Campagne and E. Leclerc, Org. Biomol. Chem., 2015, 13, 7351–7380 RSC.
- R. Cheng, Y. Sang, X. Gao, S. Zhang, X.-S. Xue and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12386–12391 CrossRef CAS PubMed.
- Y. Zhao, W. Huang, L. Zhu and J. Hu, Org. Lett., 2010, 12, 1444–1447 CrossRef CAS PubMed.
- B. Gao, Y. Zhao, M. Hu, C. Ni and J. Hu, Chem.–Eur. J., 2014, 20, 7803–7810 CrossRef CAS.
- X.-P. Wang, J.-H. Lin, J.-C. Xiao and X. Zheng, Eur. J. Org. Chem., 2014, 2014, 928–932 CrossRef CAS.
- D. G. Cox, N. Gurusamy and D. J. Burton, J. Am. Chem. Soc., 1985, 107, 2811–2812 CrossRef CAS.
- I. Nowak and M. J. Robins, Org. Lett., 2005, 7, 721–724 CrossRef CAS PubMed.
- J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC.
- J. Zheng, J.-H. Lin, J. Cai and J.-C. Xiao, Chem.–Eur. J., 2013, 19, 15261–15266 CrossRef CAS PubMed.
- K. Aikawa, W. Toya, Y. Nakamura and K. Mikami, Org. Lett., 2015, 17, 4996–4999 CrossRef CAS.
- F. Tian, G. Yan and J. Yu, Chem. Commun., 2019, 55, 13486–13505 RSC.
- X. Ma and Q. Song, Chem. Soc. Rev., 2020, 49, 9197–9219 RSC.
- G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC.
- S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS.
- M. Hu, C. Ni, L. Li, Y. Han and J. Hu, J. Am. Chem. Soc., 2015, 137, 14496–14501 CrossRef CAS PubMed.
- J. Zheng, J.-H. Lin, L.-Y. Yu, Y. Wei, X. Zheng and J.-C. Xiao, Org. Lett., 2015, 17, 6150–6153 CrossRef CAS PubMed.
- Z. Zhang, W. Yu, C. Wu, C. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2016, 55, 273–277 CrossRef CAS PubMed.
- Z. Yang, C. Pei and R. M. Koenigs, Org. Lett., 2020, 22, 7234–7238 CrossRef CAS PubMed.
- Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS.
- J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587–5590 CrossRef CAS PubMed.
- P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293–1326 CrossRef CAS.
- Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44–47 CrossRef CAS PubMed.
- X.-Y. Deng, J.-H. Lin and J.-C. Xiao, Org. Lett., 2016, 18, 4384–4387 CrossRef CAS PubMed.
- Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918–923 CrossRef CAS PubMed.
- X.-P. Fu, X.-S. Xue, X.-Y. Zhang, Y.-L. Xiao, S. Zhang, Y.-L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948–956 CrossRef CAS PubMed.
- J. B. I. Sap, C. F. Meyer, J. Ford, N. J. W. Straathof, A. B. Dürr, M. J. Lelos, S. J. Paisey, T. A. Mollner, S. M. Hell, A. A. Trabanco, C. Genicot, C. W. am Ende, R. S. Paton, M. Tredwell and V. Gouverneur, Nature, 2022, 606, 102–108 CrossRef CAS PubMed.
- X.-Y. Zhang, X.-P. Fu, S. Zhang and X. Zhang, CCS Chem., 2020, 2, 293–304 CrossRef CAS.
- Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed.
- X. Liu, H. Sheng, Y. Zhou and Q. Song, Org. Lett., 2021, 23, 2543–2547 CrossRef PubMed.
- V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036–5039 CrossRef CAS PubMed.
- A. Bosmani, A. Guarnieri-Ibáñez, S. Goudedranche, C. Besnard and J. Lacour, Angew. Chem., Int. Ed., 2018, 57, 7151–7155 CrossRef CAS PubMed.
- M.-C. Tong, X. Chen, J. Li, R. Huang, H. Tao and C.-J. Wang, Angew. Chem., Int. Ed., 2014, 53, 4680–4684 CrossRef CAS PubMed.
- M. Catellani and A. Del Rio, Russ. Chem. Bull., 1998, 47, 928–931 CrossRef CAS.
- F. Xu, K. M. Korch and D. A. Watson, Angew. Chem., Int. Ed., 2019, 58, 13448–13451 CrossRef CAS PubMed.
- Y. Yu, G. Li, L. Jiang and L. Zu, Angew. Chem., Int. Ed., 2015, 54, 12627–12631 CrossRef CAS PubMed.
- G. Zhang, V. J. Catalano and L. Zhang, J. Am. Chem. Soc., 2007, 129, 11358–11359 CrossRef CAS PubMed.
- D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef CAS PubMed.
- J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Org. Lett., 2007, 9, 4127–4129 CrossRef CAS PubMed.
- K. Douki, H. Ono, T. Taniguchi, J. Shimokawa, M. Kitamura and T. Fukuyama, J. Am. Chem. Soc., 2016, 138, 14578–14581 CrossRef CAS.
- J. Bonjoch and D. Solé, Chem. Rev., 2000, 100, 3455–3482 CrossRef CAS.
- N. Zeidan and M. Lautens, Synthesis, 2019, 51, 4137–4146 CrossRef CAS.
- D. A. Petrone, A. Yen, N. Zeidan and M. Lautens, Org. Lett., 2015, 17, 4838–4841 CrossRef CAS PubMed.
- C. Shen, R.-R. Liu, R.-J. Fan, Y.-L. Li, T.-F. Xu, J.-R. Gao and Y.-X. Jia, J. Am. Chem. Soc., 2015, 137, 4936–4939 CrossRef CAS PubMed.
- D. A. Petrone, M. Kondo, N. Zeidan and M. Lautens, Chem.–Eur. J., 2016, 22, 5684–5691 CrossRef CAS PubMed.
- R.-R. Liu, Y.-G. Wang, Y.-L. Li, B.-B. Huang, R.-X. Liang and Y.-X. Jia, Angew. Chem., Int. Ed., 2017, 56, 7475–7478 CrossRef CAS PubMed.
- N. Zeidan, T. Beisel, R. Ross and M. Lautens, Org. Lett., 2018, 20, 7332–7335 CrossRef CAS PubMed.
- C. Shen, N. Zeidan, Q. Wu, C. B. Breuers, R.-R. Liu, Y.-X. Jia and M. Lautens, Chem. Sci., 2019, 10, 3118–3122 RSC.
- J.-Q. Weng, L.-L. Xing, W.-R. Hou, R.-X. Liang and Y.-X. Jia, Org. Chem. Front., 2019, 6, 1577–1580 RSC.
- R.-R. Liu, T.-F. Xu, Y.-G. Wang, B. Xiang, J.-R. Gao and Y.-X. Jia, Chem. Commun., 2016, 52, 13664–13667 RSC.
- S. Chen, X.-X. Wu, J. Wang, X.-H. Hao, Y. Xia, Y. Shen, H. Jing and Y.-M. Liang, Org. Lett., 2016, 18, 4016–4019 CrossRef CAS PubMed.
- X. Li, B. Zhou, R.-Z. Yang, F.-M. Yang, R.-X. Liang, R.-R. Liu and Y.-X. Jia, J. Am. Chem. Soc., 2018, 140, 13945–13951 CrossRef CAS PubMed.
- R.-X. Liang, R.-Z. Yang, R.-R. Liu and Y.-X. Jia, Org. Chem. Front., 2018, 5, 1840–1843 RSC.
- H. Wang and X.-F. Wu, Org. Lett., 2019, 21, 5264–5268 CrossRef CAS.
- R.-X. Liang, K. Wang, Q. Wu, W.-J. Sheng and Y.-X. Jia, Organometallics, 2019, 38, 3927–3930 CrossRef CAS.
- R.-X. Liang, L.-J. Song, J.-B. Lu, W.-Y. Xu, C. Ding and Y.-X. Jia, Angew. Chem., Int. Ed., 2021, 60, 7412–7417 CrossRef CAS.
- L. Zhao, Z. Li, L. Chang, J. Xu, H. Yao and X. Wu, Org. Lett., 2012, 14, 2066–2069 CrossRef CAS.
- S.-B. Duan, H.-Y. Zhang, B.-Y. Hao, J. Zhao, Y.-P. Han, Y. Zhang and Y.-M. Liang, Org. Chem. Front., 2021, 8, 5895–5901 RSC.
- G. Wang, M. Z. Wei, T. X. Liu, W. W. Jin, Y. H. Zhang, B. Wang, Y. Xia and C. J. Liu, Adv. Synth. Catal., 2022, 364, 909–913 CrossRef CAS.
- R.-X. Liang and Y.-X. Jia, Acc. Chem. Res., 2022, 55, 734–745 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2101086, 2101084 and 2103217. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03169h |
| This journal is © The Royal Society of Chemistry 2022 |