
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombination of vancomycin and guanidinium-functionalized helical polymers for synergistic antibacterial activity and biofilm ablation†
Wen-Bin
Liu
b,
Run-Tan
Gao
a,
Li
Zhou
 b,
Na
Liu
a,
Zheng
Chen
a and
Zong-Quan
Wu
*a
b,
Na
Liu
a,
Zheng
Chen
a and
Zong-Quan
Wu
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun 130012, China. E-mail: zqwu@jlu.edu.cn
bDepartment of Polymer Science and Engineering, Hefei University of Technology, Hefei 230009, China
First published on 9th August 2022
Abstract
The emergence of various resistant bacteria and overuse of antibiotics have led to severe side effects. Therefore, developing efficient and safe antibacterial systems is important. Herein, well-defined antimicrobial material–helical poly(phenyl guanidinium isocyanide) block copolymers with different conformations (L-P3-van, D-P3-van, and DL-P3-van) that connect vancomycin (van) to the polymer through a disulfide bond were synthesized. The prepared antimicrobial materials exhibit broad-spectrum antimicrobial activity, low bacterial resistance, and good proteolytic stability. They also overcome the intrinsic resistance of Gram-negative bacteria to van with a 100-fold increase in antimicrobial activity. Interestingly, the conformation of the material promotes its antimicrobial activity. The left-handed helix conformation shows five-fold more antimicrobial activity than the right-handed helical conformation, thereby opening a path for the application of nanochirality in the field of antibiotics.
Introduction
Recently, considerable progress has been achieved in the study of polymeric antimicrobial systems for the prevention and elimination of biofilms.1–3 Such systems can improve biocompatibility, reduce bacterial resistance, and increase antimicrobial efficiency.4–6 Although these polymer antimicrobial systems have been extensively examined, the effect of chirality on their antimicrobial effect remains unclear. Many biological macromolecules such as amino acids, DNA, cell-penetrating peptides, and antibacterial peptides have helical structures and are closely related to vital life functions in biological systems.7–10 Therefore, helical biological macromolecules and their microenvironments are chiral in living systems. The chirality of the material affects physiological processes such as the body's immune response to viral infections.11 Most effective antibiotics, such as vancomycin (van),12 kanamycin,13 and polymyxin B,14 are chiral. From this perspective, the chirality of a polymer antimicrobial system may influence its antimicrobial effect. Moreover, different chiral carriers may behave differently when interacting with living systems.15–17 A helix is a chiral expression because left- and right-handed helices cannot overlap but are mirror images of each other.18,19 Therefore, the design and synthesis of chiral antimicrobial systems and their structure–function relationship have important significance.The formation of biofilms in bacterial infections is a major medical challenge because biofilms can isolate bacteria from the host immune response and antibiotics. The traditional strategy to treat bacterial infections is to use antimicrobials.20 Developed antimicrobials include antibiotics, antimicrobial peptides,21 cationic polymers,22,23 and other nanomaterials.24–27 However, only a few of these materials can destroy the original biofilm.28–30 Van is an effective antibiotic used against Gram-positive bacteria.31 However, its overuse has led to the emergence of resistant bacteria.32–36 Moreover, van is inherently inactive against Gram-negative bacteria because it cannot pass through the outer membrane of these bacteria.37 Therefore, studies on Gram-positive bacteria overcoming resistance because of abuse and Gram-negative bacteria overcoming intrinsic resistance to van are warranted. Guanidine derivatives have received considerable attention for their antimicrobial potential and membrane association and penetration.38–40 The most representative is the cell penetration peptides (CPPs), which are usually composed of short side chains and arginine-rich peptides.41,42 These CPPs enter the cell by direct membrane penetration; alternatively, the CPPs induce membrane multilayers and then enter the cells through membrane fusion or endocytosis.43–45
Because of the excellent antibacterial and biofilm ablation properties of guanidine derivatives and van and the many practical problems they face, we propose a synergistic combination of guanidine polymers and van to develop an ultra-efficient antimicrobial system. Partially inspired by the cell-penetrating peptides and biological helices, we designed and synthesized a number of helical poly(phenyl guanidinium isocyanide) block copolymers with van connected to the polymer through the disulfide bond (Scheme 1) to obtain antibacterial materials with broad-spectrum antibacterial activities, low resistance trends, good proteolytic stability, and structural flexibility.46,47 These antimicrobial materials not only exert good antimicrobial activity against Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus but also effectively disrupt the pre-formed biofilm by effectively penetrating biofilm barriers and disrupting bacteria surrounded by biofilms. The prepared antibacterial materials show up to 100-fold stronger activity against Gram-negative bacteria than van. This result can be attributed to the helical poly(phenyl guanidinium isocyanide) that can penetrate the outer membrane of Gram-negative bacteria and release van inside the bacteria for sterilization because of the cleavage of the disulfide bond by high concentrations of glutathione in the bacteria. Furthermore, the chirality of the material promotes antibacterial activity and biofilm disruption.
 | ||
| Scheme 1 Synthesis of antimicrobial nanoparticles. | ||
Results and discussion
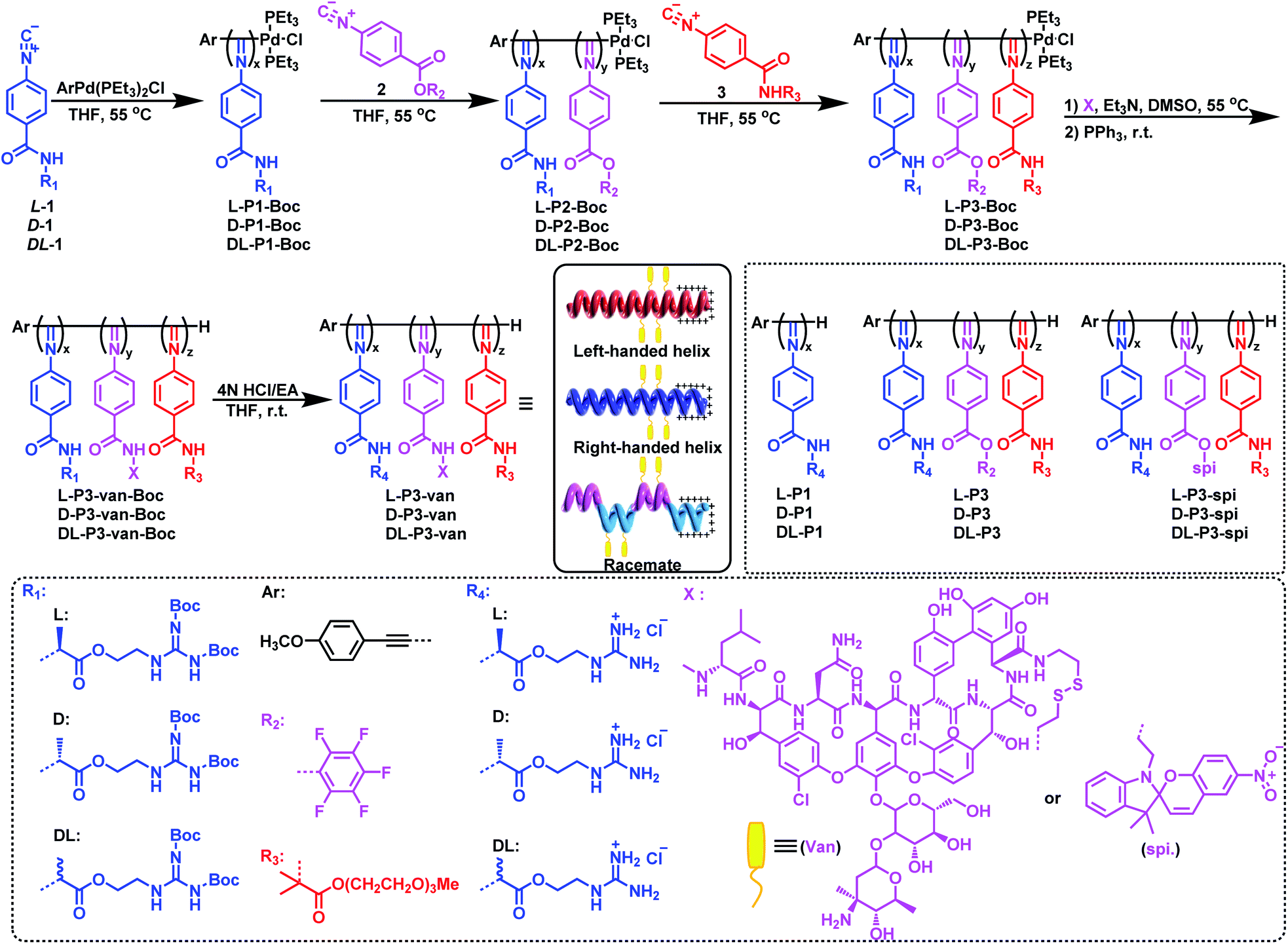
As outlined in Scheme 1, an enantiopure phenyl isocyanide monomer L-1 bearing L-guanidine substituent was polymerized by an alkyne-Pd(II) catalyst (ArPd(PEt3)2Cl) in THF at 55 °C ([L-1]0/[Pd]0 = 30) following a living polymerization mechanism (the characterization of the monomer is shown in Fig. S1–S9 in the ESI).†48 The resulting polymer (L-P1-Boc) demonstrated a single modal and narrow distributed elution trace on size exclusion chromatography (SEC), as shown in Fig. 1a. The number-average molecular weight (Mn) and its distribution (Mw/Mn) values were 11.7 kDa and 1.08, respectively. For chain extension of this polymer, the pentafluorophenyl ester functionalized phenyl isocyanide monomer 2 was used, and the ratio of 2-to-Pd(II) was fixed at ∼5. Based on the SEC results, when monomer 2 was completely consumed, the polymerization solution was treated with monomer 3 bearing oligoethylene glycol to afford a triblock copolymer. As shown in the SEC results in Fig. 1a, the Mn value of diblock copolymer L-P2-Boc increased to 12.1 kDa, whereas the Mw/Mn remained low (1.10). After copolymerization with monomer 3, the elution trace of the resulting triblock copolymer L-P3-Boc shifted to a higher Mn-region while remaining unimodal and symmetric. Based on the SEC results, the Mn and Mw/Mn values of L-P3-Boc were determined to be 34.8 kDa and 1.21, respectively. The synthetic polymers were further confirmed using 1H NMR and FT-IR analyses (Fig. S11 in the ESI†). The block ratio is 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:7 (x:y:z), which generally agrees with the proposed structure and the monomer feed ratios (Fig. S10†). In the 19F NMR spectrum of L-P3-Boc, three broad resonances at −150, −155, and −160 ppm corresponding to the pentafluorophenol ester pendants can be clearly determined (Fig. S11†). Moreover, the characteristic vibrations of C
:1:7 (x:y:z), which generally agrees with the proposed structure and the monomer feed ratios (Fig. S10†). In the 19F NMR spectrum of L-P3-Boc, three broad resonances at −150, −155, and −160 ppm corresponding to the pentafluorophenol ester pendants can be clearly determined (Fig. S11†). Moreover, the characteristic vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO bonds can be detected at 1630 and 1737 cm−1, respectively, in the FT-IR spectra of the triblock copolymer L-P3-Boc, which confirmed the formation of the expected triblock copolymer (Fig. S12†).
N and CO bonds can be detected at 1630 and 1737 cm−1, respectively, in the FT-IR spectra of the triblock copolymer L-P3-Boc, which confirmed the formation of the expected triblock copolymer (Fig. S12†).
 | ||
| Fig. 1 (a) SEC curves of homopolymers L-P1-Boc, L-P2-Boc, L-P3-Boc and L-P3-van-Boc. (b) CD and UV-vis spectra of antibacterial nanoparticles. (c) DLS, (d) stability, (e) TEM, and (f) AFM image of L-P3-van. The error bars are based on the standard deviations of three parallel tests. | ||
Using the triblock copolymer, we attempted to synthesize van and guanidine synergistic antibacterial nanoparticles using van carrying the disulfide bond (van–NH2, see the ESI† for the synthesis methods and characterization) (Scheme 1). The polymer L-P3-van-Boc was obtained by reacting van–NH2 with the pentafluorophenol benzoate pendant of the triblock copolymer in THF using triethylamine as a catalyst. The resulting polymer was confirmed using SEC. As shown in Fig. 1a, compared with that of the precursor, the SEC trace of the polymer shifted to a shorter retention time region. Both Mn and Mw/Mn of the resulting polymer were 37.6 kDa and 1.32, respectively. In the 1H NMR spectrum, the signals originating from van and cystamine were clearly observed (Fig. S14†). No 19F resonance could be determined from the 19F NMR spectra of the resulting L-P3-van-Boc (Fig. S11†). This result confirmed that almost all pentafluorophenol benzoate pendants participated in the reaction. Moreover, the characteristic vibration peaks of van were clearly visible in the FT-IR spectra of L-P3-van-Boc (Fig. S12†). Thus, the polymers carry plenty of van-units of the pendants. Subsequently, the protective Boc group was removed under acidic conditions (HCl/EA) to obtain the corresponding nano-antibacterial material L-P3-van (Scheme 1). This polymer was insoluble in organic solvents but soluble in water. The enantiomeric antipode D-P3-van and achiral DL-P3-van with similar block ratios and degree of polymerizations (DPs) of each block to those of L-P3-van were prepared using the same method (Scheme 1 and Fig. S13–S15, ESI†).
The helicity of synthetic polymers was examined using circular dichroism (CD) and UV-vis absorption spectroscopies. As shown in Fig. 1b, because of the asymmetric induction of the chiral monomer L-1, L-P1-Boc demonstrated intense CD at the absorption region of the poly(phenyl isocyanide) backbone, thus confirming the formation of a predominant left-handed helix. After copolymerization with achiral monomers 2 and 3, the CD intensities decreased because of the partial helical inversion of the newly formed blocks. However, after introducing van and removing the protective Boc group, the CD was maintained as that of the triblock copolymer, suggesting that the optical activity of the helical polyisocyanide was maintained. As anticipated, D-P3-van demonstrated a mirror CD profile to that of L-P3-van, whereas DL-P3-van was CD silent (Fig. 1b). The optical activities were confirmed using the optical rotation investigation (Table 1 and Fig. S16†). The structure was stable, with almost no observable changes in CD and UV-vis spectra at the temperature range of 5 °C to 50 °C in water (Fig. S17†).
| Samplesa | Sizeb (nm) | ε 364 (M−1 cm−1) | MIC (μM) | MBEC (μM) | HC50 (μM) | Selectivity HC50/MIC90 | |||
|---|---|---|---|---|---|---|---|---|---|
| E. coli | S. aureus | E. coli | S. aureus | RBC | E. coli | S. aureus | |||
| a The polymers were synthesized according to ESI Scheme S2. b Determined by dynamic light scattering. c Measured in water at 25 °C (c = 0.2 mg mL−1). d E. coli (O157). e S. aureus (ATCC25923). f Rat red blood cells. | |||||||||
| DL-P3-Van | 137 | — | 1.5 | 0.8 | 45 | 4 | 150 | 10 | 187.5 |
| L-P3-Van | 142 | −8.10 | 0.5 | 0.2 | 15 | 1 | 150 | 30 | 750 |
| D-P3-Van | 136 | +7.80 | 2 | 1 | 70 | 5 | 150 | 7.5 | 150 |
The nano-antimicrobial material L-P3-van demonstrated good solubility in water because of the presence of the guanidine group and oligoglycol chain. The hydrodynamic diameter (Dh) of L-P3-van in water determined by dynamic light scattering (DLS) was 142 nm (Fig. 1c). A prolonged standing study demonstrated that the Dh and polydispersity index remained almost unchanged over a month (Fig. 1d). Accordingly, the Dhs values of DL-P3-van and D-P3-van were 137 and 136 nm, respectively (Fig. S18a and d†). The morphology of L-P3-van was investigated by transmission electron microscopy (TEM) and atomic force microscopy (AFM). As shown in Fig. 1e and f, L-P3-van demonstrated spherical nanoparticles in the TEM and AFM images. The average diameters of these nanoparticles were estimated to be 110 and 115 nm, as determined by TEM and AFM, respectively; generally, these results agree with the DLS analyses. Accordingly, both TEM and AFM studies demonstrated that DL-P3-van and D-P3-van had similar spherical morphologies and sizes to those of L-P3-van due to the similar block ratios and DPs of the three polymers (Fig. S18, ESI†). The driving force for self-assembly is not very clear at the current stage, while the different solvophobic effects of the three blocks in water may have great contribution to the assembly.10 The self-assembled micelles have locally enriched positive charges, and thus favor enhanced antimicrobial activity and selectivity.49
Then, the antibacterial potential of the three polymers was evaluated against Gram-positive S. aureus (ATCC25923) and Gram-negative E. coli (O157), and the results are summarized in Tables 1 and S2 and Fig. S19 in the ESI.† The minimum inhibitory concentration (MIC) is defined as the lowest polymer concentration required to inhibit 100% bacterial growth after an incubation period of 18 h.50 For Gram-positive S. aureus, van demonstrated potent antimicrobial activity against extracellular S. aureus (MIC = 0.69 μM), which was consistent with the literature.51 Compared with the antibacterial activity of van, the antimicrobial activity of the synthetic polymers increased by ∼30–100 fold after the introduction of van. Interestingly, the helicity of polymers resulted in different antimicrobial effects, although they have similar DPs and block ratios. Compared with the antibacterial activities of DL-P3-van and D-P3-van with MIC = 0.8 and 1 μM, respectively, L-P3-van had a stronger antibacterial activity (MIC = 0.2 μM). This result can be attributed to the left-handed antibacterial polymer acting on the cell membrane faster than the antimicrobial materials with racemic and right-handed helices. As shown in Fig. 2a, the bactericidal efficiency of L-P3-van at ∼2 μM was >99%, and its antibacterial activity was comparable to that of van. Similarly, the antimicrobial activity of L-P3-van against E. coli was ∼15–40 fold higher than that of van alone. Even at higher concentrations, van does not naturally have high antibacterial activity (MIC = 50 μM) against Gram-negative bacteria. When it was introduced in the helical polymer, its antibacterial activity increased by up to 100-fold. Note that the bactericidal efficiency of L-P3-van at 5 μM exceeded 99% for E. coli (Fig. 2b). A series of copolymers with different block ratios of van to guanidinium and oligoglycol segments were prepared and evaluated against two typical bacteria, E. coli and S. aureus (Fig. S20–S24, ESI†). For both S. aureus and E. coli, the introduction of van and the guanidine groups into the copolymers was beneficial for increasing the antimicrobial activity, while the ratios of van to guanidinium and to oligoglycol moieties have a slight influence on the antibacterial properties.
 | ||
| Fig. 2 Antibacterial ability of L-P3-van against (a) S. aureus, (b) E. coli and van (2 and 25 μM) as the positive controls, respectively. PBS as the negative control. * indicates no detection of bacteria in the plate counting experiments. The error bars indicate the standard deviations of triplicate experiments. (c) Photographs obtained by culturing the sample solutions from S. aureus and E. coli incubated with PBS, DL-P3-van, L-P3-van and D-P3-van solution on individual Petri dishes and (d) corresponding fluorescence microscopic images of S. aureus and E. coli that were stained with calcein-AM/PI dyes. | ||
The hemolytic concentration-50% (HC50) of polymers was determined against freshly drawn mouse red blood cells (RBCs) to assess the toxicity toward mammalian cells.52 HC50 is defined as the lowest polymer concentration required to lyse 50% of the RBCs within an incubation period of 1 h. The highest concentration of polymer solution tested was 2000 μg mL−1.50 As summarized in Table S2,† homopolymers DL-P1, L-P1, and D-P1 were highly antibacterial against E. coli and S. aureus but extremely hemolytic (HC50 = 1.3 μM). After introducing the oligoglycol segment, the hemolytic activity of the material decreased, and the HC50 values corresponding to DL-P3, L-P3, and D-P3 were 135 μM. This result can be attributed to the oligoglycol segments resisting protein adsorption, providing stealth properties, modifying the hydrophilic/hydrophobic balance of the polymer structure, and reducing the electrostatic repulsion between the amphipathic polymer chains.53–56 Although the oligoglycol pendants are short, the polymeric backbone bearing plenty of oligoglycol pendants could resist protein adsorption.54–56 For comparison, owing to protein adsorption, L-P2 without oligoglycol pendants was precipitated when treated with S. aureus (Fig. S25, ESI†).
Bacteria with and without the presence of antibiotic polymers were cultured in Petri dishes to evaluate their antibiotic effects. As shown in Fig. 2c, DL-P3-van, L-P3-van, and D-P3-van demonstrated high sterilization effects on E. coli and S. aureus, of which the left-handed L-P3-van demonstrated the best performance. Fluorescence microscopy imaging experiments were performed to visualize the viable and inactive bacteria. Calcein-AM was used to label the viable cells with green fluorescence, whereas PI with red fluorescence merely penetrated bacteria with damaged cell membranes. As shown in Fig. 2d, nearly all S. aureus and E. coli cells were stained red with PI after the L-P3-van treatment, indicating the disintegration of bacterial membranes. However, very rare red fluorescence was observed in the phosphate-buffered saline (PBS) group. The bacteria exhibited green and red fluorescence for the other formulations of van, DL-P3-van, and D-P3-van, suggesting an incomplete bacterial killing. These results demonstrated that the antimicrobial polymers killed the bacteria by destroying their membranes and cell walls. In addition to the disruptive interactions of the guanidine group with the cell membrane, the ability of van to block the cell wall growth contributes to the increased antimicrobial capacity.57,58
The SEM observations revealed that the bacteria treated with L-P3-van, D-P3-van, and DL-P3-van were ruptured, and the morphology hardly maintained integrity, especially that of the L-P3-van-treated bacteria (Fig. 3b–d and f–h). The PBS-treated bacteria had complete morphology and a smooth surface (Fig. 3a and e). Then, the protein leakage from the bacteria was first explored using the Bradford protein assay kit. All treatments of van, DL-P3-van, L-P3-van, and D-P3-van resulted in a certain amount of protein leakage, indicating that the membrane was damaged to a certain extent (Fig. S26†). As expected, the L-P3-van-treated bacteria displayed the largest amount of protein leakage and the maximum membrane damage, which is consistent with the SEM observations.
 | ||
| Fig. 3 SEM images of (a)–(d) S. aureus and (e)–(h) E. coli treated with PBS, L-P3-van, D-P3-van and DL-P3-van. | ||
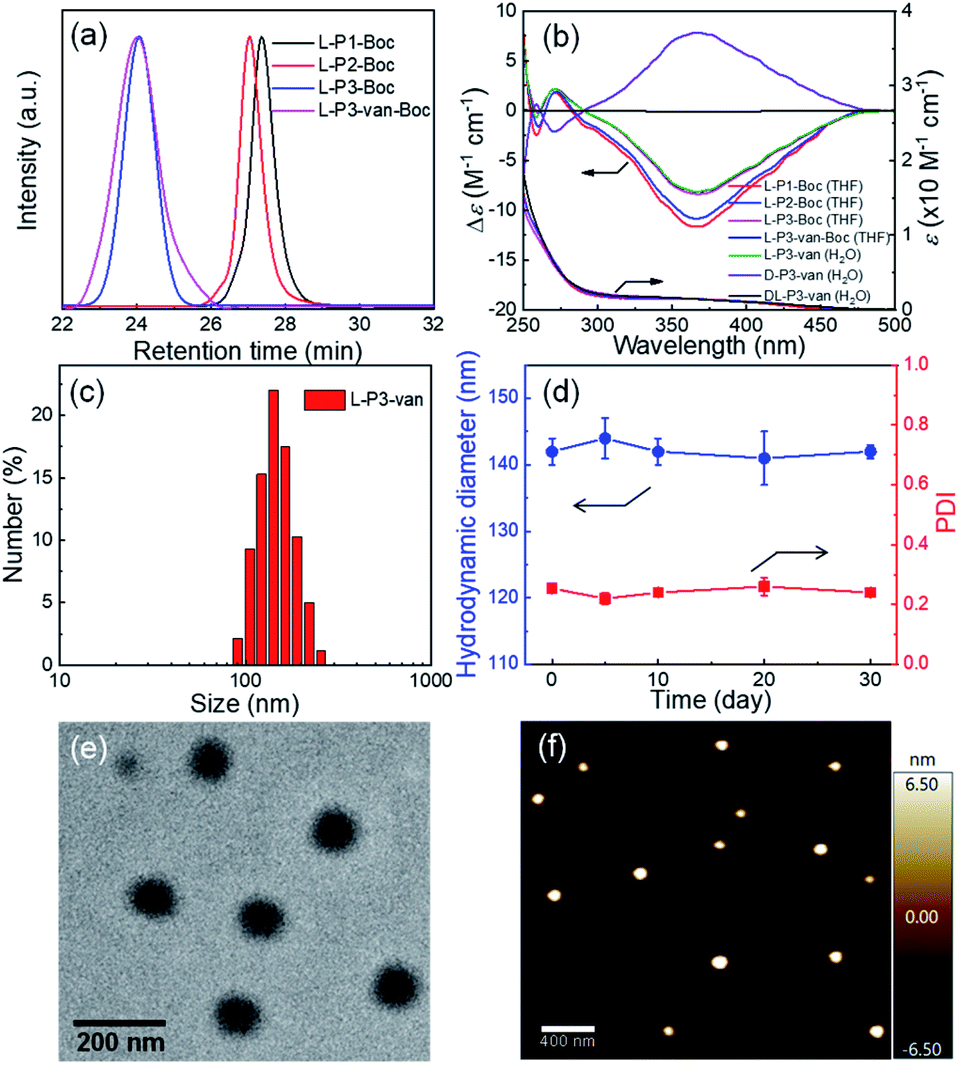
To investigate the effect of the helical conformation on the antibacterial effects of the antimicrobial materials, we introduced the fluorescein spiropyran (spi.) into the helical polymers to synthesize three fluorescent markers with different conformations (L-P3-spi, D-P3-spi, and DL-P3-spi, see the ESI† for the synthesis methods and characterization Fig. S27 and S28†). Because of the fluorescence properties of the polymer itself, antibacterial processes were monitored for different materials by confocal microscopy. As shown in Fig. 4, S. aureus was detreated with L-P3-spi, D-P3-spi, and DL-P3-spi. After 1 h, visible fluorescence was observed in the bacteria treated with L-P3-spi under confocal microscopy but not in the bacteria treated with D-P3-spi and DL-P3-spi after ∼5 h (Fig. S29, ESI†). After 7 h, a large area of S. aureus was observed by confocal microscopy and labeled by using the left-handed L-P3-spi. This result suggests that the left-handed polymer acted faster on the bacterial cell membranes than the other two polymers. These antimicrobial polymers carrying van and guanidinium pendants kill the bacteria by destroying their membrane and cell walls. Thus, the polymer acting faster on the bacteria can kill the bacteria more quickly and showed better antibacterial properties.11,59 The L-P3-van, D-P3-van, and DL-P3-van polymers have similar DPs and block ratios, and are self-assembled into micelles of similar sizes in water. The different antibacterial properties of these polymers were probably ascribed to the different chirality and handedness of the backbone, which resulted in left- and right-handed packing of the van and guanidinium pendants on L-P3-van and D-P3-van, respectively, while the van and guanidinium pendants of DL-P3-van may packed in a random way. The different arrays of the pendants thus resulted in different antibacterial properties.
 | ||
| Fig. 4 (a) Real-time monitoring of DL-P3-spi, L-P3-spi and D-P3-spi infecting S. aureus. (b) Fluorescence intensities per image for bacteria treated with DL-P3-spi, L-P3-spi and D-P3-spi, as calculated by ImageJ software. | ||
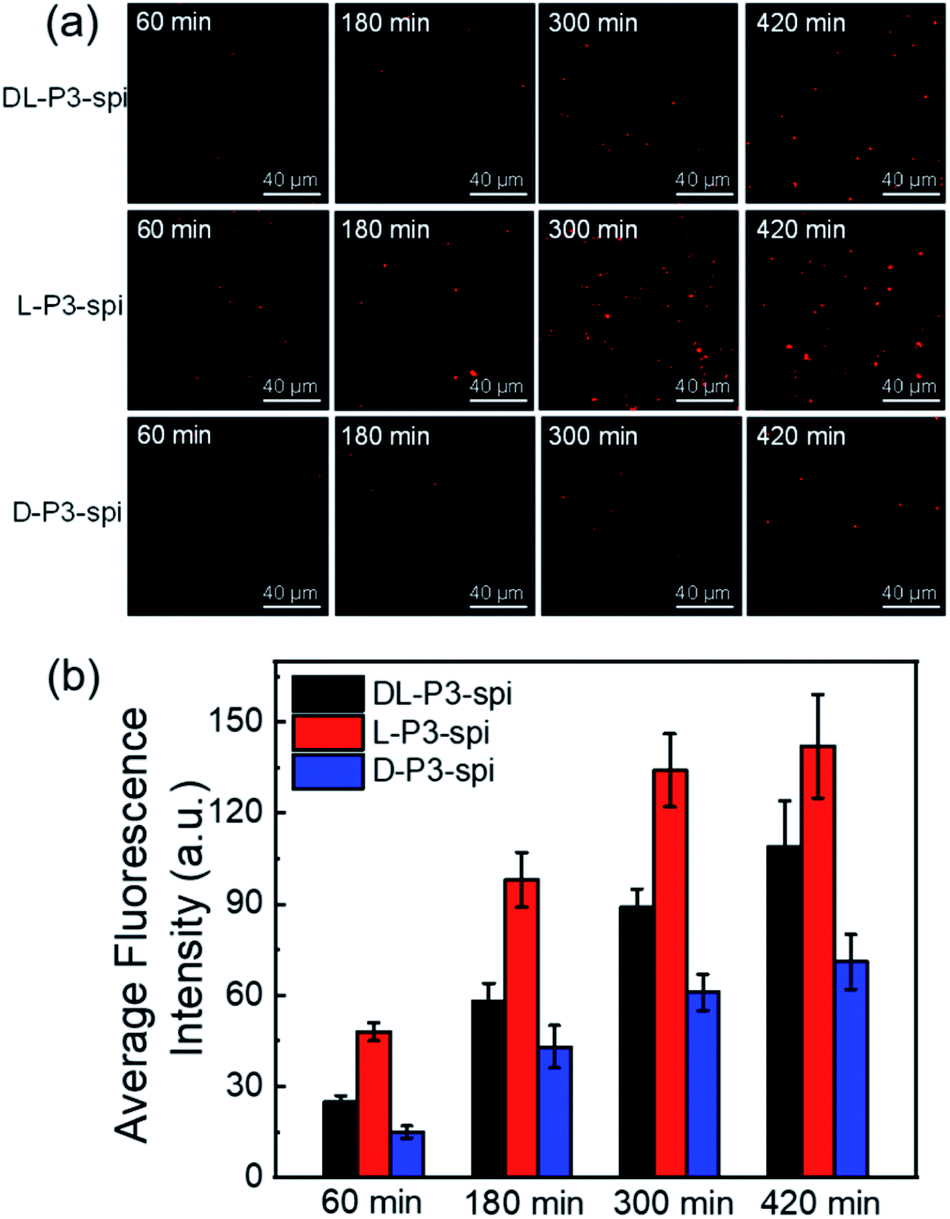
Encouraged by the performance of DL-P3-van, L-P3-van, and D-P3-van against bacterial cells, we evaluated the efficacy of DL-P3-van, L-P3-van, and D-P3-van as agents to disrupt bacterial biofilms. S. aureus and E. coli biofilms were pre-cultivated and then treated with DL-P3-van, L-P3-van, and D-P3-van for another 24 h. The resulting bacterial growth in biofilms was observed by measuring the optical density at 600 nm (OD600). PBS as a negative control and van as a positive control were employed. Crystal-violet staining and the minimum biofilm eradication concentration assay were performed to accurately evaluate the biofilm inhibition and eradication activities of DL-P3-van, L-P3-van, and D-P3-van (Fig. 5, S30 and S31 in the ESI†).
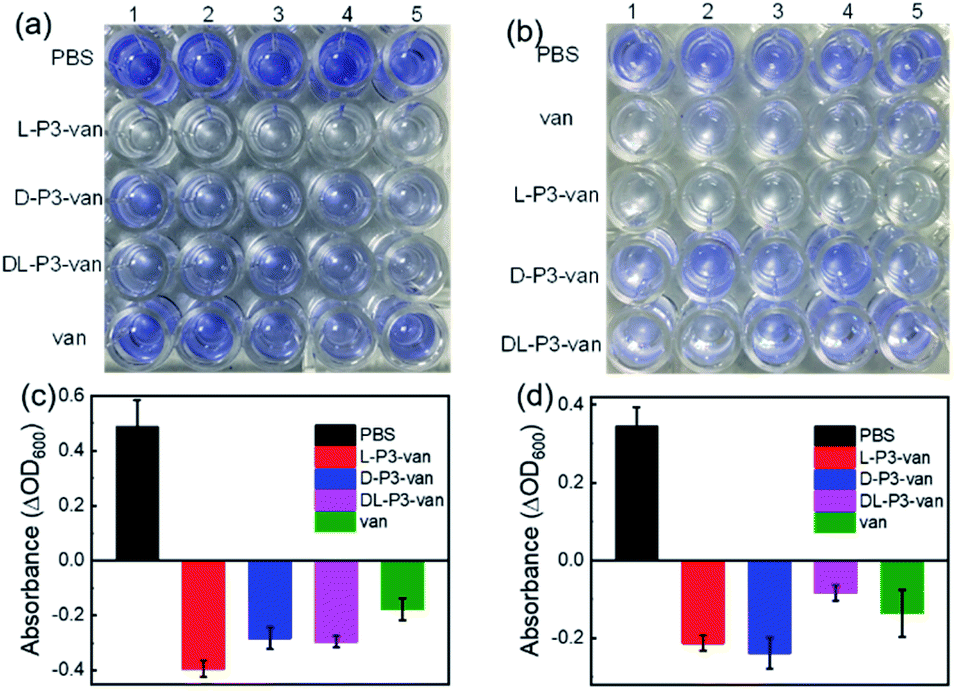 | ||
| Fig. 5 Inhibition of (a) E. coli and (b) S. aureus biofilm formation by different antimicrobial materials stained with crystal violet. Each concentration of compound was tested in five wells. (c) E. coli and (d) S. aureus growth after treatment with different antimicrobial materials for 1 day in biofilm stages. | ||
As summarized in Tables 1 and S2,† biofilm inhibition and eradication effects could be observed at 5 μM, which indicated that DL-P3-van, L-P3-van, and D-P3-van have potential to be therapeutic agents to tackle biofilm-associated infections. Although the actual mechanism of biofilm disruption is under investigation, we believe the following factors play major roles. First, the positively charged polymers could perturb the negatively charged exopolysaccharide matrix of biofilms through electrostatic and hydrophobic interactions.38–40 Second, these helical polymers can act as cell-penetrating peptides to cross the membrane into the cell interior,9,60,61 releasing van and thus hindering the synthesis of the bacterial cell wall, ultimately leading to biofilm degradation.59
Conclusions
We designed and synthesized a series of well-defined helical polymers (L-P3-van, D-P3-van, and DL-P3-van) with a redox stimulus-responsive release of van for preparing synergistic antibacterial and ablation biofilms. These antimicrobial materials demonstrated broad-spectrum antibacterial activities, low bacterial resistance, and good proteolytic stability. L-P3-Van exhibited high activity, which was 100-fold higher than that achieved by van. This result can be attributed to the fact that helical poly(phenyl guanidinium isocyanide) can penetrate the outer membrane of Gram-negative bacteria, thus releasing van into the Gram-negative bacteria for sterilization. Interestingly, the chirality of the material promoted the antibacterial activity and disruption of biofilms. Both antimicrobial and confocal microscopic experiments demonstrated that the material with a left-handed helical conformation performed better with respect to antibacterial activity and biofilm removal. To summarize, these results reveal that the prepared helical polymers are efficient and safe antibacterial systems with essential application prospects for treating bacterial infections and ablating biofilms in vivo.Data availability
The detailed experimental data associated with this work are available in the ESI.†Author contributions
W.-B. L., R.-T. G. and L. Z. performed the experiments and analyses. W.-B. L. and Z.-Q. W. wrote and edited the paper. N. L., Z. C. and Z.-Q. W. conceived the idea and supervised the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China for financial support (NSFC, No. 22071041, 21971052, 51903072, and 21871073). L. Zhou and Z.-Q. Wu appreciate the Fundamental Research Funds for the Central Universities of China (Grant No. PA2019GDPK0057, and PA2020GDJQ0028).Notes and references
- S. Eckhardt, P. S. Brunetto, J. Gagnon, M. Priebe, B. Giese and K. M. Fromm, Chem. Rev., 2013, 113, 4708–4754 CrossRef CAS PubMed.
- A. Gupta, S. Mumtaz, C.-H. Li, I. Hussain and V. M. Rotello, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- L. Gao, M. Li, S. Ehrmann, Z. Tu and R. Haag, Angew. Chem., Int. Ed., 2019, 58, 3645–3649 CrossRef CAS PubMed.
- S. Guo, Q. Huang, Y. Chen, J. Wei, J. Zheng, L. Wang, Y. Wang and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 618–623 CrossRef CAS PubMed.
- A. Antonoplis, X. Zang, M. A. Huttner, K. K. L. Chong, Y. B. Lee, J. Y. Co, M. R. Amieva, K. A. Kline, P. A. Wender and L. Cegelski, J. Am. Chem. Soc., 2018, 140, 16140–16151 CrossRef CAS PubMed.
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
- B. T. Stokke, A. Elgsaeter, D. A. Brant and S. Kitamura, Macromolecules, 1991, 24, 6349–6351 CrossRef CAS.
- Z. Li, D. Teng, R. Mao, X. Wang, Y. Hao, X. Wang and J. Wang, J. Med. Chem., 2018, 61, 7991–8000 CrossRef CAS PubMed.
- M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek Jr., C. Yao, L.-F. Chen, A. L. Ferguson, G. C. L. Wong and J. Cheng, PNAS, 2015, 112, 13155–13160 CrossRef CAS PubMed.
- L. Xu, X. Wang, W. Wang, M. Sun, W. J. Choi, J.-Y. Kim, C. Hao, S. Li, A. Qu, M. Lu, X. Wu, F. M. Colombari, W. R. Gomes, A. L. Blanco, A. F. de Moura, X. Guo, H. Kuang, N. A. Kotov and C. Xu, Nature, 2022, 601, 366–373 CrossRef CAS PubMed.
- F. Umsttter, C. Domhan, T. Hertlein, K. Ohlsen, E. Mühlberg, C. Kleist, S. Zimmermann, B. Beijer, K. D. Klika, U. Haberkorn, W. Mier and P. Uhl, Angew. Chem., Int. Ed., 2020, 59, 8823–8827 CrossRef.
- A. Brezden, M. F. Mohamed, M. Nepal, J. S. Harwood, J. Kuriakose, M. N. Seleem and J. Chmielewski, J. Am. Chem. Soc., 2016, 138, 10945–10949 CrossRef CAS PubMed.
- A. Luther, M. Urfer, M. Zahn, M. Müller, S.-Y. Wang, M. Mondal, A. Vitale, J.-B. Hartmann, T. Sharpe, F. Lo Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M.-A. Westwood, N. Desjonquères, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithié, G. Upert, A. Lederer, P. Zbinden, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452–458 CrossRef CAS PubMed.
- Y. Bao, Y. Huang, M. L. Lam, T. Xu, N. Zhu, Z. Guo, X. Cui, R. H. W. Lam and T. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 17976–17986 CrossRef CAS PubMed.
- X. Wang, C. Yan, K. Ye, Y. He, Z. Li and J. Ding, Biomaterials, 2013, 34, 2865–2874 CrossRef CAS PubMed.
- W. Liu, Y. Bao, M. L. Lam, T. Xu, K. Xie, H. S. Man, E. Y. Chan, N. Zhu, R. H. W. Lam and T. Chen, ACS Nano, 2016, 10, 7409–7417 CrossRef CAS PubMed.
- H. Huang, J. Deng and Y. Shi, Macromolecules, 2016, 49, 2948–2956 CrossRef CAS.
- C. Tsiamantas, X. de Hatten, C. Douat, B. Kauffmann, V. Maurizot, H. Ihara, M. Takafuji, N. Metzler-Nolte and I. Huc, Angew. Chem., Int. Ed., 2016, 55, 6848–6852 CrossRef CAS PubMed.
- K. Bush, P. Courvalin, G. Dantas, J. Davies, B. Eisenstein, P. Huovinen, G. A. Jacoby, R. Kishony, B. N. Kreiswirth, E. Kutter, S. A. Lerner, S. Levy, K. Lewis, O. Lomovskaya, J. H. Miller, S. Mobashery, L. J. V. Piddock, S. Projan, C. M. Thomas, A. Tomasz, P. M. Tulkens, T. R. Walsh, J. D. Watson, J. Witkowski, W. Witte, G. Wright, P. Yeh and H. I. Zgurskaya, Nat. Rev. Microbiol., 2011, 9, 894–896 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- K. Fukushima, J. P. K. Tan, P. A. Korevaar, Y. Y. Yang, J. Pitera, A. Nelson, H. Maune, D. J. Coady, J. E. Frommer, A. C. Engler, Y. Huang, K. Xu, Z. Ji, Y. Qiao, W. Fan, L. Li, N. Wiradharma, E. W. Meijer and J. L. Hedrick, ACS Nano, 2012, 6, 9191–9199 CrossRef CAS PubMed.
- J. T. Seil and T. J. Webster, Int. J. Nanomed., 2012, 7, 2767–2781 CAS.
- K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem. Int. Ed., 2013, 52, 8285–8289 ( Angew. Chem. , 2013 , 125 , 8443–8447 ) CrossRef CAS PubMed.
- I. Roy, D. Shetty, R. Hota, K. Baek, J. Kim, C. Kim, S. Kappert and K. Kim, Angew. Chem. Int. Ed., 2015, 54, 15152–15155 ( Angew. Chem. , 2015 , 127 , 15367–15370 ) CrossRef CAS PubMed.
- L.-S. Wang, A. Gupta and V. M. Rotello, ACS Infect. Dis., 2016, 2, 3–4 CrossRef CAS PubMed.
- S. I. Maffioli, in Antibiotics, ed. C. O. Gualerzi, L. Brandi, A. Fabbretti and C. L. Pon, Wiley-VCH, Weinheim, 2013, pp. 1–22 Search PubMed.
- K. Dua, S. D. Shukla, R. K. Tekade and P. M. Hansbro, Drug Delivery Transl. Res., 2017, 7, 179–187 CrossRef CAS PubMed.
- Y. Liu, H. J. Busscher, B. Zhao, Y. Li, Z. Zhang, H. C. van der Mei, Y. Ren and L. Shi, ACS Nano, 2016, 10, 4779–4789 CrossRef CAS PubMed.
- H.-Z. Lai, W.-Y. Chen, C.-Y. Wu and Y.-C. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 2046–2054 CrossRef CAS PubMed.
- F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand and D. Häbich, Angew. Chem., Int. Ed., 2006, 45, 5072–5129 CrossRef CAS PubMed.
- T. M. Coque, J. F. Tomayko, S. C. Ricke, P. C. Okhyusen and B. E. Murray, Antimicrob. Agents Chemother., 1996, 40, 2605–2609 CrossRef PubMed.
- World Health Organization, Antimicrobial Resistance: Global Report on Surveillance, 2014, https://www.who.int/drugresistance/documents/surveillancereport/en/%3e, 2014, accessed 1–10–17 Search PubMed.
- R. R. Watkins and R. A. Bonomo, Infect. Dis. Clin. North Am., 2016, 30, 313–322 CrossRef PubMed.
- K. E. Jones, N. G. Patel, M. A. Levy, A. Storeygard, D. Balk, J. L. Gittleman and P. Daszak, Nature, 2008, 451, 990–993 CrossRef CAS PubMed.
- V. Yarlagadda, G. B. Manjunath, P. Sarkar, P. Akkapeddi, K. Paramanandham, B. R. Shome, R. Ravikumar and J. Haldar, ACS Infect. Dis., 2016, 2, 132–139 CrossRef CAS PubMed.
- A. Antonoplis, X. Zang, M. A. Huttner, K. K. L. Chong, Y. B. Lee, J. Y. Co, M. R. Amieva, K. A. Kline, P. A. Wender and L. Cegelski, J. Am. Chem. Soc., 2018, 140, 16140–16151 CrossRef CAS PubMed.
- T. Böttcher, I. Kolodkin-Gal, R. Kolter, R. Losick and J. Clardy, J. Am. Chem. Soc., 2013, 135, 2927–2930 CrossRef PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- E. Lei, M. P. Pereira and S. O. Kelley, Angew. Chem., Int. Ed., 2013, 52, 9660–9663 CrossRef CAS PubMed.
- A. Brezden, M. F. Mohamed, M. Nepal, J. S. Harwood, J. Kuriakose, M. N. Seleem and J. Chmielewski, J. Am. Chem. Soc., 2016, 138, 10945–10949 CrossRef CAS PubMed.
- W. B. Kauffman, T. Fuselier, J. He and W. C. Wimley, Trends Biochem. Sci., 2015, 40, 749–764 CrossRef CAS PubMed.
- C. Allolio, A. Magarkar, P. Jurkiewicz, K. Baxova, M. Javanainen, P. E. Mason, R. Sachl, M. Cebecauer, M. Hof, D. Horinek, V. Heinz, R. Rachel, C. M. Ziegler, A. Schrofel and P. Jungwirth, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11923–11928 CrossRef CAS PubMed.
- F. Sgolastra, B. M. Deronde, J. M. Sarapas, A. Som and G. N. Tew, Acc. Chem. Res., 2013, 46, 2977–2987 CrossRef CAS PubMed.
- D. I. Andersson, D. Hughes and J. Z. Kubicek-Sutherland, Drug Resistance Updates, 2016, 26, 43–57 CrossRef CAS PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- Y.-X. Xue, J.-L. Chen, Z.-Q. Jiang, Z.-P. Yu, N. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Polym. Chem., 2014, 5, 6435–6438 RSC.
- H. Sun, Y. Hong, Y. Xi, Y. Zou, J. Gao and J. Du, Biomacromolecules, 2018, 19, 1701–1720 CrossRef CAS PubMed.
- A. Punia, A. Mancuso, P. Banerjee and N.-L. Yang, ACS Macro Lett., 2015, 4, 426–430 CrossRef CAS PubMed.
- R. O. Darouiche and R. J. Hamill, Antimicrob. Agents Chemother., 1994, 38, 1059–1064 CrossRef CAS PubMed.
- P. Sahariah, B. E. Benediktssdottir, M. A. Hjalmarsdottir, O. E. Sigurjonsson, K. K. Sorensen, M. B. Thygesen, K. J. Jensen and M. Masson, Biomacromolecules, 2015, 16, 1449–1460 CrossRef CAS PubMed.
- S. Zhao, W. Huang, C. Wang, Y. Wang, Y. Zhang, Z. Ye, J. Zhang, L. Deng and A. Dong, Biomacromolecules, 2020, 21, 5269–5281 CrossRef CAS PubMed.
- T.-K. Nguyen, S. Lam, K. K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed.
- B. Zhang, M. Li, M. Lin, X. Yang and J. Sun, Biomater. Sci., 2020, 8, 6969–6977 RSC.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- C. Watanakunakorn, Clin. Infect. Dis., 1981, 3, S210–S215 CrossRef.
- N. E. Allen, J. N. Hobbes and T. I. Nicas, Antimicrob. Agents Chemother., 1996, 40, 2356–2362 CrossRef CAS PubMed.
- Y.-F. Zhang, X.-P. Hou, G. Wu, R.-C. Li, X.-W. Qiao, H. T. Tien and A. Ottova, Electrochem. Commun., 1999, 1, 238–241 CrossRef CAS.
- J. M. Hyman, E. I. Geihe, B. M. Trantow, B. Parvin and P. A. Wender, PNAS, 2012, 109, 13225–13230 CrossRef CAS PubMed.
- J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03419k |
| This journal is © The Royal Society of Chemistry 2022 |