
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of the mechanism of NO reduction with ammonia (SCR) on zeolite catalysts†
Konstantin
Khivantsev‡
 *a,
Ja-Hun
Kwak‡
*b,
Nicholas R.
Jaegers
*a,
Iskra Z.
Koleva‡
c,
Georgi N.
Vayssilov
c,
Miroslaw A.
Derewinski
ad,
Yong
Wang
ae,
Hristiyan A.
Aleksandrov‡
*c and
Janos
Szanyi‡
*a
*a,
Ja-Hun
Kwak‡
*b,
Nicholas R.
Jaegers
*a,
Iskra Z.
Koleva‡
c,
Georgi N.
Vayssilov
c,
Miroslaw A.
Derewinski
ad,
Yong
Wang
ae,
Hristiyan A.
Aleksandrov‡
*c and
Janos
Szanyi‡
*a
aPacific Northwest National Laboratory Richland, WA 99352, USA. E-mail: Konstantin.Khivantsev@pnnl.gov; NJaegers@berkeley.edu; Janos.Szanyi@pnnl.gov
bUlsan National Institute of Science and Technology (UNIST), South Korea. E-mail: JHKwak@unist.ac.kr
cFaculty of Chemistry and Pharmacy, University of Sofia, 1126 Sofia, Bulgaria. E-mail: HAA@chem.uni-sofia.bg
dJ. Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, Krakow 30-239, Poland
eThe Gene and Linda Voil and School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA 99164, USA
First published on 10th August 2022
Abstract
Cu/zeolites efficiently catalyze selective reduction of environmentally harmful nitric oxide with ammonia. Despite over a decade of research, the exact NO reduction steps remain unknown. Herein, using a combined spectroscopic, catalytic and DFT approach, we show that nitrosyl ions (NO+) in zeolitic micropores are the key intermediates for NO reduction. Remarkably, they react with ammonia even below room temperature producing molecular nitrogen (the reaction central to turning the NO pollutant to benign nitrogen) through the intermediacy of the diazo N2H+ cation. Experiments with isotopically labeled N-compounds confirm our proposed reaction path. No copper is required for N2 formation to occur during this step. However, at temperatures below 100 °C, when NO+ reacts with NH3, the bare Brønsted acid site becomes occupied by NH3 to form strongly bound NH4+, and consequently, this stops the catalytic cycle, because NO+ cannot form on NH4-zeolites when their H+ sites are already occupied by NH4+. On the other hand, we show that the reaction becomes catalytic on H–zeolites at temperatures when some ammonia desorption can occur (>120 °C). We suggest that the role of Cu(II) ions in Cu/zeolite catalysts for low-temperature NO reduction is to produce abundant NO+ by the reaction: Cu(II) + NO → Cu(I)⋯NO+. NO+ then reacts with ammonia to produce nitrogen and water. Furthermore, when Cu(I) gets re-oxidized, the catalytic cycle can then continue. Our findings provide novel understanding of the hitherto unknown steps of the SCR mechanism pertinent to N–N coupling. The observed chemistry of Cu ions in zeolites bears striking resemblance to the copper-containing denitrification and annamox enzymes, which catalyze transformation of NOx species to N2, via di-azo compounds.
Air pollution is one of the main health and environmental concerns in our (post)industrial society.1–3 Worsened air quality during industrial expansion is attributed to a great extent to toxic nitric oxide (NO) gas, where nearly 55% of the global emissions are due to transportation exhaust. The ability of Cu/zeolites to turn NO to N2 in the presence of ammonia was first discovered in Japan in the 1960s4 for the Cu/FAU zeolite. However, the FAU framework is less stable than the corresponding SSZ-13 and BEA zeolites.5 Within the last decade, the ammonia selective catalytic reduction (SCR) technology was implemented on the large scale5–18 for diesel engines on the basis of Cu(Fe) in BEA and SSZ-13 zeolites.
Some advances were achieved towards the goal of understanding the rate-limiting steps of the SCR mechanism.5–19 Cu(II) ions are present in Cu/zeolite materials as well as Cu(II)–OH ions.5–19,24 The rate-limiting step for low-temperature SCR, for low copper loaded materials (Cu loading below 0.5 wt%), was shown to be the re-oxidation of reduced Cu(I)(NH3)2 complexes via the formation of transient (NH3)2Cu(II)–O2–Cu(II)(NH3)2 dimers.16,17 However, the steps involved in the exact mechanism of NO reduction to N2 have remained unknown,5–19 and the proposed DFT steps were shown hard to prove/observe experimentally.
In this study, we selectively formed NO+ ions in H-BEA zeolite (typical helium ion microscopy images of H-BEA are shown in Fig. S0†) by the reaction of NO with O2 (Fig. 1A).20–22 NO+ is formed through the reaction depicted in eqn (1):
| 2NO + ½O2 + 2 H–zeolite → 2NO+/zeolite− + H2O | (1) |
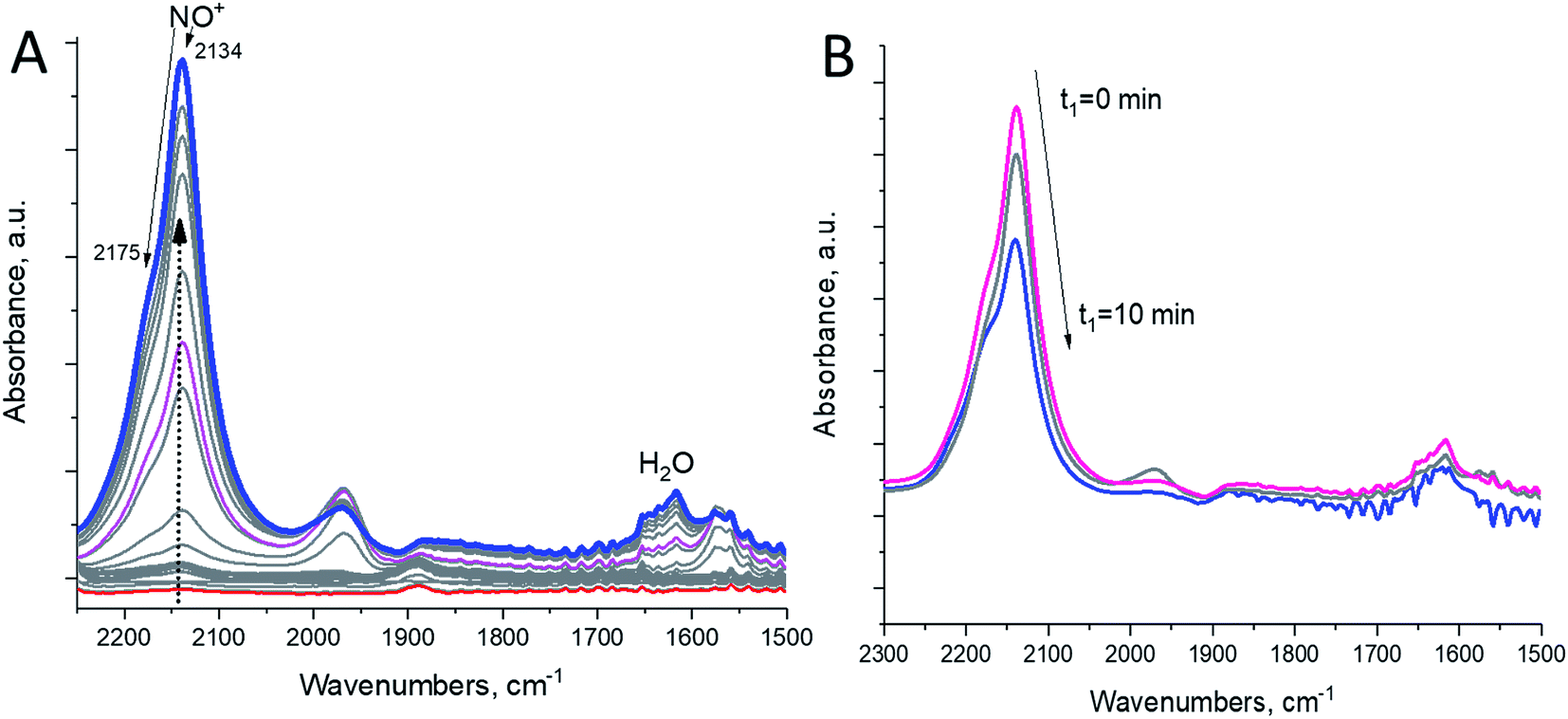 | ||
| Fig. 1 H-BEA zeolite with Si/Al ∼ 15. (A, left spectrum) In situ FTIR during the first sequential NO adsorption (0.1 Torr equilibrium pressure), followed by sequential O2 addition (0.17 Torr total eq. pressure – when O2 is added, NO+ begins to develop in significant amounts); T = 20 °C. (B, right spectrum) FTIR spectra collected after pulling high vacuum 10−8 Torr from time = 0 to time = 10 minutes at 150 °C. Spectra were recorded after cooling back to 20 °C. | ||
NO+ occupies two different cationic positions with the corresponding N–O stretching frequencies at ∼2133 and ∼2175 cm−1. The same type of chemistry also occurs on H-SSZ-13 (Fig. S1†). Note that adsorption of NO2 also produces NO+ but with stoichiometric NO3− amounts due to N2O4 disproportionation (Fig. S2†). Our DFT calculations further corroborate the described chemistry vide infra.
Evacuation to high vacuum (∼10−7 Torr) (with quick heating to 150 °C) allows the removal of excessive NO and O2, leaving the zeolite with NO+ adsorbed in it (Fig. 1B).
We exposed the NO+/zeolite to 15N-labeled ammonia at room temperature (∼20 °C) (Fig. 2A). Ammonia first occupies Brønsted acid sites forming NH4+ complexes which interact with NH3 in the pores, generating (NH3)x cluster networks interacting with NH4+ ions.23,26 During this, NO+ reacts with ammonia, as evidenced by the swiftly diminishing NO+ band, eventually leaving no new visible NO stretches as the excess of 15NH3 produces the complex bands typical for NH4+–(NH3)x clusters in zeolites in the 2250–1750 cm−1 region24 (Fig. 2A).
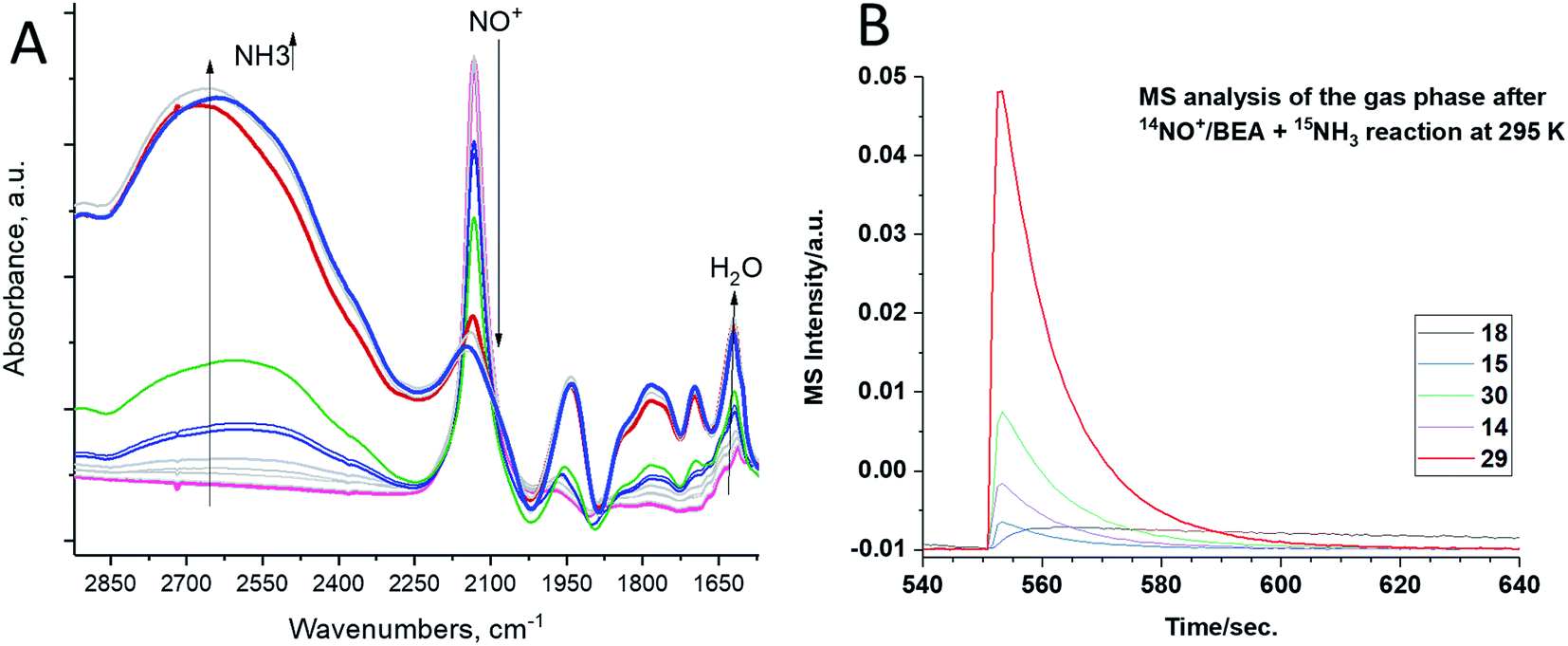 | ||
| Fig. 2 H-BEA zeolite with Si/Al ∼ 15. (A, left spectrum) In situ sequential FTIR during 15NH3 adsorption (total equilibrium pressure 0.020 Torr) on the NO+/H-BEA sample at 20 °C. Ammonia reacts with NO+. The gas phase effluent from the reaction (T = 20 °C) was analyzed with a mass spectrometer attached to an IR cell. (B) shows formation of 14N15N dinitrogen with the characteristic m/z = 29 signal. | ||
The analysis of the gas-phase product by mass-spectrometry reveals a major peak at 29 amu per charge, corresponding to 14N–15N molecules (Fig. 2B). Thus, NO+ reacts with ammonia to form molecular nitrogen at room temperature on H-BEA. Eqn (2) and (3) can be used to describe this process. Note that NO+ does not necessarily have to be bound to zeolite in the presence of NH3 and can initially become solvated by NH3 (it is well-known that ammonia has propensity to solvate cationic species in and outside zeolites;16,17,24 our DFT calculations in Table 1 further confirm strong favorability of NO+ solvation by ammonia with an energy gain of 95 kJ mol−1); the designation zeolite–NO+ is just a representation of the NO+/H–zeolite system in which NO+ may be solvated or semi-solvated by ammonia molecules.
| Zeolite–N14O+ + N15H3 → zeolite–N14O(15NH3) → [zeolite–N14(OH)N15H2] → zeolite–[14/15N2H+] + H2O | (2) |
| [Zeolite–14/15N2H+] → zeolite-H + N14–N15 | (3) |
| Reaction | ΔE, kJ mol−1 | E a, kJ mol−1 |
|---|---|---|
| a Alternative reaction paths: marked in red color in Fig. 7B. b Marked in red color in Fig. 9. c Marked in blue color in Fig. 9. d Marked in red color in Fig. 10. e Marked in blue color in Fig. 10. | ||
| Direct participation of the chabazite | ||
| NO+/Zeo + NH3(g) → NO+(NH3)/Zeo | −97 | — |
| NO+(NH3)/Zeo → NONH2/H+–Zeo | −7 | 5 |
| NONH2/H+–Zeo → NONH2_rotated1/H+–Zeo | −31 | — |
NONH2_rotated1/H+–Zeo → HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH/H+–Zeo NOH/H+–Zeo |
12 | 13 |
| HNNOH/H+–Zeo → HNNOH_rotated1/H+–Zeo |
18 | — |
| HNNOH_rotated1 → (N2 + H2O)/H+–Zeo |
−225 | 8 |
| HNNOH/H+–Zeo → HNNOH_rotated2/H+–Zeoa |
29 | — |
| HNNOH_rotated2/H+–Zeo → (NNH+ + H2O)/Zeoa |
−8 | 14 |
| (NNH+ + H2O)/Zeo → (NNH+_rotated + H2O)/Zeoa | −21 | — |
| (NNH+_rotated + H2O)/Zeo → (N2 + H2O)/H+–Zeoa | −207 | 2 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| No direct participation of the chabazite at some reaction steps | ||
| NO+/Zeo + NH3→ NO+(NH3)/Zeo | −95 | |
| NO+(NH3)/Zeo → NONH2_rotated3/H+–Zeo | −40 | 131 |
| NONH2_rotated3/H+–Zeo → HNNOH_rotated3/H+–Zeo |
41 | 148 |
| HNNOH_rotated3/H+–Zeo → (N2 + H2O)/H+–Zeo | −236 | — |
|
||
| PhNH 2 transformation in the pores of Zeo/NO + | ||
| NO+/Zeo + PhNH2(g) → (PhNH2–NO+)/Zeo | −186 | — |
| (PhNH2–NO+)/Zeo → PhNHNO/H+–Zeo |
5 | 31 |
| PhNHNO/H+–Zeo → PhNHNOH+/Zeo |
−49 | 3 |
| PhNHNOH+/Zeo → PhNNOH/H+–Zeo |
57 | 65 |
| PhNNOH/H+–Zeo → (PhNN+ + H2O)/Zeo |
−69 | 11 |
| (PhNN+ + H2O)/Zeo → PhNN+/Zeo + H2O(g) |
26 | — |
| PhNHNOH+/Zeo → (PhNN+ + H2O)/Zeob |
−12 | 158 |
| PhNHNO/H+–Zeo → PhNNOH_rotated/H+–Zeoc |
−4 | 1 |
| PhNNOH_rotated/H+–Zeo → (PhNN+ + H2O)/Zeoc |
−57 | 23 |
|
||
| Interaction of BF 4 /NO + with NH 3 | ||
| NO+/BF4− + NH3(g) → NO+(NH3)/BF4− | −91 | — |
| NO+(NH3)/BF4− → NONH2/BF3–HF | 15 | 16 |
| NONH2/BF3–HF → NONH2_rotated1/BF3–HF | −42 | — |
| NONH2_rotated1/BF3–HF → HNNOH/BF3–HF |
−3 | 2 |
| HNNOH/BF3–HF → HNNOH_rotated1/BF3–HF |
31 | — |
| HNNOH/BF3–HF_rotated1 → (N2 + H2O)/BF3–HF |
−221 | 1 |
| HNNOH/BF3–HF → HNNOH_rotated2/BF3–HF d |
44 | — |
| HNNOH_rotated2/BF3–HF → (NNH+ + H2O)/BF4−d |
−17 | 16 |
| NONH2/BF3–HF → NONH2_rotated2/BF3–HFe | −25 | — |
| NONH2_rotated2/BF3–HF → HNNOH_rotated3/BF3–HFe |
17 | 126 |
The hydronium diazonium cation (N2H+) is extremely unstable. Its immediate decomposition to N2 and H–zeolite drives the reaction forward. Despite this, we find that at sufficiently high molecular N2 pressure in the cell (∼15 Torr), we can observe a small N–N stretch of the –HN2+ complex at 2334 cm−1 even at room temperature (Fig. 3A), which was previously shown to form at low temperatures upon N2 interaction with Brønsted acid sites (Fig. 3B).25–27
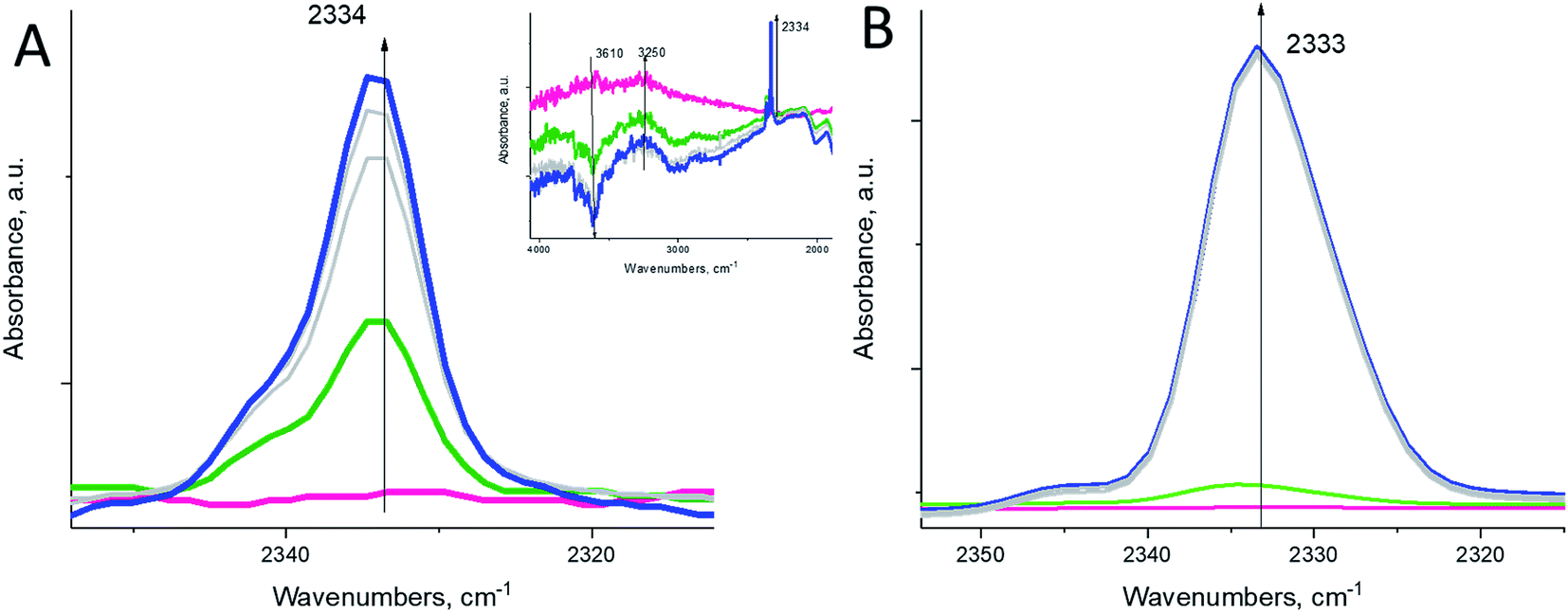 | ||
| Fig. 3 (A, left spectrum) In situ FTIR during sequential dinitrogen N2 adsorption (equilibrium pressure ∼ 15 Torr) on H-BEA at ∼23 °C. A very unstable zeolite–[HN2+] complex can be observed even at room temperature, but it requires elevated N2 pressure. The inset shows that N2 interacts with Brønsted acid sites of Si–OH–Al zeolite groups (OH stretch of such groups is located at ∼3610 cm−1). (B, right spectrum) In situ FTIR during adsorption of only ∼0.1 Torr N2 on the same H-BEA sample (equilibrium pressure is 0.001 Torr) at 77 K produces N–N stretches of N2 adsorbed on Brønsted acid sites. Note that in this case, even low equilibrium pressure of nitrogen produces intense N–N stretches. | ||
To provide proof that the reaction proceeds through a diazo compound, we chose aniline PhNH2, an equivalent of the ammonia molecule but with 1 hydrogen atom substituted by a phenyl group, and it reacted with NO+ in the BEA zeolite. The phenyl group stabilizes the formation of PhN2+ salts (phenyl diazonium salts) through the mesomeric effect, and unlike alkyl diazonium salts, aryl diazonium compounds are stable and characterized by N–N stretches in the ∼2250–2300 cm−1 region, more specifically ∼2270 cm−1 for phenyl diazonium in solution.28 Indeed, we monitored the reaction of NO+ and PhNH2, spectroscopically: the intensity of the NO+ band diminished, and a new N–N stretch appeared at ∼2270 cm−1, corresponding to the N–N vibration of the Ph–N2+ fragment (Fig. 4). The reaction of NO+ with NH3 is more sluggish than with NH3, consistent with the higher activation barrier calculated for PhN2+ formation (Table 1). This is the first observation of a diazo-salt stabilized on a solid support (zeolite).
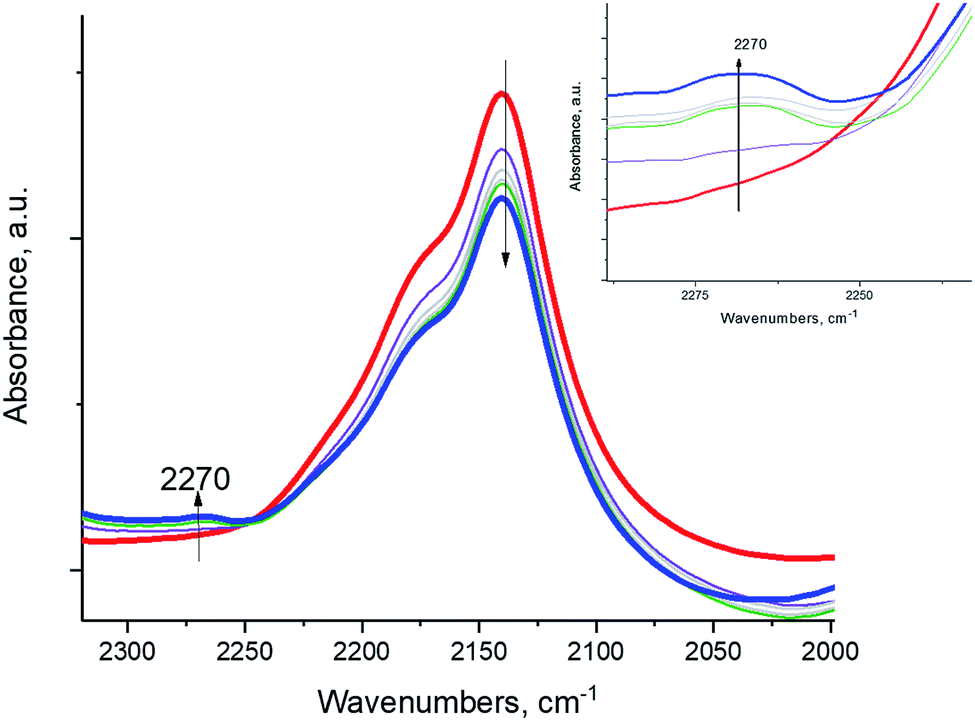 | ||
| Fig. 4 In-situ FTIR during sequential ∼0.3 Torr aniline PhNH2 adsorption (equilibrium pressure ∼ 0.03 Torr) NO+/H-BEA. | ||
NO+ in H-SSZ-13 (with Si/Al ∼ 12; typical HAADF-STEM images of this sample are shown in Fig. S7†) reacts similarly with NH3 (Fig. 5), with concomitant N2 evolution (confirmed by mass spectrometry):
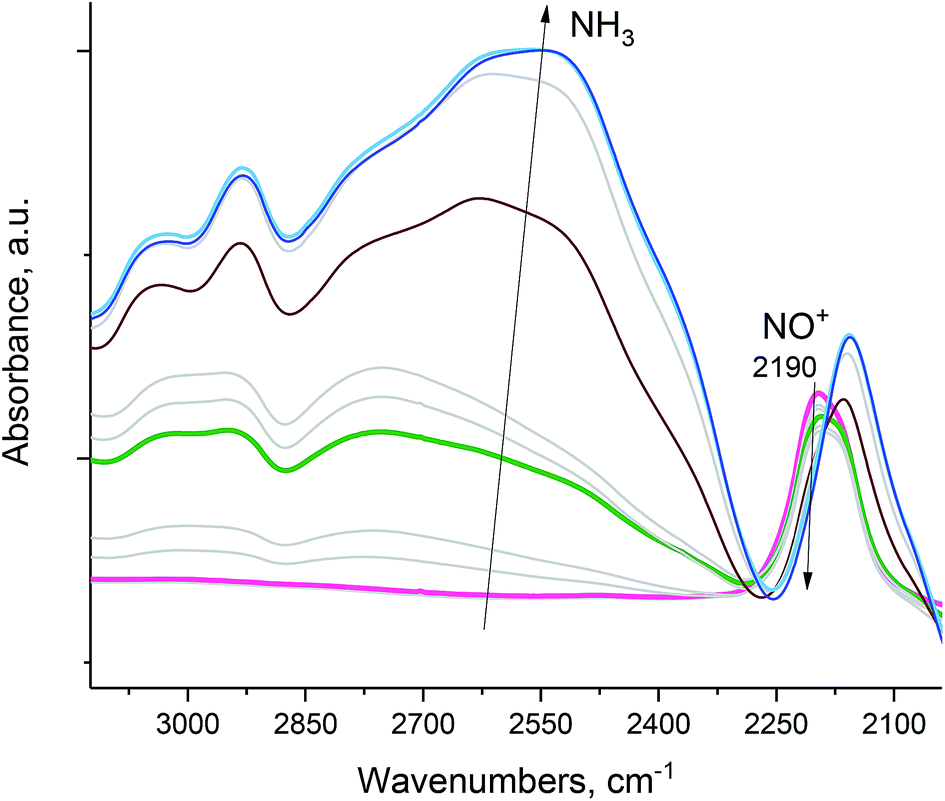 | ||
| Fig. 5 H-SSZ-13 zeolite with Si/Al ∼ 12. In situ sequential FTIR during 14NH3 adsorption (total equilibrium pressure ∼ 0.15 Torr) on the NO+/H-SSZ-13 sample at 20 °C. Ammonia reacts with NO+ as evidenced by the disappearance of the NO+ stretch. The intense band at ∼2110 cm−1 is due to zeolite interactions with NH3. Pulling vacuum on the sample at 120 °C produces spectra showing no NO+ stretches (Fig. S3†), consistent with the complete reaction of NO+. | ||
As such, NO+ is the critical intermediate species in the conversion of NO in these zeolites. Copper is not required to observe the NO+ reactivity with NH3. Moreover, we reacted inorganic nitrosyl salt NO+ with 15NH3 and observed 14N–15N in the gas-phase (consistent with our findings for NO+ in zeolites; this reaction takes place vigorously even at a temperature as low −50 °C) (eqn (4)):
| 14NO[BF4] + 15NH3 (at −50 °C) → 14N15N (gas) + H2O + H[BF4] | (4) |
This reaction (eqn (4)) most likely proceeds through the N2H+ intermediate as well.
For NO+ in the zeolite, once it reacts with NH3 with the release of N2, the Brønsted acid is free and immediately interacts with ammonia to produce NH4+. This latter process “kills” the reactivity as the NO+ species can no longer be produced due to the necessity of Brønsted acid sites, as evidenced by FTIR (Fig. S4†); indeed, no NO+ evolves above trace amounts upon the NO + O2 reaction on the NH4–zeolite (Fig. S4†). Only at elevated temperatures, when some NH3 can desorb and free up a portion of Brønsted acid sites to re-form NO+, can the reaction proceed catalytically which we show in Fig. S5.† The bare H-BEA zeolite is catalytically active for NO reduction with NH3 in dry streams (see Fig. S5† for reactivity at 200 and 150 °C with time-on-stream) (measurable activity is observed). NO+ is formed in zeolite through eqn (1) (see the earlier discussion in the manuscript).
With these new data, we can now further explain the possible role of Cu in zeolites for SCR. As is well-established in the literature, Cu(II) ions are required for the continuous catalytic reaction to proceed.5–19 In the presence of NO, Cu(II) ions can produce NO+via 1-electron reduction of Cu(II) (analogous chemistry is observed, for example, for Pd(II) in zeolites30 where Pd(II) was shown to reduce to Pd(I) by NO pulses with the concomitant formation of NO+) (eqn (5)):
| Zeo2Cu(II) + NO → Zeo1Cu(I)⋯NO+–Zeo1 | (5) |
Fig. 6A shows spectroscopic evidence of Cu(II) reduction by NO (when Cu(II) is first reduced to Cu(I) prior to NO adsorption, very little NO+ formation occurs because electron transfer does not take place in the absence of Cu(II) as demonstrated in Fig. 6B).
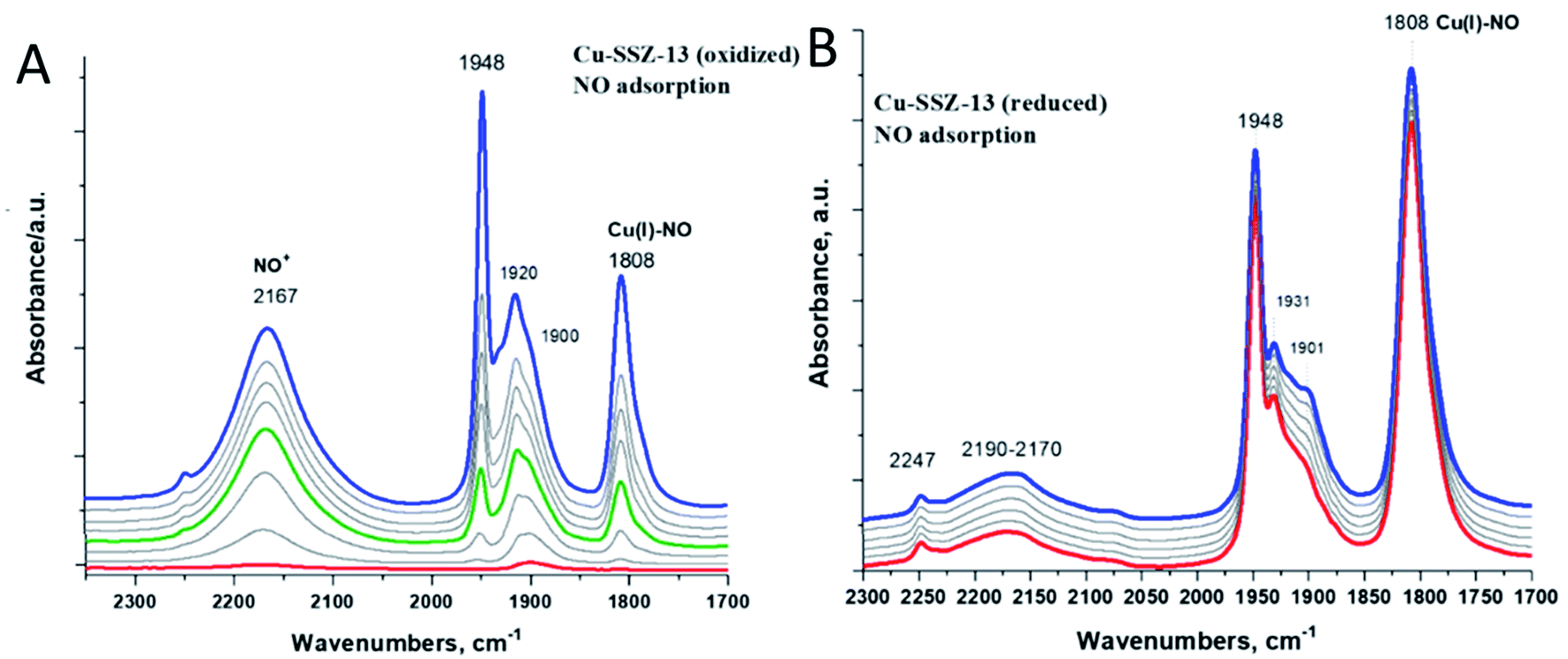 | ||
| Fig. 6 1 wt% Cu/H-SSZ-13 sample with Si/Al ∼ 12. (A) In situ FTIR during 1 Torr NO adsorption on the pre-oxidized sample (pre-oxidized in O2 at 300 °C). NO+ and Cu(I)–NO evolve simultaneously from Cu(II) reduction by NO. (B) The same sample (tablet) was pre-reduced in the IR cell (at 300 °C): 1 Torr NO adsorption (same equilibrium pressure) after reduction. | ||
We have previously been able to confirm by solid-state 15N NMR studies that NO indeed is capable of reducing Cu(II) to Cu(I) in SSZ-13: the resulting complex had Cu(I) and NO+ in proximity to each other, with NO+ located side-on towards a copper ion.19 Similar chemistry is observed for Pd(II) in zeolites.29
The mechanism of NO+–zeolite formation from the H+–zeolite and NO and/or NO2 has been investigated experimentally22,34 and theoretically.35 It was shown35 that the barrier for NO+ formation on H+-CHA from NO2 is only 15 kJ mol−1, while the process is endothermic by only 2 kJ mol−1. Our novel experimental findings for NO+ in zeolites prompted us to investigate the proposed reaction steps with density functional theory (DFT) calculations. First, we investigated two pathways for selective catalytic reduction of NO by the ammonia via formation of the NO+ species in the zeolite: (1) with the direct (Fig. 7, Table 1) participation of the zeolite and (2) without (Fig. 8, Table 1) the direct zeolite participation at some reaction steps. Both pathways start with adsorption of NH3 to NO+/Zeo with a binding energy of ammonia of ∼97 kJ mol−1 and subsequent formation of nitrosamine via transfer of one of the H atoms from the ammonia to an O center from the AlO4 tetrahedron.
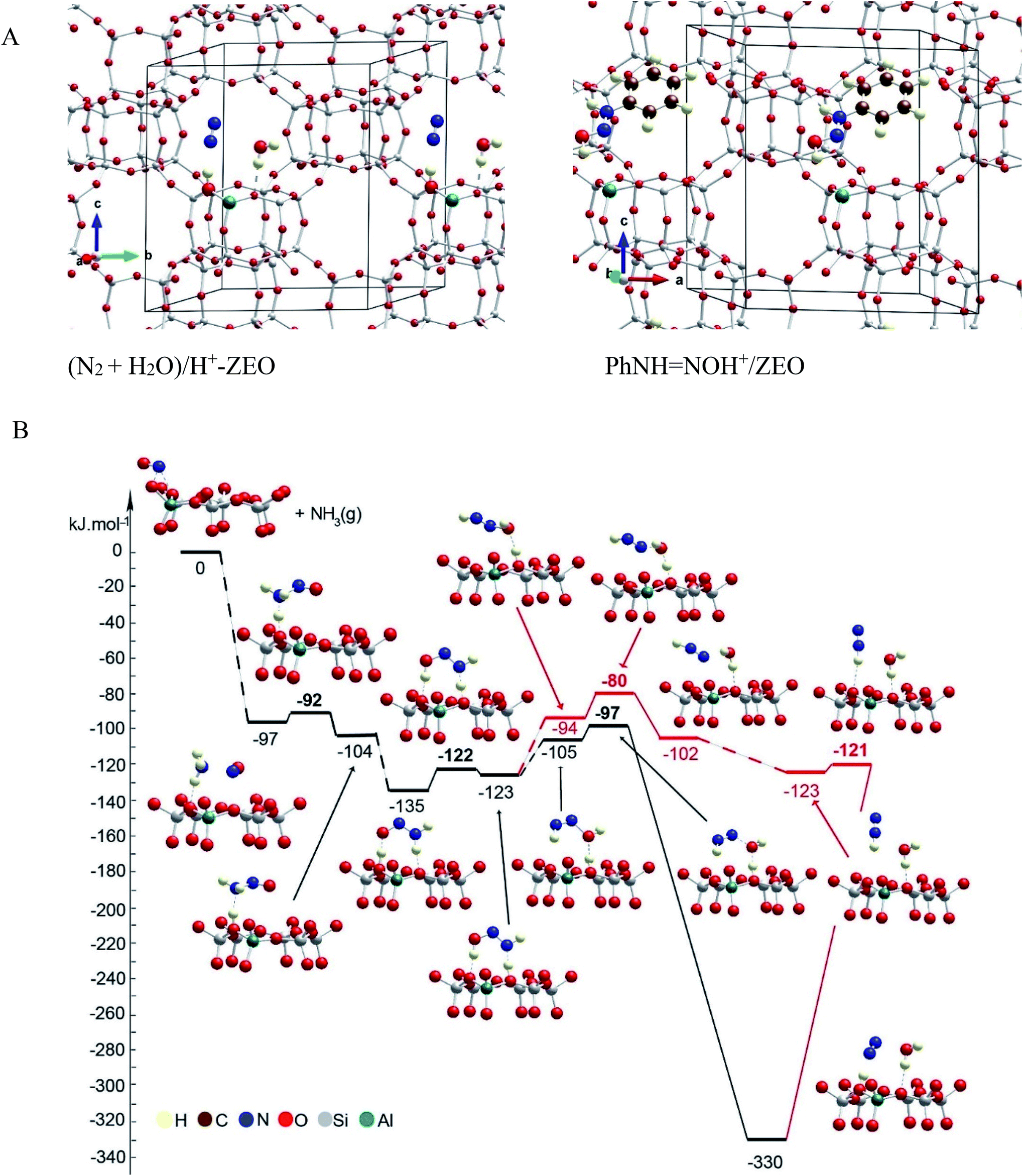 | ||
| Fig. 7 (A) General location of the species in the pores of the unit cell of chabazite. Color coding: H – yellow; N – blue; C – brown; O – red; Al – green; Si – gray. (B) Energy diagram and optimized models of reaction steps with the direct participation of the chabazite. Alternative reaction paths are shown in different colors. The models are visualized with the VESTA program.33 | ||
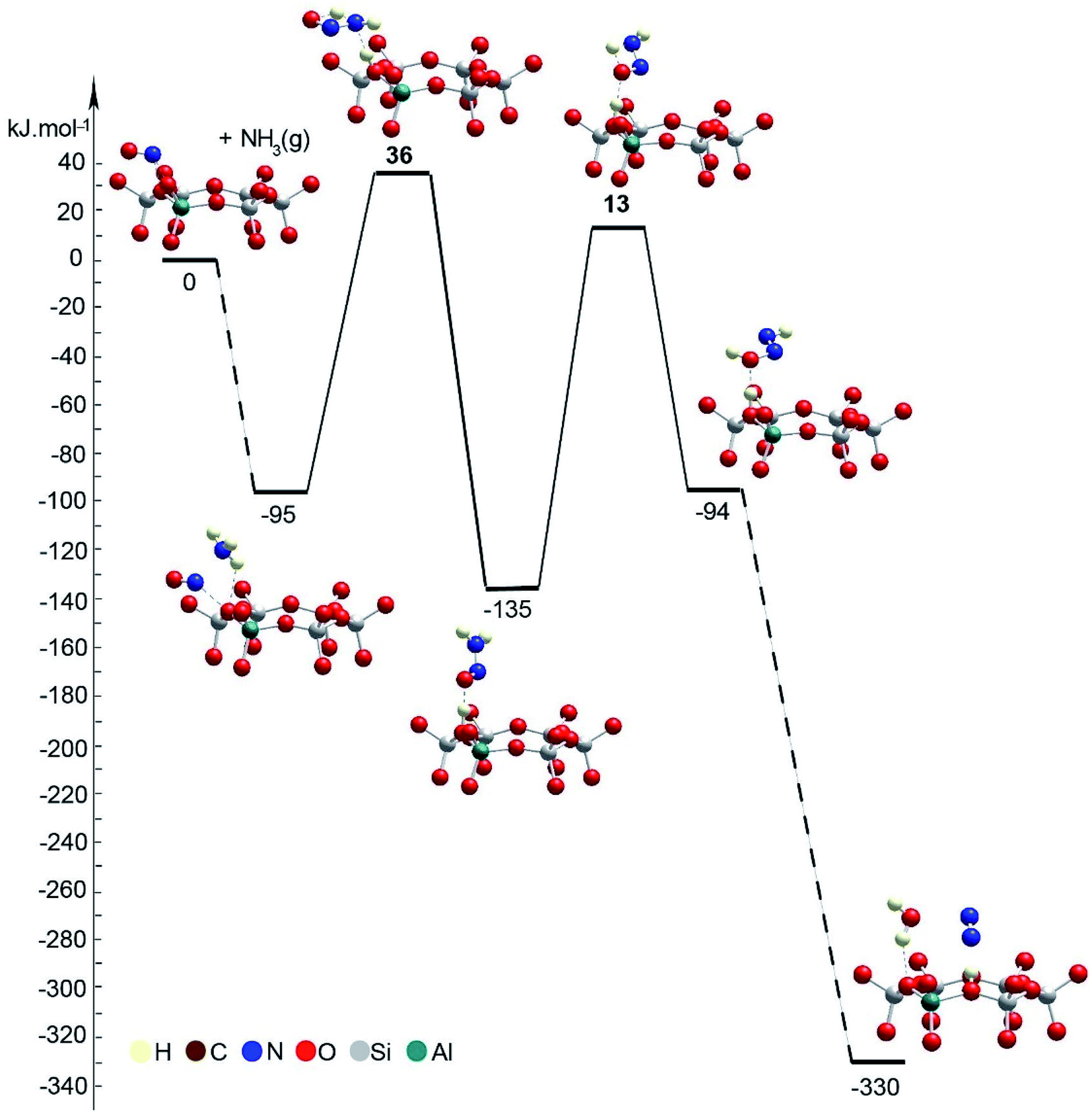 | ||
| Fig. 8 Energy diagram and optimized models of reaction steps with no direct participation of the chabazite at some reaction steps. The models are visualized with the VESTA program.33 | ||
When the reaction step occurs via direct H transfer, the energy barrier is only 5 kJ mol−1 (Fig. 7B, Table 1). We also found another transition state (TS) structure with elongated N–H distance (Fig. 8, Table 1), and it is less stable by more than 120 kJ mol−1 than the previous one, despite the fact that the zeolite participates in the reaction step since one of the H atoms from NH3 is transferred to the zeolite as a proton. Further, one of the H atoms of the NH2 group of the nitrosamine should migrate to the O center of the same molecule thus forming a HONNH molecule. This can be done via H transfer occurring in the gas phase without the participation of the zeolite. In this case however, the reaction step is endothermic by 41 kJ mol−1, while the barrier is as high as 148 kJ mol−1. Alternatively, the process can occur stepwise with the direct participation of the zeolite support. In this case initially the molecule of nitrosamine reorientates so that two hydrogen bonds are formed between the guest molecule and support: the N–H fragment with the O center from a zeolite and a zeolite proton with the O center from the nitrosamine. The new configuration is more stable by 31 kJ mol−1 than the previous one.
Further, via a synchronous transition state structure, the H atom from the NH fragment moves to the zeolite O center, and simultaneously, the zeolite proton migrates to the O atom from the guest molecule. The energy barrier is only 13 kJ mol−1, while the reaction step is only slightly endothermic by 12 kJ mol−1. Similar energetics were also reported previously.36 In the next step, the HONNH molecule should be converted into the final products: H2O and N2. This can be done again via direct H transfer in the gas phase from the N to the O atom with no direct participation of the zeolite support. Based on the previous knowledge, it is expected that the reaction step will have a high barrier. Alternatively, we considered the process with the participation of the zeolite, as two reaction pathways were considered: concerted and stepwise pathways, as in both cases, initially, the guest molecule changes its position with respect to the zeolite support as one of the hydrogen bonds, NH⋯O, breaks. The new structures are less stable by 18 and 29 kJ mol−1 than the final state structure of the previous reaction step. In the concerted mechanism, in one step, the zeolite proton moves to the OH group from the HONNH species, thus forming a water molecule, while the H bound to the N atom moves to an O zeolite center forming a N2 molecule. The reaction step is strongly exothermic, −225 kJ mol−1, with a very low barrier, 8 kJ mol−1. The high exothermicity36,37 and the low barrier36 were also inferred in previous theoretical studies. Alternatively, in the stepwise mechanism, H2O and NNH+ can be formed first, overcoming a low barrier of 14 kJ mol−1 as the reaction step is slightly exothermic, −8 kJ mol−1. Next, NNH+ reorients so that a hydrogen bond is formed with the zeolite O center. In the final step, H moves to the zeolite O center; this bridging OH group and N2 molecule are formed. This step is essentially barrierless and strongly exothermic, −207 kJ mol−1. These DFT results confirm favorability of NO+ interaction with NH3 to form N2 basically with little to no barriers, consistent with the experimentally observed low-temperature reactivity of these species. Similar mechanisms of NH2NO decomposition were reported36,37 as they considered additional steps of transfer of the zeolite proton from one basic O center to another assisted by the O or N atom in the HNNOH, which seems to be the highest ones according to the latter study. According to us, such steps can be omitted as shown by the mechanism proposed by us.
Next, we modeled the reduction process using aniline PhNH2 interacting with the NO+ species positioned as a charge-compensating cation in chabazite (Fig. 9, Table 1).
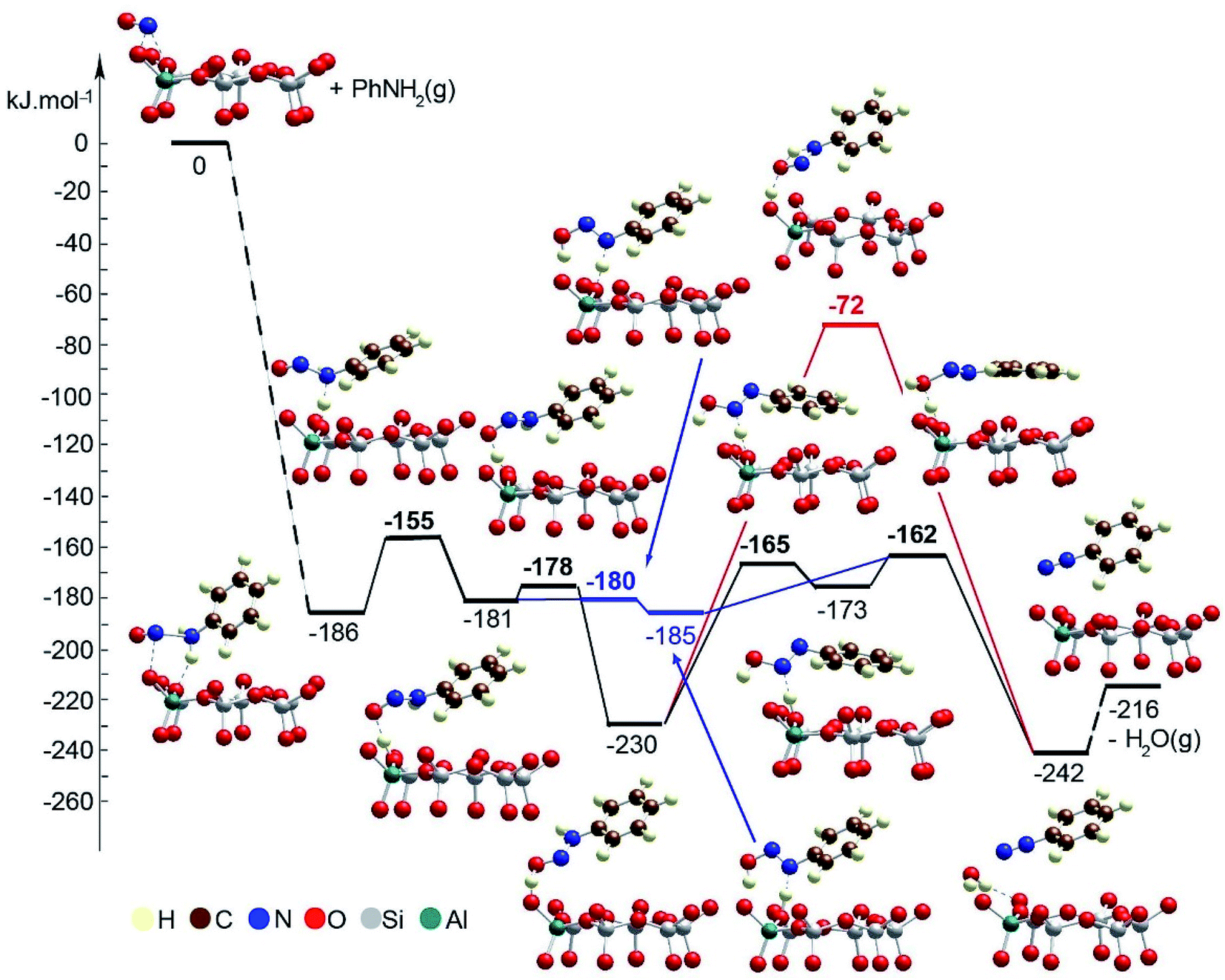 | ||
| Fig. 9 Energy diagram and optimized models of the reaction steps for the PhNH2 transformation in the pores of Zeo/NO+. The models are visualized with the VESTA program.33 | ||
The adsorption energy of aniline to NO+ species is −186 kJ mol−1. Further, one of the H atoms of the amino groups migrates to the O center from the zeolite, thus forming ONNHPh coordinated to the zeolite proton. This reaction step is slightly endothermic by 5 kJ mol−1, and its energy barrier is 31 kJ mol−1. From the latter structure, one can obtain a benzenediazonium cation, PhNN+, via two alternative pathways:
– In the more plausible pathway, the HONNPh structure is formed via a synchronous TS structure in which a zeolite proton migrates onto the O atom from the organic molecule, while the H atom from the NH fragment moves to a zeolite O center. The reaction step is essentially barrierless and very slightly exothermic, −4 kJ mol−1. Further, the zeolite proton interacts with the OH group from the organic molecule which leads to formation of PhNN+ and a water molecule. The barrier for this reaction step is 23 kJ mol−1, and it is exothermic by 57 kJ mol−1.
– The alternative pathway requires overcoming of higher barriers than in the first one. In the first step, the zeolite proton migrates to the O center from the organic molecule as PhNHNOH+ is formed. The process is essentially barrierless and exothermic by 49 kJ mol−1. At the final stage, a PhNN+ cation should be formed, as we considered two possible reaction pathways. In the concerted mechanism, in one step, the O center forms bonds with both H centers which are initially positioned at the zeolite O center and the N atom from the organic molecule. The second H transfer is in the gas phase without direct participation of the zeolite which leads to an unstable transition state structure, and thus, the barrier is 158 kJ mol−1, and reaction energy is only slightly exothermic by 12 kJ mol−1. In the stepwise mechanism, a H atom bound to the N center is transferred to an O zeolite center forming a HONNPh molecule. This reaction step is endothermic by 57 kJ mol−1, and the barrier is 65 kJ mol−1. Finally, the zeolite proton interacts with the OH group which leads to formation of H2O and a PhNN+ cation. This step requires overcoming of a very low barrier, 11 kJ mol−1, and it is exothermic by 69 kJ mol−1.
Furthermore, because we observed that the NO+ reaction with ammonia is not exclusive to zeolite and that inorganic nitrosyl salts (such as NO[BF4]) react with ammonia to form molecular nitrogen at a temperature as low as −50 °C, two pathways for the SCR reaction with ammonia on BF4−/NO+ were investigated: (1) with and (2) without the direct participation of the boron fluoride complex (Fig. 10, Table 1). Both schemes start with adsorption of ammonia onto the BF4−/NO+ substrate forming the H3NNO+ species, as the binding energy is −91 kJ mol−1. In the first step, one of the H atoms of H3NNO+ moves to the BF4− anion. The reaction step is slightly endothermic by 15 kJ mol−1 and requires overcoming a barrier of 16 kJ mol−1. In the final structure, the nitrosamine and hydrogen fluoride are produced and bound by a hydrogen bond between H from the BF3–HF complex and the N from the amino group. Further, a rearrangement of the complex occurs, so that the O atom interacts with the proton from the BF3–HF complex. This complex is more stable by 25 kJ mol−1 than the previous one, while if a second hydrogen bond is formed, a further stabilization of 17 kJ mol−1 is achieved. In the next step, nitrosamine transforms into HNNOH. This can be achieved without direct participation of the BF3–HF substrate as one of the H atoms migrates from the amino group to the O atom. This requires overcoming of a very high barrier, 126 kJ mol−1, as the step is slightly endothermic, 17 kJ mol−1. Alternatively, HNNOH can be formed via a synchronous transition state structure with the participation of the BF3–HF substrate. Synchronously, the proton from HF migrates to the O center from the nitrosamine molecule, while one of the H atoms from the amino group migrates back to another F− center. In this way, the process is essentially barrierless and energetically neutral. Further, there are three possibilities for the HNNOH molecule to be transformed into N2 and H2O: (1) migration of H from NH to the O center without participation of the BF3–HF complex (this way was not modeled since it is expected that such a reaction step will occur with a very high barrier); (2) via a synchronous transition state structure where H2O and N2 are formed in one step; (3) a concerted mechanism where in the first step, H2O and NNH+ are formed, and afterwards, N2 is formed as the H+ migrates to the BF4− moiety. The second concerted mechanism is the most probable as it is essentially barrierless and highly exothermic by 221 kJ mol−1.
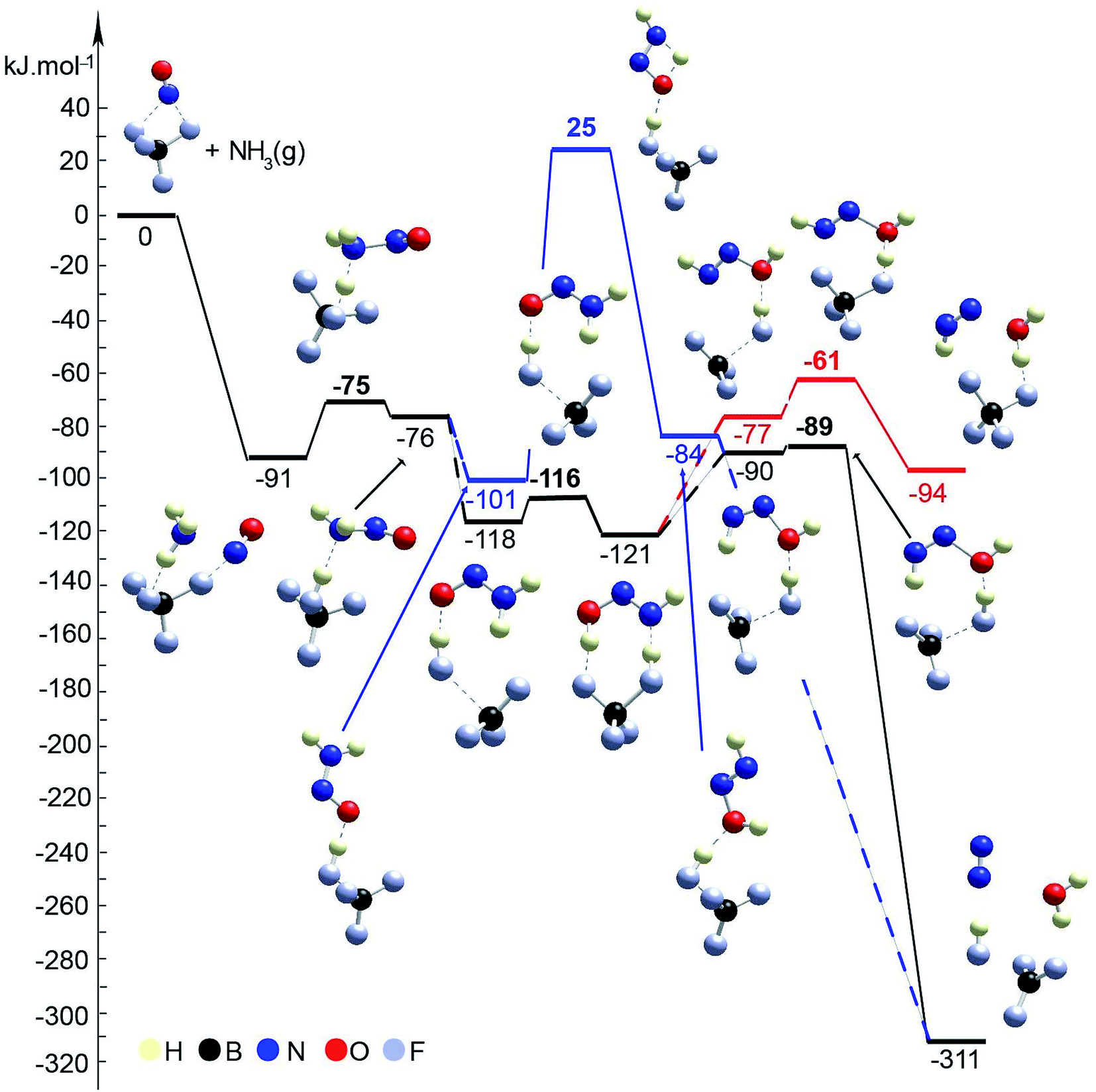 | ||
| Fig. 10 Energy diagram and optimized models of reaction steps for the interaction of BF4/NO+ with NH3 (with and without direct participation of BF4−). The models are visualized with the VESTA program.33 | ||
In order to assess the favorability of Cu(II) reduction by NO in SSZ-13, we considered the reduction of Cu2+ located in six and eight membered rings of a CHA type zeolite structure containing two Al centers by NO (the structures are shown in Fig. 11). First, we modeled formation of a Cu2+(NO) complex in the zeolite, which is exothermic by −145 and −203 kJ mol−1, respectively. In the second step, the complex converts to two cationic species (Cu+ and NO+); each of them compensates for one Al center. This step is endothermic by 31 and 43 kJ mol−1. Thus, the overall exothermicity of the reduction of Cu2+ by NO to Cu+ and NO+ is −114 and −160 kJ mol−1, respectively. The second reaction step seems to be also kinetically feasible, as the calculated barrier for the case of Cu2+ located in the six membered ring of CHA is 62 kJ mol−1. These DFT results are fully consistent with our experimental findings.
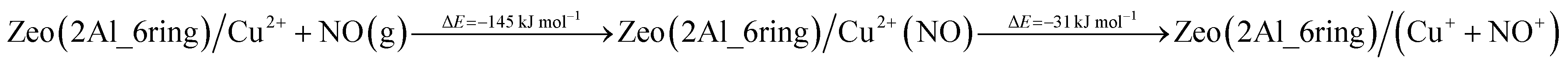
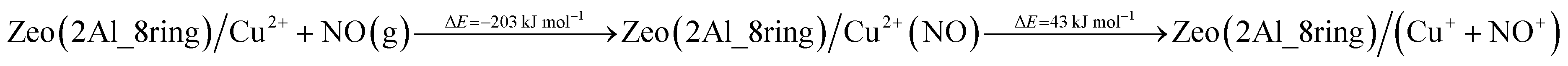
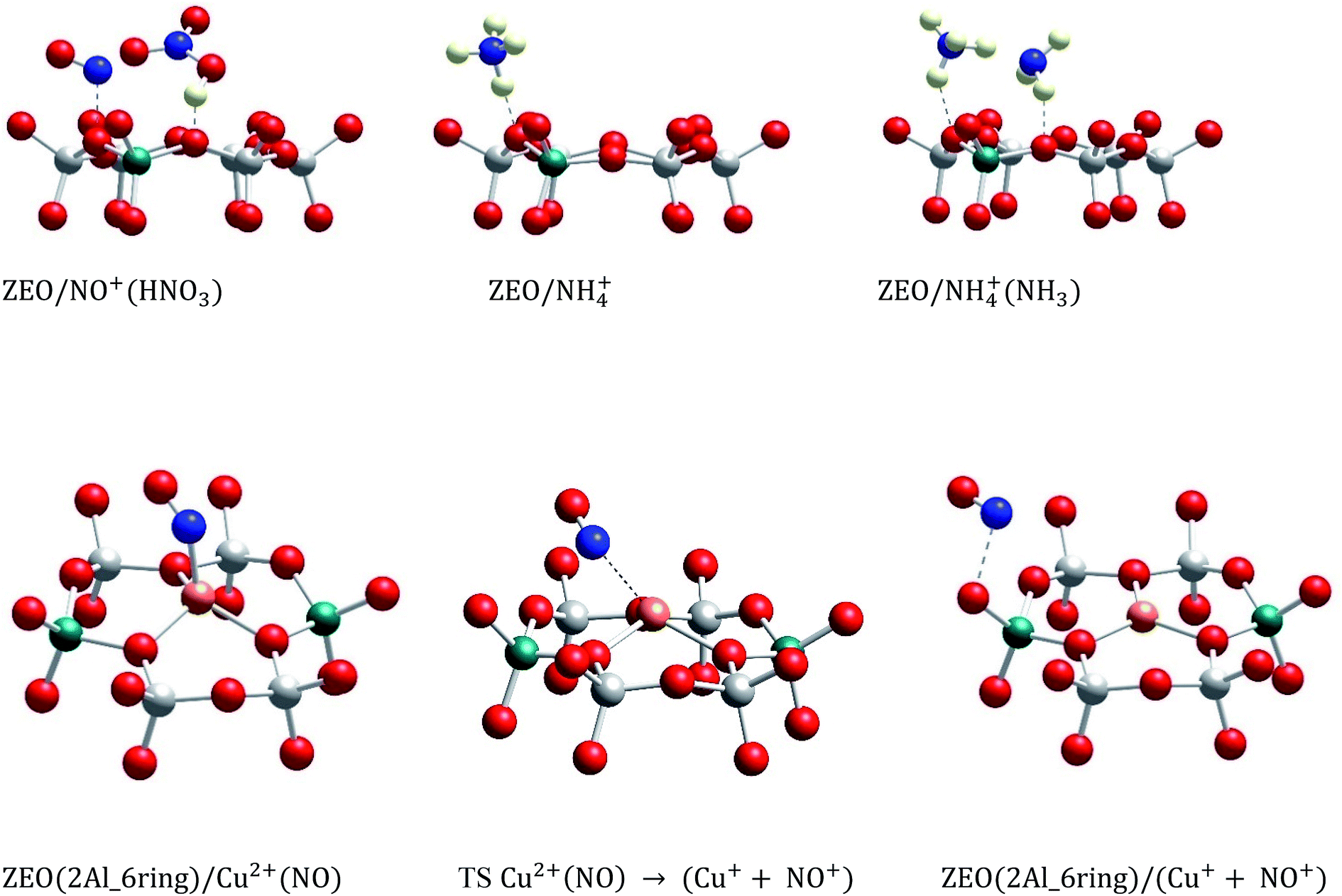 | ||
| Fig. 11 Optimized structures of selected models. The models are visualized with the VESTA program.33 | ||
Additionally, we modeled the steps regarding NO+ formation and NH3 adsorption in the zeolite which are all highly favorable and exothermic. In agreement with earlier experimental studies for NH3 adsorption, interaction of NH4+ ions with NH3 is also very favorable.24
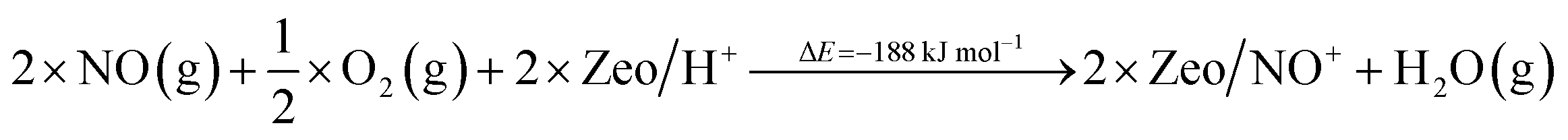 | (6) |
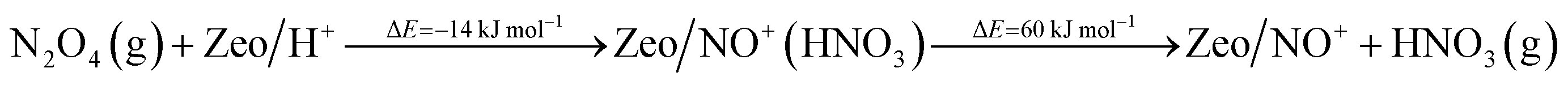 | (7) |
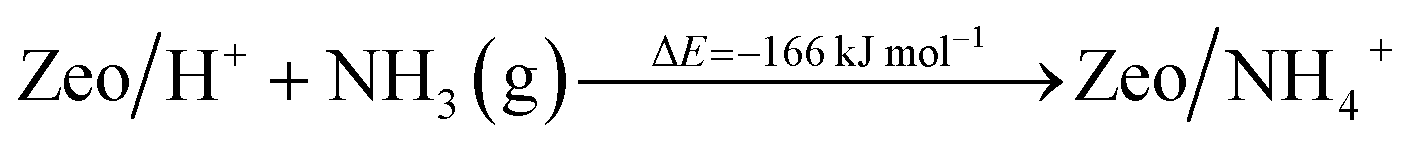 | (8) |
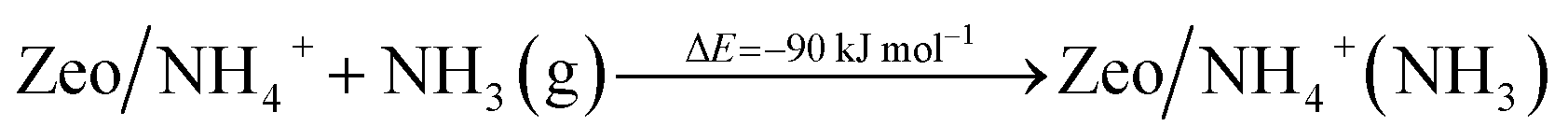 | (9) |
To summarize the DFT results, we investigated two pathways for the NO reduction reaction with ammonia on both substrates (zeolite/NO+ and BF4−/NO+): (1) with and (2) without the direct participation of the substrate. When the reaction occurs on the zeolite/NO+ system with the participation of the zeolite (Fig. 7), the barriers are very low (all of them are below 20 kJ mol−1), manifesting that the reaction can occur at very low temperatures, in line with our experimental results. However, if the zeolite does not participate in the catalytic process directly, the barriers are >130 kJ mol−1 (Fig. 8). According to our calculations, nitrosamine (NH2NO) can be formed as an intermediate (see Fig. 7), but the barrier for its transformation is very low. Similarly, the NNH+ species can be formed as well (Fig. 7), which we prove experimentally through studies with the phenyl diazonium cation (that is more stable than NNH+ shown in Fig. 4). The results with the other substrate, BF4−/NO+, are similar (Fig. 10). When NH3 is decomposed with the participation of the HF–BF3 substrate, the barriers are very low, while if the substrate does not participate in the reaction directly, some of the barriers become higher than 120 kJ mol−1. Similar results are also found for the aniline reduction (Fig. 9), where PhNN+ can be formed via the catalytic role of the zeolite as the highest barrier in the initial rate-limiting step is only 31 kJ mol−1.
Based on these combined theoretical and experimental data, we can suggest that the role of copper is to promote NO+ formation since Brønsted acid sites get occupied by NH4+ after NO+ reacts with NH3 on the bare H–zeolite and cannot contribute to NO+ formation as we show herein. The resulting NO+ reacts with ammonia to reform N2. The Cu(I)(NH3)2 complex, in turn, gets re-oxidized back to Cu(II) with oxygen.16,17 Notably, this bears striking resemblance to the recent advances in enzymatic chemistry of Cu-containing enzymes for denitrification and annamox (anaerobic ammonia oxidation to nitrogen) processes. Only in recent decades, the pioneering studies of Murphy and co-workers (for denitrification)31 and Kartal, Strous, and co-workers (for annamox)32 revealed the central role of the Cu(I)⋯NO+ intermediate and diazo-compounds in these processes. Our data point to the presence of a similar active site for the Cu–zeolite system (used industrially in vehicles) and Cu-enzymes, occurring in nature.
We note that our findings represent the first observation of the potential intermediates of NO reduction with ammonia (SCR) responsible for N–N bond formation for zeolites and copper–zeolite systems and lay out the strategy to investigate vehicle-relevant SCR under more complex gas feed conditions (that include water and hydrocarbons, in addition to oxygen, NO and ammonia); under these complex vehicle-relevant gas conditions, additional mechanistic pathways, leading to N–N coupling, may also be operative.
Data availability
The data are available in the main text and the ESI.†Author contributions
KK conceived the project, performed most synthesis, catalytic, infra-red experiments, analyzed data and wrote the manuscript, KK and NRJ obtained funding through the QuickStarter LDRD program at PNNL. NRJ, JHK, MAD, JS performed synthesis and spectroscopy experiments, analyzed data and contributed valuable discussion. HAA, IZK, GNV perfromed all DFT calculations, discussed the data and co-wrote the DFT section of the manuscript. KK, MAD, NRJ, JHK, IZK, HAA, GNV, YW and JS discussed all the data and the final manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
KK and NRJ performed research described in this paper as a part of the Quickstarter Initiative at Pacific Northwest National Laboratory. It was conducted under the Laboratory Directed Research and Development (LDRD) Program at PNNL, a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy (DOE). IZK is thankful for the financial support received from the program “Young scientists and Postdoctoral candidates” of the Bulgarian Ministry of Education and Science, MCD No. 95/20.12.2019. HAA is grateful for the support by the European Regional Development Fund and the Operational Program “Science and Education for Smart Growth” under contract UNITe No. BG05M2OP001-1.001-0004-C01 (2018–2023). GNV acknowledges the support of the project EXTREME, funded by the Bulgarian Ministry of Education and Science, D01-76/30.03.2021, through the programme “European Scientific Networks”. The research at PNNL was supported by the U.S. Department of Energy, Energy Efficiency and Renewable Energy, Vehicle Technology Office. The experiments were conducted in the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research at Pacific Northwest National Laboratory (PNNL). PNNL is a multi-program national laboratory operated for the DOE by Battelle Memorial Institute. We acknowledge the support of CLEERS (Crosscut Lean Exhaust Emissions Reduction Simulations). CLEERS is an initiative funded by the U.S. Department of Energy’s (DOE) Vehicle Technologies Office to support the development of accurate tools for use in the design, calibration, and control of next generation engine/emission control systems that maximize efficiency while complying with emission regulations. The previous version of this manuscript was published as a pre-print on the pre-print server ChemRxiv on Oct 23rd, 2020, https://doi.org/10.26434/chemrxiv.13134770.v1.References
- Royal College of Paediatrics and Child Health, Every breath we take—the lifelong impact of air pollution, Royal College of Paediatrics and Child Health, London, 2016 Search PubMed.
- W. M. H. Sachtler, Catalysis from Art to Science, in Surface Chemistry and Catalysis. Fundamental and Applied Catalysis, ed. A. F. Carley, P. R. Davies, G. J. Hutchings and M. S. Spencer, Springer, Boston, MA, 2002 Search PubMed.
- M. K. Khair and W. A. Majewski, Diesel emissions and their control; SAE International, Warrendale, 2006 Search PubMed.
- T. Seiyama, T. Arakawa, T. Matsuda, N. Yamazoe and Y. Takita, Catalytic reduction of nitric oxide with ammonia over transition metal ion-exchanged zeolites, Chem. Lett., 1975, 781 CrossRef CAS.
- S. I. Zones, Zeolite SSZ-13 and its method of preparation, US Pat., 4 544 538, 1985.
- J.-H. Kwak, R. G. Tonkyn, D. H. Kim, J. Szanyi and C. H. Peden, Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3, J. Catal., 2010, 275, 187–190 CrossRef CAS.
- I. Bull, A. Moini, G. Koermer, J. Patchett, W. Jaglowski and S. Roth, Zeolite catalyst with improved NOx reduction in SCR, US Pat., US20070134146A1, 2010.
- D. W. Fickel and R. F. Lobo, Copper Coordination in Cu-SSZ-13 and Cu-SSZ-16 Investigated by Variable-Temperature XRD, J. Phys. Chem. C, 2010, 114, 1633–1640 CrossRef CAS.
- J. H. Kwak, D. Tran, S. D. Burton, J. Szanyi, J. H. Lee and C. H. F. Peden, Effects of hydrothermal aging on NH3-SCR reaction over Cu/zeolites, J. Catal., 2012, 203 CrossRef CAS.
- J. H. Kwak, D. Tran, J. Szanyi, C. H. F. Peden and J. H. Lee, The Effect of Copper Loading on the Selective Catalytic Reduction of Nitric Oxide by Ammonia Over Cu-SSZ-13, Catal. Lett., 2012, 142, 295 CrossRef CAS.
- S. J. Schmieg, S. H. Oh, C. H. Kim, D. B. Brown, J. H. Lee, C. H. F. Peden and D. H. Kim, Thermal durability of Cu-CHA NH3-SCR catalysts for diesel NOx reduction, Catal. Today, 2012, 184, 252 CrossRef CAS.
- J. H. Kwak, H. Y. Zhu, J. H. Lee, C. H. F. Peden and J. Szanyi, Two different cationic positions in Cu-SSZ-13?, Chem. Commun., 2012, 48, 4758 RSC.
- F. Gao, J. H. Kwak, J. Szanyi and C. H. F. Peden, Current Understanding of Cu-Exchanged Chabazite Molecular Sieves for Use as Commercial Diesel Engine DeNOx Catalysts, Top. Catal., 2013, 56, 1441–1459 CrossRef CAS.
- S. T. Korhonen, D. W. Fickel, R. F. Lobo, B. M. Weckhuysen and A. M. Beale, Isolated Cu2+ ions: active sites for selective catalytic reduction of NO, Chem. Commun., 2010, 47, 800–802 RSC.
- F. Gao, E. D. Walter, E. M. Karp, J. Y. Luo, R. G. Tonkyn, J. H. Kwak, J. Szanyi and C. H. F. Peden, Synthesis and Evaluation of Cu-SAPO-34 Catalysts for Ammonia Selective Catalytic Reduction. 1. Aqueous Solution Ion Exchange, J. Catal., 2013, 300, 20–29 CrossRef CAS.
- J. H. Kwak, T. Varga, C. H. F. Peden, F. Gao, J. C. Hanson and J. Szanyi, Following the movement of Cu ions in a SSZ-13 zeolite during dehydration, reduction and adsorption: A combined in situ TP-XRD, XANES/DRIFTS study, J. Catal., 2014, 314, 83–93 CrossRef CAS.
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. F. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed.
- F. Gao, E. D. Walter, M. Kollar, Y. Wang, J. Szanyi and C. H. F. Peden, Understanding ammonia selective catalytic reduction kinetics over Cu/SSZ-13 from motion of the Cu ions, J. Catal., 2014, 319, 1–14 CrossRef CAS.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials, Chem. Soc. Rev., 2015, 44, 7371–7405 RSC.
- J. H. Kwak, J. H. Lee, S. D. Burton, A. S. Lipton, C. H. F. Peden and J. Szanyi, A common intermediate for N2 formation in enzymes and zeolites: side-on Cu-nitrosyl complexes, Angew. Chem., Int. Ed., 2013, 52, 9985–9989 CrossRef CAS PubMed.
- K. I. Hadjiivanov, Identification of Neutral and Charged NxOy Surface Species by IR Spectroscopy, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS.
- K. Hadjiivanov, J. Saussey, J. L. Freysz and J. C. Lavalley, FT-IR study of NO + O2 co-adsorption on H-ZSM-5: re-assignment of the 2133 cm−1 band to NO+ species, Catal. Lett., 1998, 52, 103–108 CrossRef CAS.
- J. Szanyi and M. T. Paffett, The Adsorption of NO and Reaction of NO with O2 on H-, NaH-, CuH-, and Cu-ZSM-5: An in situ FTIR Investigation, J. Catal., 1996, 164, 232–245 CrossRef CAS.
- A. Zecchina, L. Marchese, S. Bordiga, C. Paze and E. Gianotti, Vibrational Spectroscopy of NH4+ Ions in Zeolitic Materials: An IR Study, J. Phys. Chem. B, 1997, 101, 10128 CrossRef CAS.
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, Structure and Reactivity of Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-Ion-Exchanged Chabazite Catalyst for NH3-Mediated Selective Catalytic Reduction, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed.
- K. Chakarova and K. Hadjiivanov, H-Bonding of Zeolite Hydroxyls with Weak Bases: FTIR Study of CO and N2 Adsorption on H-D-ZSM-5, J. Phys. Chem. C, 2011, 115, 4806 CrossRef CAS.
- D. Perra, N. Drenchev, K. Chakarova, M. G. Cutrufello and K. Hadjiivanov, Remarkable acid strength of ammonium ions in zeolites: FTIR study of low-temperature CO adsorption on NH4-FER, RSC Adv., 2014, 4, 56183–56187 RSC.
- L. A. Kazitsyna, B. S. Kikot and O. A. Reutov, Infrared absorption spectra of solutions of diazonium salts in the region of 2200–2300 cm−1, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1964, 13, 894 CrossRef.
- J. Szanyi, J. H. Kwak and C. H. F. Peden, The Effect of Water on the Adsorption of NO2 in Na– and Ba–Y, FAU Zeolites: A Combined FTIR and TPD Investigation, J. Phys. Chem. B, 2012, 108(12), 3746–3753 CrossRef.
- K. Khivantsev, N. R. Jaegers, I. Z. Koleva, H. A. Aleksandrov, L. Kovarik, M. Engelhard, F. Gao, Y. Wang, G. N. Vayssilov and J. Szanyi, Stabilization of Super Electrophilic Pd+2 Cations in Small-Pore SSZ-13 Zeolite, J. Phys. Chem. C, 2020, 124, 309–321 CrossRef CAS.
- E. I. Tocheva, F. I. Rosell, G. Mauk and M. E. P. Murphy, Side-on copper-nitrosyl coordination by nitrite reductase, Science, 2004, 7, 867–870 CrossRef PubMed.
- B. Kartal, W. J. Maalcke, N. M. de Almeida, I. Cirpus, J. Gloerich, W. Geerts, H. J. M. Op den Camp, H. R. Harhangi, E. M. Janssen-Megens, K.-J. Francoijs, H. G. Stunnenberg, J. T. Keltjens, M. S. M. Jetten and M. Strous, Molecular mechanism of anaerobic ammonium oxidation, Nature, 2011, 479, 127–130 CrossRef CAS PubMed.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- K. Khivantsev, N. R. Jaegers, H. A. Aleksandrov, L. Kovarik, M. A. Derewinski, Y. Wang, G. N. Vayssilov and J. Szanyi, Biomimetic CO oxidation below −100 °C by a nitrate-containing metal-free microporous system, Nat. Commun., 2021, 12, 6033 CrossRef CAS PubMed.
- S. Yasumura, C. Liu, T. Toyao, Z. Maeno and K. Shimizu, Lean NOx Capture and Reduction by NH3via NO+ Intermediates over H-CHA at Room Temperature, J. Phys. Chem. C, 2021, 125, 1913–1922 CrossRef CAS.
- Y. Mao, Z. Wang, H.-F. Wang and P. Hu, Understanding Catalytic Reactions over Zeolites: A Density Functional Theory Study of Selective Catalytic Reduction of NOx by NH3 over Cu-SAPO-34, ACS Catal., 2016, 6, 7882–7891 CrossRef CAS.
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA, ACS Catal., 2020, 10(10), 5646–5656 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00350c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |