
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of anion polarizability on ion pairing in microhydrated salt clusters†
Arghya
Chakraborty
 a,
Thomas
Brumme
ac,
Sonja
Schmahl
a,
Hendrik
Weiske
a,
Carsten
Baldauf
*b and
Knut R.
Asmis
*a
a,
Thomas
Brumme
ac,
Sonja
Schmahl
a,
Hendrik
Weiske
a,
Carsten
Baldauf
*b and
Knut R.
Asmis
*a
aWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstrasse 2, D-04103 Leipzig, Germany. E-mail: knut.asmis@uni-leipzig.de
bFritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin, Germany. E-mail: baldauf@fhi-berlin.mpg.de
cTheoretische Chemie, Technische Universität Dresden, Dresden, Germany
First published on 30th September 2022
Abstract
Despite longstanding interest in the mechanism of salt dissolution in aqueous media, a molecular level understanding remains incomplete. Here, cryogenic ion trap vibrational action spectroscopy is combined with electronic structure calculations to track salt hydration in a gas phase model system one water molecule at a time. The infrared photodissociation spectra of microhydrated lithium dihalide anions [LiXX′(H2O)n]− (XX′ = I2, ClI and Cl2; n = 1–3) in the OH stretching region (3800–2800 cm−1) provide a detailed picture of how anion polarizability influences the competition among ion–ion, ion–water and water–water interactions. While exclusively contact ion pairs are observed for n = 1, the formation of solvent-shared ion pairs, identified by markedly red-shifted OH stretching bands (<3200 cm−1), originating from the bridging water molecules, is favored already for n = 2. For n = 3, Li+ reaches its maximum coordination number of four only in [LiI2(H2O)3]−, in accordance with the hard and soft Lewis acid and base principle. Water–water hydrogen bond formation leads to a different solvent-shared ion pair motif in [LiI2(H2O)3]− and network formation even restabilizes the contact ion pair motif in [LiCl2(H2O)3]−. Structural assignments are exclusively possible after the consideration of anharmonic effects. Molecular dynamics simulations confirm that the significance of large amplitude motion (of the water molecules) increases with increasing anion polarizability and that needs to be considered already at cryogenic temperatures.
1. Introduction
Salt ions strongly disrupt hydrogen-bond networks in water impacting a multitude of biochemical, industrial, and atmospheric processes such as the stability of proteins, homogeneous catalysis or aerosol formation.1–3 Thus, it is instructive to study the underlying mechanisms of the salt–water interaction on a molecular level. The course of salt dissolution, i.e. the weakening and ultimately the breaking of ionic bonds by interposing water molecules, is typically discussed considering transformations from contact (CIP) over solvent-shared (SIP) to solvent-separated ion pairs (2SIP).4,5 Recent studies with dielectric relaxation,4,6 infrared,7 neutron diffraction and X-ray absorption spectroscopy8,9 have provided evidence for CIP, SIP and 2SIP motifs in salt solution,3 but the mechanistic details of the process are still elusive from experiment and currently only available from computational studies. A step forward in this context is the characterization of salt pairs according to the cooperative or anticooperative effects10 of the respective counterion on specific ion–water interactions.11 But studies in the aqueous phase – due to the highly fluxional nature of hydrogen-bond networks and the heterogeneity of the local hydration shells in water – notoriously fail to capture the specificity and the spatial extent of ion–water interactions as is evident from X-ray and neutron diffraction investigations on dilute LiCl and NaCl solutions.9 On the computational side, it has become evident over the last two decades that the reliable description of salt dissolution, in general, and ion pairing, in particular, requires considering solvent polarization effects.12–17Archetypical models of the first hydration shell, i.e. salt ions with a well-defined number of water molecules, can be isolated in the form of microhydrated gas-phase clusters and then selectively probed using spectroscopic tools. This reduction of complexity allows us to monitor different types of interactions at the molecular level. As theory and experiment advance in parallel, experimental outcomes can be used to test available theories on salt hydration.18 The experimental investigation on neutral alkali halide salt–water complexes, which was started in the condensed phase by matrix isolation infrared spectroscopy (MI-IR) in the 1970s,19 has now been extended to the gas-phase with Fourier-transform microwave,20 femto-second pump–probe,21 and helium droplet IR spectroscopy techniques.22,23
However, mass selection of neutral clusters remains challenging and therefore obtaining size-specific interaction details is currently only viable with ionic species. Anion photoelectron spectroscopy (APES) was employed to characterize systems like LiI−(H2O)0–6, NaCl−(H2O)0–6, CsI−(H2O)0–6, NaSO4−(H2O)0–4, and NaBO2−(H2O)0–4.24–27 LiI was found prone to solvent induced charge separation as can be expected from Pearson’s hard and soft acid base (HSAB) theory.28 ‘Hard’ ions feature a high ionic potential (charge/ionic radii) and are weakly polarizable. ‘Soft’ refers to ions with large ionic radii and small charges, which are highly polarizable. However, unambiguous structural assignments are more difficult to obtain solely from APES studies.
In this regard, infrared photodissociation (IRPD) spectroscopy is a more versatile tool for structure characterization. Reports on microhydrated (NaSO4−)2, Mg2SO42+, Ni2(SO4)32+, CaOH−, MgOH− or MgNO3+ yield insight into the effect of stepwise hydration on ion–ion, ion–water and water–water interactions.29–33 The formation of ionic hydrogen bonds (IHBs) is accompanied by characteristic shifts of the corresponding O–H stretching frequencies.34 In our recent study on [LiCl2(H2O)n]− (n = 1–3), we established that SIP vs. CIP formation can be distinguished based on the extent of the observed frequency shifts of the hydrogen-bonded O–H stretching oscillators.35
In the present work, we extend our study to include iodide anions and focus on the microhydration of archetypical salt–water anions of the type [LiXX′(H2O)n]− with XX′ = I2, ClI and Cl2 and n = 1–3. We investigate these systems in the gas-phase at cryogenic temperatures, which enables us to dissect the interaction between salt ions and a few water molecules as a function of anion polarizability. Note that the iodide anion is roughly twice as polarizable as the chloride anion.36 We focus on characterizing ion-pairing in light of Collin’s “law of matching water affinity” which states that hard–soft (vs. hard–hard and soft–soft) ion combinations, like Li+/I−, are more prone to solvent-induced charge separation.28,37 Methodologically, we utilize vibrational action spectroscopy of D2-tagged [LiXX′(H2O)1–3]− anions complemented by ab initio simulations. The inclusion of anharmonic effects is crucial for a reliable interpretation of the experimental vibrational spectra of these systems,35 and we perform this here both in a static and in a dynamic fashion.
2. Results and analysis
2.1 IRPD spectroscopy
Fig. 1 shows the vibrational action spectra of [LiI2(H2O)1–3]− (a–c), [LiClI(H2O)1–3]− (d–f) and [LiCl2(H2O)1–3]− (g–i) in the O–H stretching region (3800–2800 cm−1). The data for [LiCl2(H2O)n]− are from ref. 35. All IRPD spectra were recorded using D2-tagging (D2-loss) to ensure probing (a) in the linear absorption region and (b) colder ions. An exception is the spectrum of [LiClI(H2O)]− (trace d), for which the D2-tagged ion-signal levels were too low and therefore the H2O-loss channel from the untagged anion was monitored. Spectra obtained with monitoring D2 and H2O-loss are very similar (except the band due to D2 stretching) for these monohydrates as shown for [LiI2(H2O)]− and [LiCl2(H2O)]− in Fig. S1 in the ESI.† The position and assignments of the observed IRPD bands are summarized in Table S1 (ESI†). In order to evaluate the influence of the nature of the two halide anions on the course of microhydration of these ion pairs, we first focus on the lithium diiodide anion (LiI2)−, and then discuss the mixed halide system (LiClI)−, after which we compare the present results to those obtained for microhydrated lithium dichloride anions [LiCl2(H2O)1–3]− that have been discussed in detail previously.35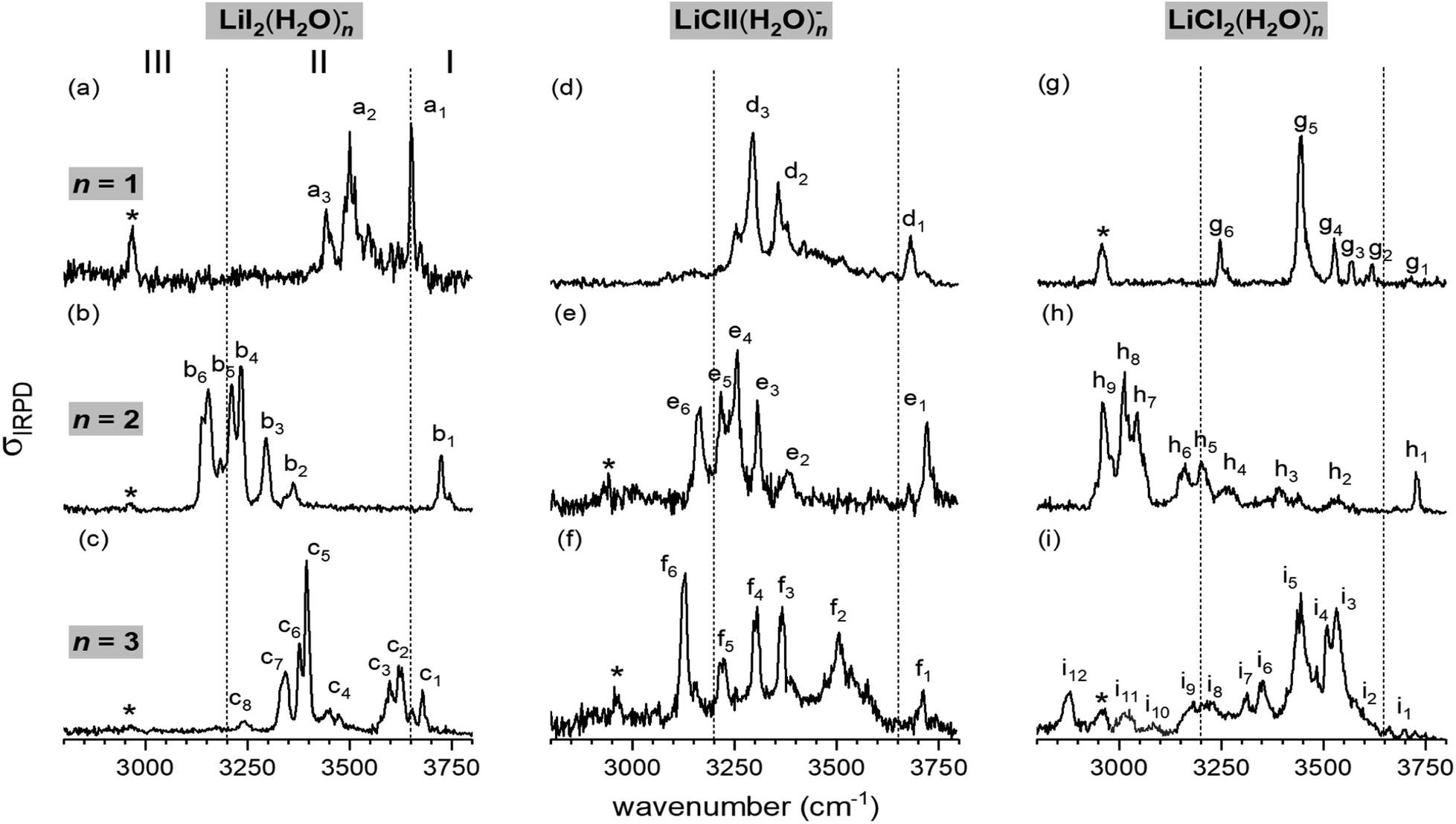 | ||
| Fig. 1 Vibrational action spectra of the [LiXX′(H2O)n]− anions with XX′= I2, ClI, and Cl2 from left to right and n = 1–3 (top to bottom) in the spectral region of O–H stretching modes measured at an ion trap temperature of 13 K. All IRPD spectra were recorded using D2-tagging (D2-loss) to ensure probing in the linear absorption region, except for [LiClI(H2O)]− (trace d), for which ion signal levels were too low and therefore the H2O-loss channel from the untagged anion was monitored. Bands attributed to the excitation of the messenger molecule (D2-stretching vibration) are denoted by asterisks. The spectra for [LiCl2(H2O)n]− are from ref. 35. | ||
The characteristic H2O binding motifs are encoded in the IRPD band pattern, in particular, in the relative red-shifts of the fundamental O–H stretching excitations with respect to those in the free water molecule.30,35 The three previously defined spectral regions for [LiCl2(H2O)1–3]− anions prove useful for a qualitative discussion of the measured spectra and therefore we make use of these also for the present systems.35 The excitation of free O–H oscillators is typically found in region I (>3650 cm−1). Region II (3650–3200 cm−1) covers O–H oscillators involved in water–water as well as weaker halide–water hydrogen bonds. Region III (<3200 cm−1) is characteristic of strong halide–water interactions. Note that the excitation of the nominally IR-inactive D2 stretching mode, which gains IR-activity upon polarization, is observed around 2960 cm−1 in many of the IRPD spectra (denoted by an asterisk), blue-shifted by roughly 25–35 cm−1 from the value for free D2.38
In the hydrogen-bonded O–H stretching region (<3650 cm−1), a clear dependence of the extent of the O–H stretching frequency red-shift on the degree of microhydration is observed. For n = 1, the red-shift is modest and bands are only observed above 3450 cm−1, indicating weaker iodide–water hydrogen bonds. The most pronounced red-shift is observed for n = 2. This is the only spectrum that exhibits absorption in region III, which is indicative of strong iodide–water hydrogen-bonding and therefore implies the presence of a solvent-shared ion pair.35 For n = 3, the red-shift is intermediate, with the most intense IR signal found at 3400 cm−1. The comparison of this spectrum to the reported spectra of Li+(H2O)3 and I−(H2O)3 (see Fig. S2 in the ESI†) reveals no clear similarities.34,39,40 Hence, a characteristic structure motif like a water trimer ring, characterized by a sharp doublet feature at ∼3550 cm−1, is probably not present in [LiI2(H2O)3]−.
The n = 2 spectra, on the other hand, show greater similarity, suggesting similar structures, i.e. the solvent-shared structure containing two bridging water molecules previously identified for [LiCl2(H2O)2]−. The markedly red-shifted bands in region III in combination with a rather intense free O–H stretching band around or even above 3700 cm−1 can be attributed to two decoupled O–H oscillators for each water molecule, one bridging the Li+/X− ion pair and the other dangling freely. The position of the most red-shifted band in the spectrum of [LiClI(H2O)2]− (e6) coincides with the corresponding band in the [LiI2(H2O)2]− spectrum (b6), while band h9 in the [LiCl2(H2O)2]− spectrum is found to be red-shifted roughly 200 cm−1 more. Hence, the water molecules bridge the Li+/I− rather than the Li+/Cl− ion pair in [LiClI(H2O)2]−. Note that the present IRPD spectra of the LiI2 and LiClI species show no absorption in the upper part of region II, indicating that water–water hydrogen bonds are not present and that, in contrast to [LiCl2(H2O)2]−, again only a single isomer (i.e. containing two bridging water molecules) is present in the iodide-containing systems.
The n = 3 spectra are markedly different from each other. [LiI2(H2O)3]− seems to yield the simplest spectrum, hinting at a structure of higher symmetry. The spectrum of [LiClI(H2O)3]− extends over a broader spectral range with the most intense IRPD band (f6) located in region III, indicating that also for n = 3 a solvent-shared motif, in which the Li+/I− ion pair is bridged, is preferred. IR2MS2 experiments for [LiI2(H2O)3]− and [LiClI(H2O)3]− (see Fig. S3†) demonstrate that only a single stable isomer is present. This is in contrast to the [LiCl2(H2O)3]− spectrum, which was assigned to three isomers, two solvent-shared ones that predominantly absorb below 3200 cm−1, characteristic for a bridged Li+/Cl− ion pair, and one intact core structure contributing mainly in the 3400–3600 cm−1 region.35 The role of isomers is discussed together with the results of the AIMD simulations in the following sections.
2.2 Microhydration motifs and energetics
In order to assign the spectra, we performed a minimum-energy structure search using the MP2 method in combination with the def2TZVP basis set. All structures and energies are shown in Fig. S4–S6 in the ESI.† Bare lithium dihalide anions are found to adopt a linear (X–Li–X′) structure of either D∞h (X = X′) or C∞v (X ≠ X′) symmetry (see Table S2a and S2b† for MP2 bond distances). Characteristic microhydration motifs (top), relevant for the assignment of the IRPD spectra, and their relative zero-point energy corrected energies ΔE0 (bottom) are plotted in Fig. 2. We make use of the same m.bw.i nomenclature scheme for the isomers as introduced in our previous publication on [LiCl2(H2O)1–3]−.35 Here, m refers to the number of water molecules, bw to the number of interion bridging water molecules and i to the number of water–water hydrogen bonds. Additionally, the letters a–z were employed in the ascending order to differentiate between multiple (m.bw.i) isomers according to ΔE0 for the LiCl2 species. In order to facilitate the comparison of microhydration motifs over all systems studied here, we use the same letter/motif combinations for the LiClI and LiI2 species, even though these then do not necessarily reflect the order (with respect to energy) for these particular systems anymore. In addition, water molecules are labeled according to their hydrogen bond donating (D) and accepting (A) capacities in the hydrogen-bonded network.41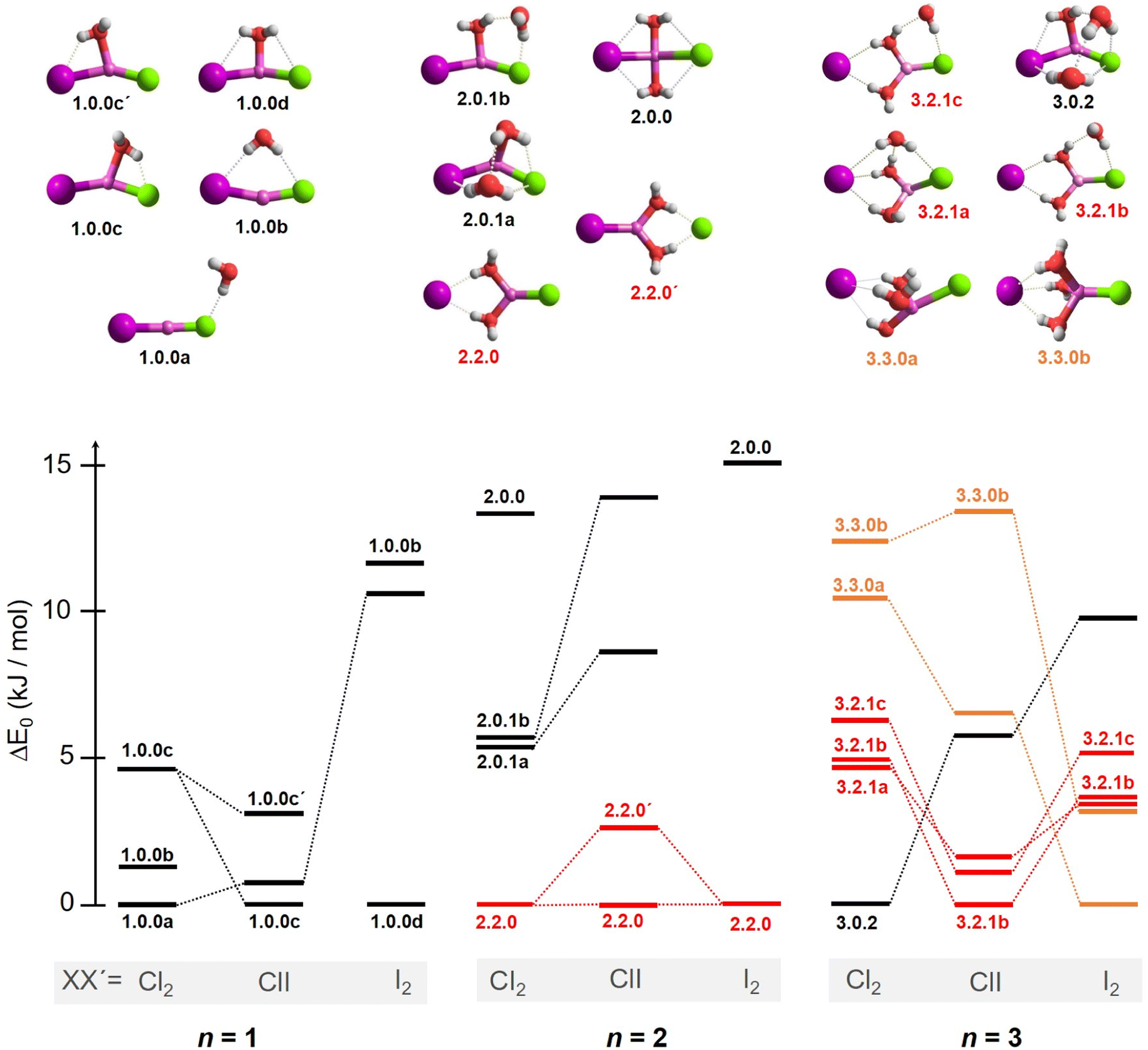 | ||
| Fig. 2 Top panel: relevant structural motifs for [LiClI(H2O)n]− anions in the size range n = 1–3, representative for all [LiXX′(H2O)n]− systems studied here. Bottom panel: zero-point energy corrected relative energies ΔE0 (see the Computational methods section) for [LiXX′(H2O)1–3]− with XX′= Cl2, ClI, I2. Similar structural motifs for a certain cluster size are connected with lines and distinctive motifs for n > 1 are colour-coded to guide the eye. A prime symbol (e.g. 1.0.0c vs. 1.0.0c′ or 2.2.0 vs. 2.2.0′) is used for the mixed-halide anions XX′ = ClI to denote related motifs, in which exclusively the position of two halide anions is switched. | ||
For n = 1, we find only intact core structures (CIP/CIP), in which the Li–X distances are elongated by less than 4% compared to the bare structures. The water either interacts exclusive with a single halide (1.0.0a), with two halides (1.0.0b), with Li and one halide (1.0.0c and 1.0.0c′) or substantially with all three ions (1.0.0d). The energetic ordering of these motifs depends intimately on the nature of the halides (see the bottom part of Fig. 2) and each system indeed exhibits a different global minimum structure, i.e., 1.0.0a, 1.0.0c and 1.0.0d for XX′ = Cl2, ClI and I2, respectively.
In contrast, the lowest energy motif for n = 2, the SIP/CIP configuration 2.2.0 in which one of the halides interacts with two bridging water molecules, is the same for all three systems. In the SIP moiety, the interion distance is roughly 1.7 times longer than in the bare ion core. For the mixed halide system, the hydration of the more polarizable halide (2.2.0 vs. 2.2.0′) is energetically favored. Intact-core CIP/CIP structures with (2.0.1a/b) and without (2.0.0) inter-water hydrogen bonds are higher in energy and this energy gap increases with increasing polarizability of the halides.
For n = 3, halide-dependent differences in the lowest-energy microhydration motif are most obtrusive. The dichloride system prefers an intact-core CIP/CIP configuration (3.0.2) that is stabilized by extended water–water and strong anion–water hydrogen bonds. In contrast, the highest degree of Li-solvation involving three bridging D-H2Os is apparent for the diiodide system (3.3.0). The mixed halide system prefers a slightly different SIP/CIP configuration (3.2.1) containing only two bridging water molecules between the Li+/I− ion pair as well as a third water molecule (AD-H2O) strongly interacting with the chloride. This 3.2.1 motif is found to be the next most stable structure for dichloride and diiodide systems.
2.3 Assignment of the IRPD spectra
After this more general discussion of the predicted microhydration motifs, we now proceed with the structural assignment based on a detailed comparison of the experimental and computed vibrational spectra. An unambiguous spectral assignment requires the consideration of anharmonic effects for these systems.35 These can be included either in a static (VPT2/MP2 method) or in a dynamic fashion (AIMD simulations). In this section, we focus on the VPT2/MP2 results (see Section 2.4 for the analysis of the AIMD results). For each system, we first discuss the relative energies of low-energy isomers. Subsequently, we compare the VPT2/MP2 vibrational spectra with the experimental ones. A cosine similarity score (S) analysis42,43 is employed to evaluate the agreement in order to consolidate the structure assignments.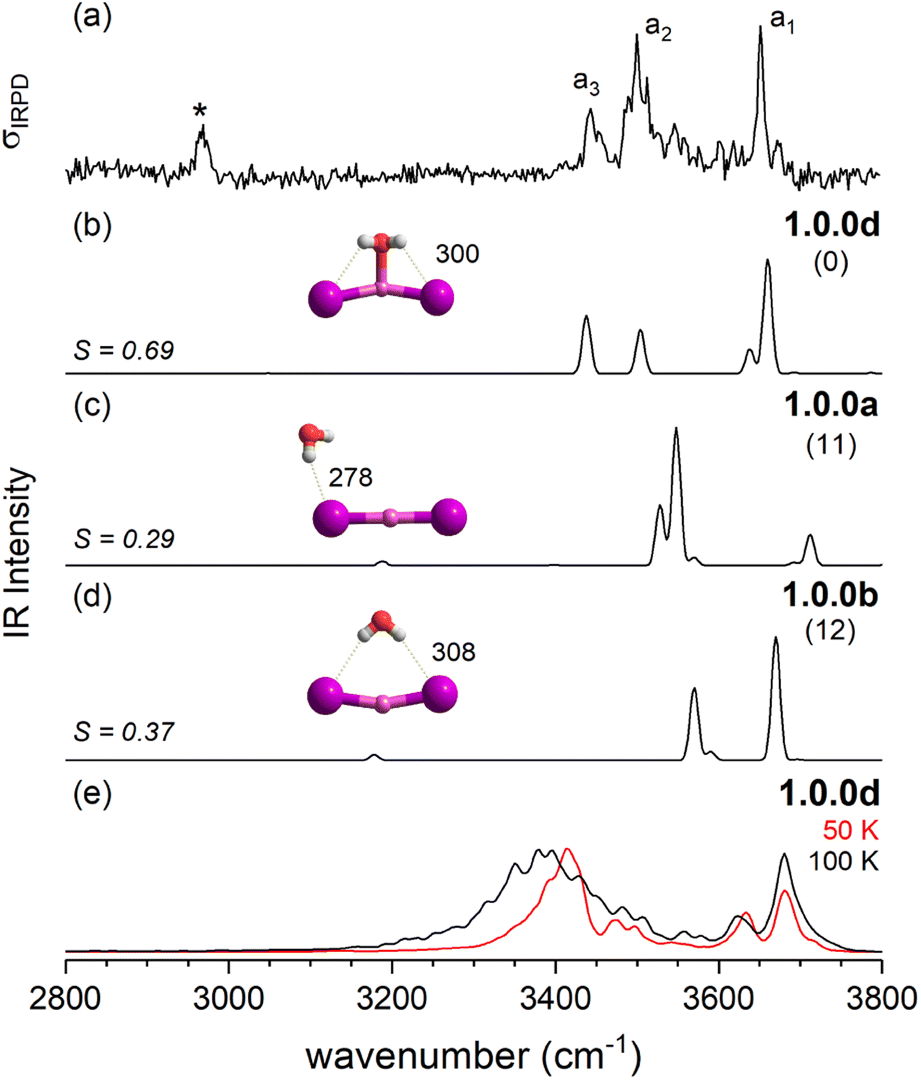 | ||
| Fig. 3 Comparison of the IRPD spectrum of the D2-tagged [LiI2(H2O)]− (trace a) to VPT2/MP2 static anharmonic (traces b–d) and dynamic anharmonic spectra (e). The IRPD band due to D2-stretching mode is denoted by an asterisk. VPT2 spectra along with corresponding cosine similarity scores (S) of three most stable isomers 1.0.0d (trace b), 1.0.0a (c) and 1.0.0b (d), their structures, ΔE0 (in kJ mol−1) and characteristic bond lengths (in pm) are shown. The AIMD spectra at 50 and 100 K were obtained with 1.0.0d as the starting structure. | ||
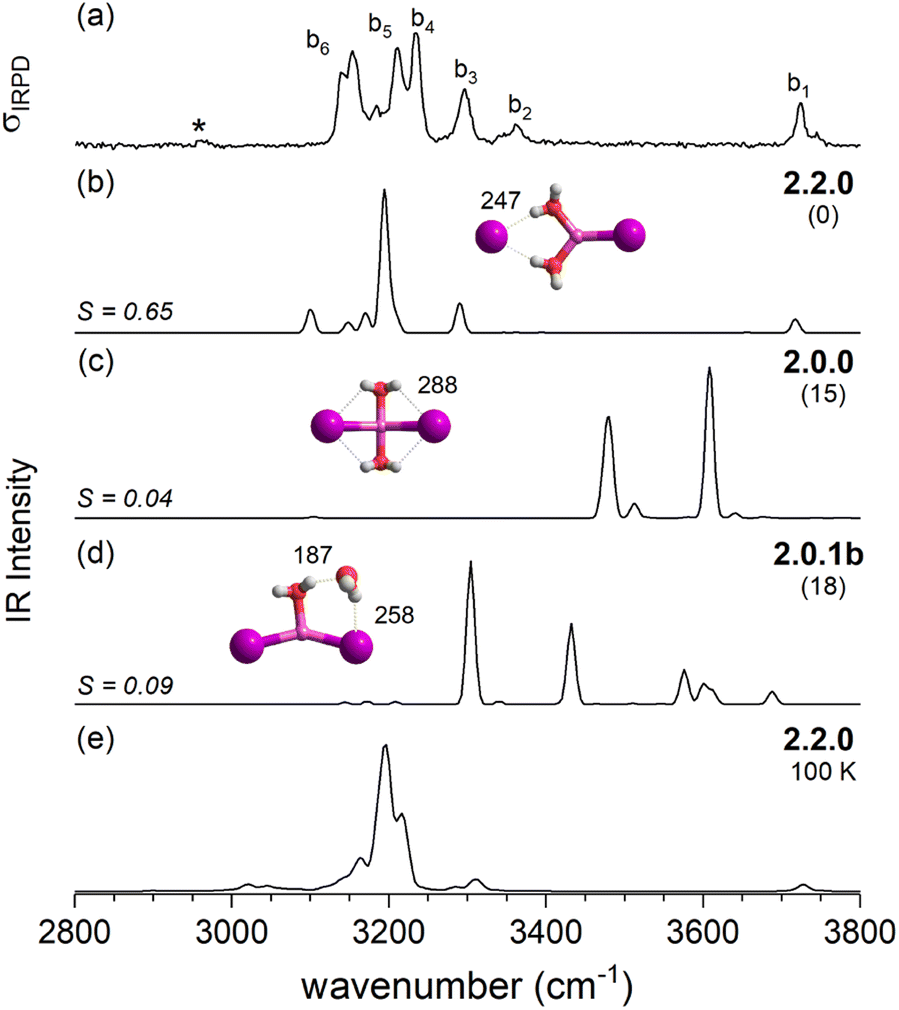 | ||
| Fig. 4 Comparison of the IRPD spectrum of the D2-tagged [LiI2(H2O)2]− (trace a) to VPT2/MP2 static anharmonic (traces b–d) and dynamic anharmonic spectra (e). VPT2 spectra along with the corresponding cosine similarity score (S) of three most stable isomers 2.2.0 (trace b), 2.0.0 (c) and 2.0.1b (d), ΔE0 (in kJ mol−1) and characteristic bond lengths (in pm) are shown. The AIMD spectrum was obtained exploring structure 2.2.0 at 100 K. The IRPD band due to D2-stretching mode is denoted by an asterisk. | ||
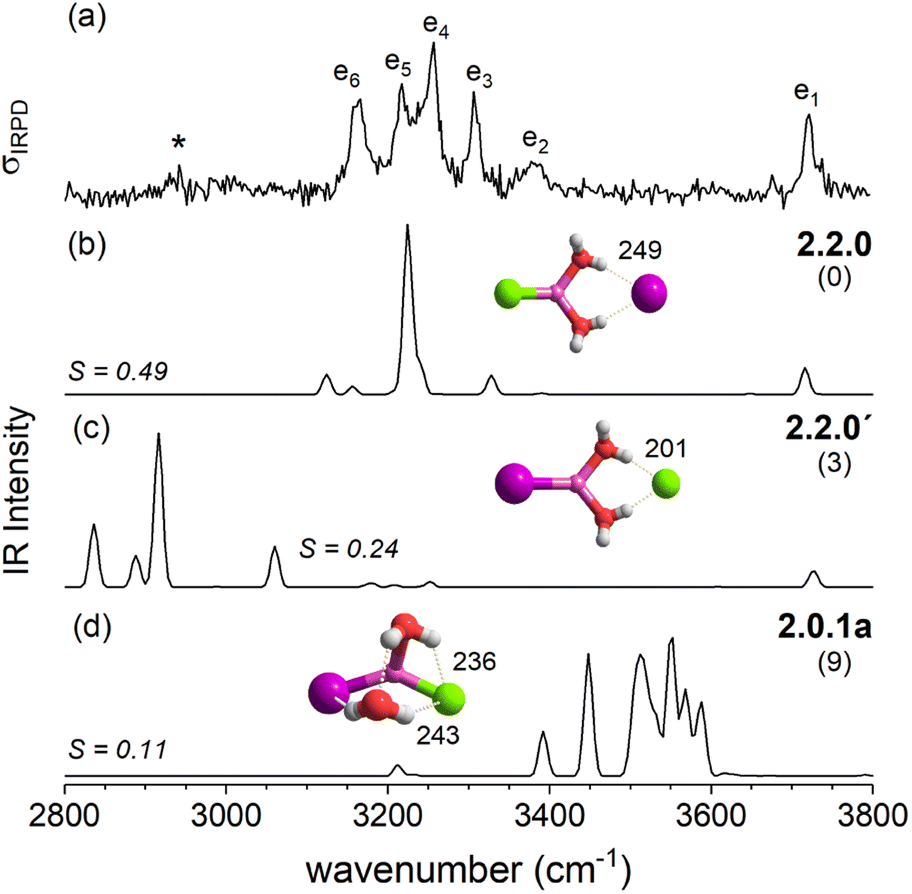
Fig. 4 compares the IRPD spectrum of D2-tagged [LiI2(H2O)2]− (a) to the VPT2/MP2 spectra of the three most stable isomers (b–d). The corresponding IR spectra show key differences and the most reasonable agreement with the experimental spectrum in terms of absorption regions and most dominant peak positions is obtained for the global-minimum energy structure 2.2.0 (S = 0.65). The similarity score for 2.2.0 is much higher than those for other low-energy isomers like 2.0.0 (S = 0.04) and 2.0.1b (S = 0.09).
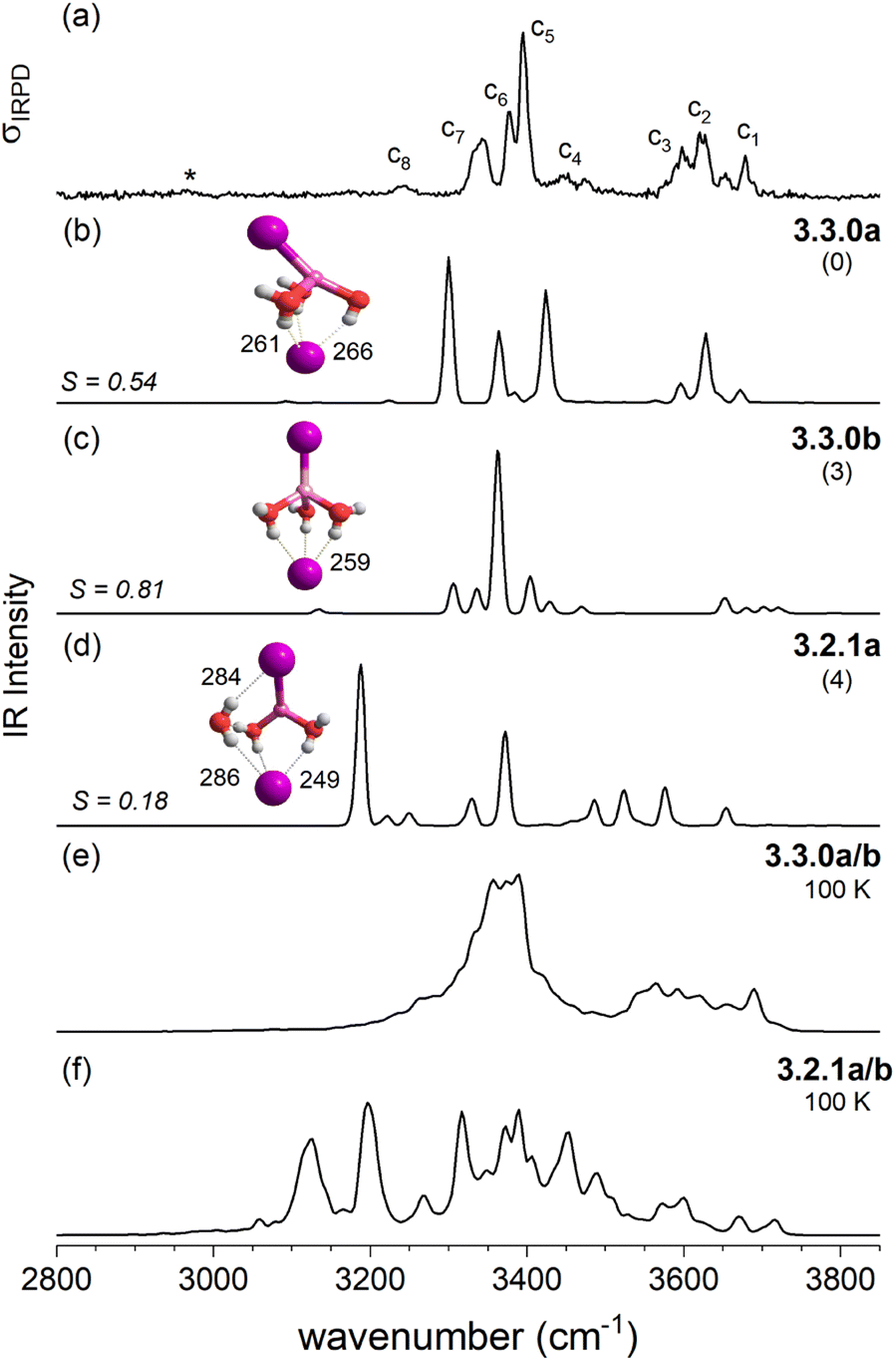 | ||
| Fig. 5 Comparison of the IRPD spectrum of D2-tagged [LiI2(H2O)3]− (trace a) to VPT2/MP2 static (traces b–d) and dynamic anharmonic spectra (traces e and f). VPT2 spectra along with the corresponding cosine similarity score (S) of the three most stable isomers 3.3.0a (trace b), 3.3.0b (c) and 3.2.1b (d), structures, ΔE0 (in kJ mol−1) and characteristic bond lengths (in pm) are shown. The AIMD spectrum obtained at 100 K exploring structures 3.3.0a/b and 3.2.1a is presented in traces e and f, respectively. | ||
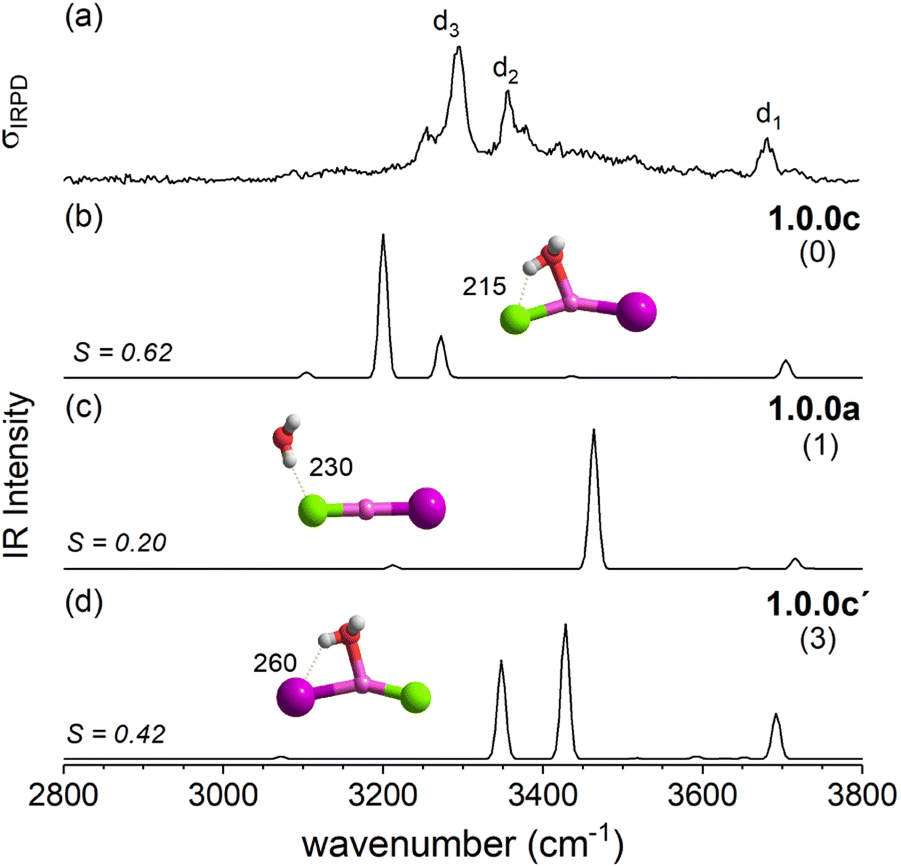 | ||
| Fig. 6 Comparison of the IRPD spectrum (trace a) of [LiClI(H2O)]− to the VPT2/MP2 anharmonic spectra of the three lowest energy isomers 1.0.0c, 1.0.0a and 1.0.0c′ as presented in traces b–d, respectively. Cosine similarity score (S), structures, ZPE-corrected relative energies ΔE0 (kJ mol−1) and characteristic bond lengths (pm) are also shown. For D2-tagged [LiClI(H2O)]− insufficient photoinduced D2-loss was observed and therefore the H2O-loss channel was monitored. | ||
The IRPD spectrum of [LiClI(H2O)]− is compared to the VPT2/MP2 anharmonic spectra of 1.0.0c (S = 0.62), 1.0.0a (S = 0.20) and 1.0.0c′(S = 0.42) in Fig. 6. The best agreement is observed for the lowest energy isomer 1.0.0c. While the similarity score is significantly larger than for the other two isomers, it remains rather small, mainly since the red-shifts of the band pair d2/d3 are significantly overestimated. Interestingly, the relative band intensities agree better. Hence, bands d1 and d3 correspond to the excitation of the free and the hydrogen-bonded O–H stretching fundamentals, respectively. d2 is due to the combination of d1 and the lowest energy rocking motion involving Li and O units (60 cm−1).
 | ||
| Fig. 7 Comparison of the IRPD spectrum (trace a) of D2-tagged [LiClI(H2O)2]− to the VPT2/MP2 anharmonic spectra of the two lowest energy solvent-shared core structures 2.2.0 and 2.2.0′ and intact-core structure 2.0.1a presented in traces b–d, respectively. Cosine similarity score (S), structures, ZPE-corrected relative energies ΔE0 (kJ mol−1) and characteristic bond lengths (pm) are also shown. | ||
The IRPD spectrum of D2-tagged [LiClI(H2O)2]− is compared to the VPT2/MP2 anharmonic spectra of 2.2.0, 2.2.0′ and 2.0.1a in Fig. 7. The three computed spectra are quite different from each other and reasonable agreement with experiment is only found for the spectrum of the minimum energy structure 2.2.0 (S = 0.49). Similar to n = 1, the simulated spectrum appears simpler than the experimental one. Indeed, VPT2 calculations predict a number of combination bands that could account for the additional features in the experimental spectrum; however their intensities are predicted too small. Note that similar discrepancies between experimental and the predicted spectra were observed for the corresponding structure 2.2.0 of [LiI2(H2O)2]− (see Fig. 4a and b). In both cases, the increase in experimental IR intensities is attributed to structural dynamics (see Section 2.4).
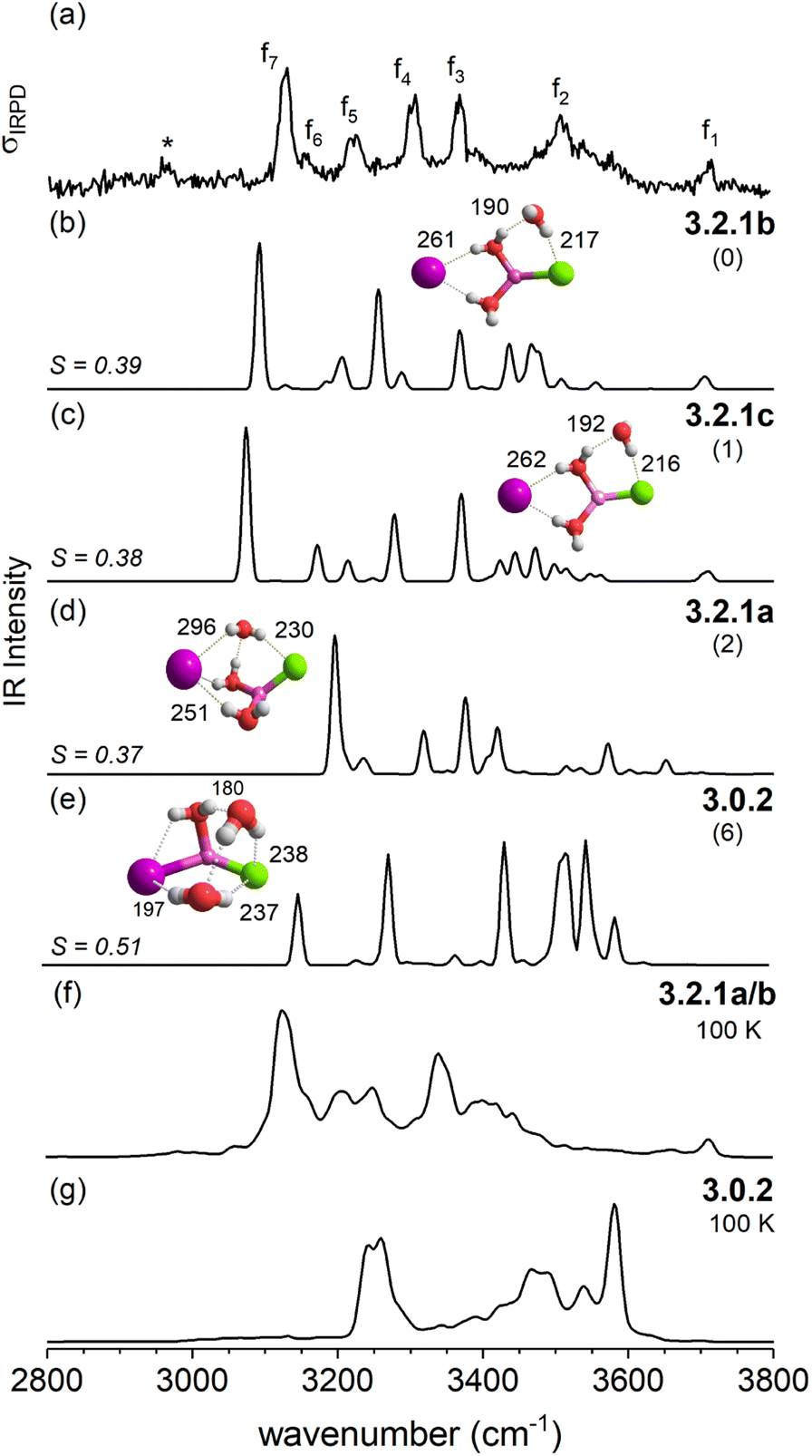 | ||
| Fig. 8 Comparison of the IRPD spectrum of D2-tagged [LiClI(H2O)3]− (trace a) to VPT2/MP2 static anharmonic (traces b–e) and dynamic anharmonic spectra (f and g). VPT2 spectra of three most stable solvent-shared structures 3.2.1b (trace b), 3.2.1c (c), and 3.2.1a (d) and the intact core structure 3.0.2 (e), corresponding ΔE0 (in kJ mol−1) and characteristic bond lengths (in pm) are shown. The AIMD spectra were obtained from MD simulations at 100 K. The band for the polarized D2 tag in trace a is marked with an asterisk. | ||
The IRPD spectrum of D2-tagged [LiClI(H2O)3]− is compared to the VPT2/MP2 anharmonic spectra of the three lowest energy isomers 3.2.1a, 3.2.1b and 3.2.1c in Fig. 8. Since the three structures are rather similar, it is not surprising that the three simulated IR spectra and their associated S values are rather similar (and relatively small) too. Indeed, none of the three spectra reproduces all experimentally observed features satisfactorily. Notably, the IR2MS2 and one-color IRPD spectra are similar and show no evidence for the presence of multiple isomers. However, fast interconversion between these three isomers is again possible, since this would only involve the reorientation of the H2O molecule hydrogen-bonded to chloride.
2.4 Analysis of structural dynamics
With AIMD simulations, we investigated the dynamics of selected [LiXX′(H2O)n]− anions in order to study the stability and dynamic interconversion of different structure motifs and also to derive vibrational spectra, which are compared to the spectra of the static structures in order to elucidate the impact of anharmonicities and structural rigidity/diversity on the spectral assignment.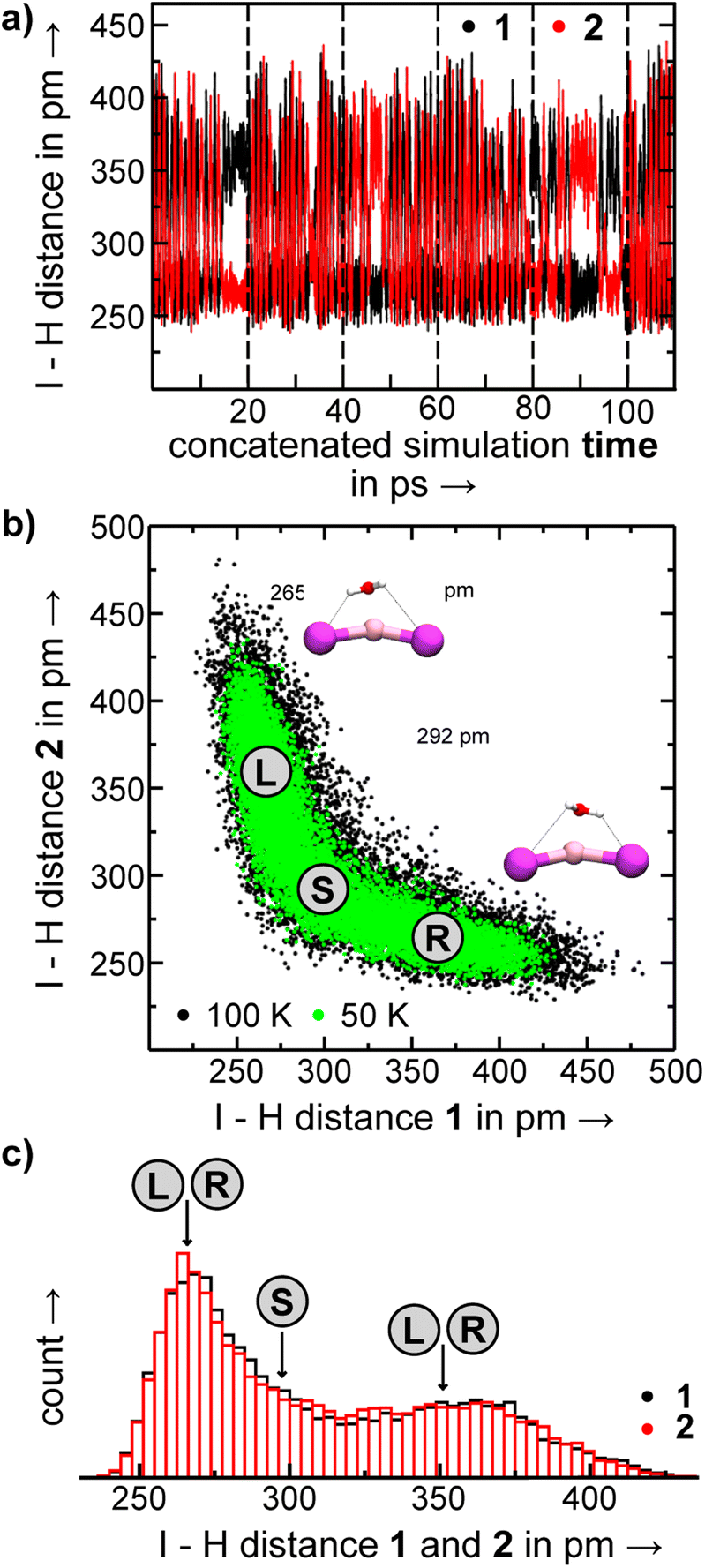 | ||
| Fig. 9 In the MD simulations of [LiI2(H2O)]− we observe oscillator between structures where the water molecule leans toward one of the iodine anions, while the symmetric structure 1.0.0d is not stable. (a) I–H distances versus time for the concatenated constant energy MD simulations of [LiI2(H2O)]−. (b) Comparison of MD frames plotted according to their I–H distances from trajectories at 50 K and at 100 K and representative structures with the water molecule leaning to either side (L and R) and the symmetric structure (S). (c and d) Histograms of the two Li–H distances. | ||
We visualize the structural dynamics in the constant energy trajectories of [LiI2(H2O)3]−, [LiClI(H2O)3]−, and [LiI2(H2O)3]− as well as representative structure snapshots in Fig. 10. In order to represent “a map of the structure space”, we choose to representative coordinates, namely the distance between the anions and the largest angle formed by the three water oxygens, and these are highlighted in the structures depicted in Fig. 10 by orange and blue lines, respectively. These coordinates allow us to distinguish the different isomers from each other. For [LiI2(H2O)3]−, for example, we see that solvent-separated structures are clearly distinguished from structures with an intact ion core (3.0.2) by an anion distance above 525 pm. Since all three plots in Fig. 10 share the same axis scaling, we can now directly see the effect of exchanging anions. By reducing ionic radii going from the diiodide to chloride–iodide to dichloride systems, we see a general shift to the left, i.e., to lower anion distances. The indicated border between water separated structures and intact ion core structures shifts from 525 pm (Fig. 10a) to below 500 pm (Fig. 10b) to 450 pm (Fig. 10c). Fig. 10 also exemplifies which anion has the dominant role in these salt–water complexes: the presence of the “harder” chloride anion is the reason why the mixed system [LiClI(H2O)3]− shares more features with the dichloride than with diiodide system. In the intact-core motif 3.0.2, the three water molecules form a hydrogen-bonded network and only slightly dislocate the cation out of the straight line between the two anions. Interestingly, this motif is the one experimentally assigned to the [LiCl2(H2O)3]− system with the hard–hard Li–Cl interactions. In the following, we will take a closer look at the individual structural motifs.
 | ||
| Fig. 10 Visualization of representative constant energy trajectories for (a) [LiI2(H2O)3]−, (b) [LiClI(H2O)3]−, and (c) [LiCl2(H2O)3]−. The dots represent frames (every 10 fs) from different NVE trajectories that are colored based on their starting structure. The axes of the plots allow for a good overview of the overall available structure space. In the displayed structure snapshots, the x-axis dimension (the anion distance) is illustrated by an orange dotted line and the y-axis dimension (the largest angle between the three water oxygens) is highlighted by a blue dotted line. A bond is drawn between lithium and the anions, if they are close enough. We also draw a vertical dashed line to separate the portion of structure space where water does not break the cation–anion interaction (left of the dashed line) from the region where water breaks this interaction (right of the dashed line). Isomer 3.0.2 (black dots) does not leave the local minimum at 100 K and during the simulation time, while in contrast, alternative structures like different types of 3.2.1 interconvert. In the case of [LiI2(H2O)3]−, isomer 3.3.0 is not stable at 100 K and transforms into 3.2.1a eventually (see panel a). | ||
As discussed above in Section 2.3, the VPT2/MP2 anharmonic spectra for the structural motifs 3.3.0 and 3.2.1 show the best agreement for [LiI2(H2O)3]− and [LiClI(H2O)3]−, respectively and this holds also for the AIMD spectra as shown in Fig. 5 and 8. In the case of the diiodide, 3.3.0b converts to the lower-symmetry isomer 3.3.0a (cf., Fig. 2). In a concerted movement, one water rotates and aligns its dipole with a second water molecule, and the iodide closest to the Li+ moves in the same direction, thus aligning the corresponding dipole antiparallel. In the MD simulations at 100 K, we observe the transformation of 3.3.0a to 3.2.1a eventually. In the constant energy trajectories that are being partly visualized in Fig. 10, we observe multiple crossings only from the 3.3.0 to the 3.2.1 basin and never back.
An interesting effect can be seen for the 3.2.1 motifs. 3.2.1a and b differ only slightly in energy (cf.Fig. 2 and Table S2b†) and interconvert frequently in the constant energy and constant temperature MD simulations; in Fig. 10, this can be seen by the cyan coloured clouds of dots extending widely diagonal in the upper right corner of the individual plots. For the mixed halide salt system, since none of the structures 3.2.1a/b/c reproduces all experimentally observed features satisfactorily (cf.Fig. 8), this fast interconversion seems probable even in the experiments. Also, for the dichloride system the best agreement between experiment and AIMD can be found for the spectra including both the 3.2.1a/b motifs (cf. Fig. S15†). However, the barrier from 3.2.1c to 3.2.1a/b is apparently high. Motif 3.2.1c is plotted (green dots) for [LiClI(H2O)3]− and [LiCl2(H2O)3]−. The respective green clouds overlap with the cyan clouds and the “heavy” atom structures look almost identical. A detailed view on the structures reveals the difference in the orientation of one of the water molecules. A transition between the 3.2.1c and 3.2.1a/b basins never occurs in our AIMD simulations since the dipole rearrangement due to the needed 180° rotation of one water molecule (cf.Fig. 8) costs too much energy. For the mixed chloride/iodide system, the 3.2.1b motif is slightly lower in energy changing its position with 3.2.1a compared to [LiI2(H2O)3]−.
In the 3.0.2 motif, the interactions between the Li cation and the two anions are essentially unperturbed by the water molecules. In the plots in Fig. 10, the respective black dot clouds remain isolated and appear rather well defined in comparison to alternative structural motifs that spread wider in structure space and that also interconvert. The dot clouds also appear to shrink from Fig. 10a–c. To take a clearer look we compute root mean square fluctuations over AIMD trajectories and plot the respective distributions in Fig. 11. The overlay illustrates that the green distribution representing [LiCl2(H2O)3]− is more narrow than the others; in particular [LiI2(H2O)3]− (blue) is characterized by a comparably long tail. We interpret this as a result of the stronger anion–water interaction due to the harder chloride anions in [LiCl2(H2O)3]−.
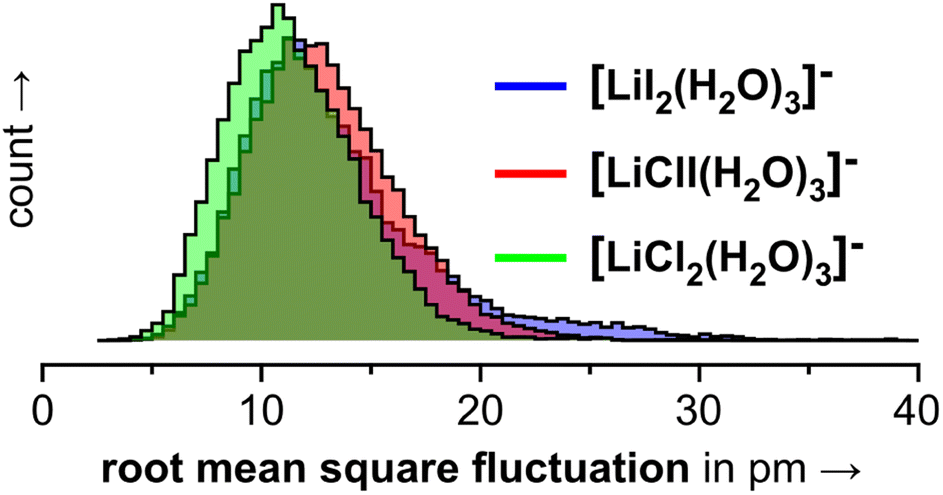 | ||
| Fig. 11 Root mean square fluctuations (RMSFs) for the individual frames of the 3.0.2 simulations shown in Fig. 10. | ||
3. Discussion
We now focus on the bigger picture that emerges from our microhydration study, namely the changes in the stability of CIP vs. SIP structures as the number of water molecules is increased, how this depends on the nature of the halide anions involved and in what way this may manifest itself in the corresponding vibrational spectra of the solution phase species.In the microhydrated lithium dihalide anions studied here, we observed a competition of several driving forces (i) interion interactions can be viewed based on the hard and soft acid and base (HSAB) concept. The interaction of the hard Li cation is stronger with the harder chloride anion than with the softer iodide anion, and hence the Li–I bond is severed more readily than the Li–Cl bond. (ii) The Li cation strives to fill its first coordination sphere with up to four ligands.44,45 (iii) Anionic binding sites have a tendency to form IHBs, the strength of which correlates with the proton affinity (PA) of the halide.34 Since the PA of chloride (1396 kJ mol−1) is more than 80 kJ mol−1 higher than that of iodide (1315 kJ mol−1), chloride also forms significantly stronger IHBs than iodide does. Note that the IHB strength also manifests itself directly in its IR signature in the form of a characteristic red-shift of the corresponding O–H stretching frequencies (see also Fig. 1). (iv) Besides interacting with the anions and cations, water molecules also form hydrogen-bonded networks. Such networks contribute substantially to the stability of the cluster, in particular, when the ionic core structure can accomodate the hydrogen bonds without significantly interfering with their directional nature. (v) Finally, the alignment of the water dipoles along the direction of cation–anion ion pairs leads to further stabilization. Summarizing, the interactions at play in salt–water complexes, in the order of decreasing bond strengths, are:37,46 hard ion–hard ion > hard ion–water > water–water > soft ion–water > soft ion–soft ion.
Fig. 12 summarizes the structures that were assigned in the present and our previous study35 on lithium dihalide anions microhydrated with up to three water molecules. For n = 1, we mainly observe the competition between two driving forces that determine the relative isomer stabilities, in particular the stability of isomers 1.0.0a/b vs. 1.0.0c/d. In 1.0.0a, the ionic core remains quasi-linear as in the bare anion and is further stabilized by the formation of a single, almost linear IHB. In 1.0.0b, two weaker IHBs are formed. Such isomers are preferred, when only hard ions are involved and both are observed for [LiCl2(H2O)]−. Conversely, in isomers 1.0.0c/d the water O-atom binds to the Li cation, increasing its coordination number to three. In addition, a weaker hydrogen bond is formed with a more electronegative anion. Interestingly, the mixed halide system also prefers the latter structure, because the weaker Li–I interaction (reflected in the relatively larger Li–I distance, Table S2a†) allows a shorter Li⋯OH2 distance and hence stronger cation–water interaction, which can also be viewed as an initiation of solvent induced charge separation. In principle, this observation accords with Collins “law of matching affinities”,37 which states that salt pairs of widely different ionic radii are prone to dissolve. Rationally, hard anions tend to form more compact, rigid structures with Li+, while the structures involving two soft anions are more flexible and indeed their spectra show evidence for large amplitude motion as indicated by the arrow in the case of the [LiI2(H2O)]− structure, 1.0.0d, in Fig. 12.
 | ||
| Fig. 12 Assigned MP2/def2TZVP minimum-energy structures of [LiXX′(H2O)n]− anions. Large amplitude motion indicated by AIMD analysis is represented by an arrow. | ||
With the addition of a second water molecule, water–water hydrogen bond interactions, in principle, become possible. However, as seen in Fig. 12, the dominant isomer in all three systems is 2.2.0, demonstrating that at least two water molecules are necessary to form a SIP in the gas phase and this motif's stability is independent of anion polarizability. Note that in the case of the dichloride also a CIP isomer, 2.0.1a, is observed experimentally, but with lower abundance. This CIP isomer contains a strained water–water hydrogen bond. Once the constraints are relaxed, e.g., by addition of a third water molecule, the corresponding CIP motif returns to its position as the most stable isomer in the dichloride system.
In contrast, the SIP motif persists as the most stable one in the iodide-containing systems for n = 3. These systems are characterized by a Li cation with a complete first coordination sphere, composed of one halide anion and three D-H2Os (3.3.0). Similar to [Li(H2O)3]+, water–water hydrogen bond formation is not observed in [LiI2(H2O)3]−.40 In the mixed halide system, [LiClI(H2O)3]−, however, an AD-H2O is present that does not directly interact with the Li cation (3.2.1), driven by the formation of a strong IHB involving the chloride anion. On similar grounds, the CIP motif 3.0.2 in [LiCl2(H2O)3]− is restabilized. Here, the extended hydrogen-bonded network involves three water molecules along with two moderately strong chloride IHBs from an ADD-H2O, resulting in a compact cluster with no available H-bond donors. Hence, while already two water molecules are sufficient to create a stable SIP, we see that three water molecules are needed for dynamic interconversion between (sub)-motifs already at low temperatures (Fig. 12).
4. Summary and conclusions
In this work, we studied microsolvation of lithium dihalide anions with one to three water molecules by means of a combination of gas phase spectroscopy and computational studies. In the process of structural assignments, we observe an interplay of different interaction types that is at first glance surprisingly complex. We observe that the SIP formation starts already with two water molecules, but that in particular for the hard Li+/Cl− ion pair, the cation–anion interaction is competitive to the water-ion interactions. Iodide systems with large, more polarizable anions allow for richer dynamics of water in the clusters even at cryogenic temperature. While we explored solvent-shared motifs here, we would expect the earliest onset of solvent-separated ion pairs for the lithium diiodide system. Besides ion-pair and hydrogen-bond interactions, we find that dipole arrangements play a crucial structure defining role.Regarding the computation of vibrational spectra, we can confirm that anharmonicity needs to be considered when treating microhydrated salt systems. The VPT2 method produces useful IR spectra. However, when larger amplitude motions come into play, spectra derived from the AIMD simulations provide better matches.
Our present study focuses on the early onset of solvation of electrolytes. Kirby and Jungwirth16 described in their “charge scaling manifesto” approaches to model such systems in molecular simulations: the macroscopic view assumes a continuum of a given polarizability modeled by a dielectric constant, while the microscopic view is based on the explicit interaction of – in a force field approach – atoms with partial charges. Electronic polarization is modelled in the macroscopic view only as a mean field while it is completely neglected in standard force fields. There are various approaches to include polarizability into classical force-field simulations besides the charge-scaling approach advocated for in the Kirby–Jungwirth paper.47
For the charge scaling approach, the “right” scaling factor has to be defined, which is challenging, as it is unclear how much of the charge shielding due to an environment is already parametrized into the partial charges. Alternative approaches to explicitly include polarization (and even charge transfer) into force fields require extensive reference data to (re)parametrize the extended force field, e.g., by using ab initio data sets.48,49
The surgical control over the cluster composition, the IR spectra containing structural information, and the possibility to assign these structures by combination with first principles theory, suggest such studies also as a basis to help improve molecular simulations: on the one hand, the scaling behavior from the micro- to the macroscopic view can be investigated by studying clusters with continuously increasing numbers of solvent molecules, i.e., up to more than several hundred water molecules.50 The parameters of new polarizable force fields may be tuned to reproduce the vibrational spectra of well-designed charged clusters that represent challenging molecular and interaction motifs of relevance in molecular simulations of, for example, peptides and proteins.
5. Methods
5.1 Experimental methods
IRPD experiments were performed on the Leipzig 6 K ring-electrode trap triple mass spectrometer described previously.51 Microhydrated lithium dihalide anions, [LiXX′(H2O)n]− with XX′ = I2, ClI, Cl2 were generated by a nanospray ion source from the corresponding 10 mM lithium halide solutions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/acetonitrile mixture. The beam of anions is skimmed, thermalized to room temperature in a helium-filled radio-frequency (RF) ion-guide, and then mass-selected using a quadrupole mass filter. The mass-selected [LiXX′(H2O)n]− ions are trapped in a RF ring-electrode ion trap held at a temperature in-between 6 and 300 K using a closed-cycle helium cryostat where they are thermalized to the ambient temperature of the trap and messenger tagged with D2 (He used for bare ions). These microhydrated salt anions are found to be tagged efficiently to D2via three-body collisions52 at an ion trap temperature of 13 K. The quadrupole mass spectra exhibiting the generation of these hydrated clusters can be found in Fig. S7–S9 in the ESI.†
:1 water/acetonitrile mixture. The beam of anions is skimmed, thermalized to room temperature in a helium-filled radio-frequency (RF) ion-guide, and then mass-selected using a quadrupole mass filter. The mass-selected [LiXX′(H2O)n]− ions are trapped in a RF ring-electrode ion trap held at a temperature in-between 6 and 300 K using a closed-cycle helium cryostat where they are thermalized to the ambient temperature of the trap and messenger tagged with D2 (He used for bare ions). These microhydrated salt anions are found to be tagged efficiently to D2via three-body collisions52 at an ion trap temperature of 13 K. The quadrupole mass spectra exhibiting the generation of these hydrated clusters can be found in Fig. S7–S9 in the ESI.†
IRPD spectra are recorded using the IR1MS2 technique, which allows for a background-free photofragment detection.53,54 To this end, all ions (bare and messenger-tagged) are extracted from the ion trap every 100 ms and focused both temporally and spatially into the center of the extraction region of an orthogonally mounted double-focusing reflection time-of-flight (TOF) tandem photofragmentation mass spectrometer. Using a first set of 30 μs long high voltage (HV) pulses, the ions are accelerated into the reflectron stage, spread out in space according to their mass-to-charge ratio (m/z) and are refocused in the initial extraction region. Prior to reacceleration towards the MCP detector, ions with a particular m/z value are irradiated with a properly timed, widely wavelength tunable (750–7000 cm−1) IR laser pulse (bandwidth: ∼3.5 cm−1), supplied by an optical parametric oscillator/amplifier (LaserVision: OPO/OPA/AgGaSe2) laser system.55 IRPD spectra are recorded by continuously scanning the laser wavelength monitored online using a HighFinesse WS6-600 wavelength meter with a scan speed such that an averaged TOF mass spectrum (over 100 laser shots) is obtained every 2 cm−1. Typically, at least such five scans are measured and averaged and the photodissociation cross section σIRPD is determined as described previously.51,56
Double-resonance IR2MS2 spectra using the ion-dip technique are obtained by employing two tunable IR lasers in a pump–probe approach and tandem mass selection stages. Briefly, the method is sensitive to laser-induced population changes of isomers that are maintained longer than the delay between the pump and probe laser pulses (∼10–20 μs). The IR spectra of isomers interconverting faster than this timescale cannot be isolated using this method. Details regarding the IR2MS2 technique can be found elsewhere.43,53
5.2 Computational methods
The DFT ab initio molecular dynamics (AIMD) simulations were performed using the FHI-aims code68 together with iPI.69 For multiple low-energy conformers, constant temperature MD simulations at T = 100 K, and for testing also at T = 50 K, were run for 20 ps with a 0.5 fs time step. The velocity rescaling thermostat presented by Bussi, Donadio, and Parrinello was used.70 After an equilibration period of approximately 10 ps, 11 individual structures were extracted every 1 ps. These 11 frames per structure served as starting structures for 10 ps long constant energy MD simulations with a step size of 0.5 fs. The electric dipole moment of the cluster was calculated every 1 fs step and used to compute IR spectra based on the dipole–dipole autocorrelation function.71 The best agreement with the experimental spectra has been found after redshifting the computed spectra by 210 cm−1.
Data availability
Gaussian files on geometry optimization and anharmonic spectra can be found with the DOI: 10.17172/NOMAD/2022.03.18-1. Trajectories for all NVT and NVE calculations can be found with the DOI: 10.17172/NOMAD/2022.05.23-1Author contributions
KRA and CB designed the study, coordinated the project and supervised the research. AC and SS recorded experimental data. AC has performed static computer simulations such as geometry optimization, harmonic and anharmonic spectra calculation. The analysis and interpretation of these data were performed by AC and KRA. AIMD simulations and their interpretation were made by TB and CB. HW, TB and CB performed and analyzed the benchmark calculations for the selection of the exchange–correlation functional and the dispersion correction used in AIMD simulations. All authors were involved in the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the German Research Foundation (DFG) as part of the individual research Grant No. 321027174 (AS133/3-1 “Spectroscopic Characterization of Salt Dissolution in Microhydrated Cluster Ions and at the Water/Vapor Interface”). The authors are thankful to Alexandra Giermann (Leipzig University) for her assistance in preparing and handling solutions. KRA acknowledges instrumental support from the Fritz Haber Institute of the Max Planck Society. TB and CB gratefully acknowledge computational resources provided by the Max Planck Computing and Data Facility.Notes and references
- Y. Marcus, Chem. Rev., 2009, 109, 1346 CrossRef CAS PubMed.
- D. J. Tobias and J. C. Hemminger, Science, 2008, 319, 1197 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts, Chem. Rev., 2003, 103, 4801 CrossRef CAS PubMed.
- R. Buchner, S. G. Capewell, G. Hefter and P. M. May, J. Phys. Chem. B, 1999, 103, 1185 CrossRef CAS.
- N. F. A. van der Vegt, K. Haldrup, S. Roke, J. Zheng, M. Lund and H. J. Bakker, Chem. Rev., 2016, 116, 7626 CrossRef CAS PubMed.
- E. Levy, A. Puzenko, U. Kaatze, P. Ben Ishai and Y. Feldman, J. Chem. Phys., 2012, 136, 114503 CrossRef PubMed.
- H. Bian, X. Wen, J. Li, H. Chen, S. Han, X. Sun, J. Song, W. Zhuang and J. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 4737 CrossRef CAS.
- I. Juurinen, T. Pylkkänen, K. O. Ruotsalainen, C. J. Sahle, G. Monaco, K. Hämäläinen, S. Huotari and M. Hakala, J. Phys. Chem. B, 2013, 117, 16506 CrossRef CAS PubMed.
- I. Harsányi, L. Temleitner, B. Beuneu and L. Pusztai, J. Mol. Liq., 2012, 165, 94 CrossRef.
- A. La Sauza-de Vega, T. Rocha-Rinza and J. M. Guevara-Vela, ChemPhysChem, 2021, 22, 1269 CrossRef PubMed.
- S. T. van der Post and H. J. Bakker, Phys. Chem. Chem. Phys., 2012, 14, 6280 RSC.
- S. Yoo, Y. A. Lei and X. C. Zeng, J. Chem. Phys., 2003, 119, 6083 CrossRef CAS.
- C. J. Fennell, A. Bizjak, V. Vlachy and K. A. Dill, J. Phys. Chem. B, 2009, 113, 6782 CrossRef CAS PubMed.
- E. Pluhařová, P. E. Mason and P. Jungwirth, J. Phys. Chem. A, 2013, 117, 11766 CrossRef PubMed.
- A. Vargas-Caamal, J. L. Cabellos, F. Ortiz-Chi, H. S. Rzepa, A. Restrepo and G. Merino, Chem. – Eur. J., 2016, 22, 2812 CrossRef CAS PubMed.
- B. J. Kirby and P. Jungwirth, J. Phys. Chem. Lett., 2019, 10, 7531 CrossRef CAS PubMed.
- P. Jungwirth and D. J. Tobias, Chem. Rev., 2006, 106, 1259 CrossRef CAS PubMed.
- D. Laage, T. Elsaesser and J. T. Hynes, Chem. Rev., 2017, 117, 10694 CrossRef CAS PubMed.
- B. S. Ault, J. Am. Chem. Soc., 1978, 100, 2426 CrossRef CAS.
- A. Mizoguchi, Y. Ohshima and Y. Endo, J. Chem. Phys., 2011, 135, 64307 CrossRef PubMed.
- G. Grégoire, M. Mons, I. Dimicoli, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard and D. Solgadi, J. Chem. Phys., 2000, 112, 8794 CrossRef.
- J. A. Davies, M. Mugglestone, S. Yang and A. M. Ellis, J. Phys. Chem. A, 2020, 124, 6528 CrossRef CAS PubMed.
- J. Tandy, C. Feng, A. Boatwright, G. Sarma, A. M. Sadoon, A. Shirley, N. D. N. Rodrigues, E. M. Cunningham, S. Yang and A. M. Ellis, J. Chem. Phys., 2016, 144, 121103 CrossRef PubMed.
- R.-Z. Li, C.-W. Liu, Y. Q. Gao, H. Jiang, H.-G. Xu and W.-J. Zheng, J. Am. Chem. Soc., 2013, 135, 5190 CrossRef CAS PubMed.
- G.-L. Hou, C.-W. Liu, R.-Z. Li, H.-G. Xu, Y. Q. Gao and W.-J. Zheng, J. Phys. Chem. Lett., 2017, 8, 13 CrossRef CAS PubMed.
- X.-B. Wang, H.-K. Woo, B. Jagoda-Cwiklik, P. Jungwirth and L.-S. Wang, Phys. Chem. Chem. Phys., 2006, 8, 4294 RSC.
- Y. Feng, M. Cheng, X.-Y. Kong, H.-G. Xu and W.-J. Zheng, Phys. Chem. Chem. Phys., 2011, 13, 15865 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- T. Wende, N. Heine, T. I. Yacovitch, K. R. Asmis, D. M. Neumark and L. Jiang, Phys. Chem. Chem. Phys., 2016, 18, 267 RSC.
- J. W. DePalma, P. J. Kelleher, C. J. Johnson, J. A. Fournier and M. A. Johnson, J. Phys. Chem. A, 2015, 119, 8294 CrossRef CAS PubMed.
- D. Schröder, L. Duchácková, J. Tarábek, M. Karwowska, K. J. Fijalkowski, M. Oncák and P. Slavícek, J. Am. Chem. Soc., 2011, 133, 2444 CrossRef PubMed.
- C. J. Johnson, L. C. Dzugan, A. B. Wolk, C. M. Leavitt, J. A. Fournier, A. B. McCoy and M. A. Johnson, J. Phys. Chem. A, 2014, 118, 7590 CrossRef CAS PubMed.
- L. Jiang, T. Wende, R. Bergmann, G. Meijer and K. R. Asmis, J. Am. Chem. Soc., 2010, 132, 7398 CrossRef CAS PubMed.
- W. H. Robertson and M. A. Johnson, Annu. Rev. Phys. Chem., 2003, 54, 173 CrossRef CAS PubMed.
- A. Chakraborty, S. Schmahl and K. R. Asmis, ChemPhysChem, 2021, 22, 1036 CrossRef CAS PubMed.
- V. G. Solomonik, J. Struct. Chem., 1979, 19, 860 CrossRef.
- K. D. Collins, Q. Rev. Biophys., 2019, 52, e11 CrossRef CAS PubMed.
- G. D. Dickenson, M. L. Niu, E. J. Salumbides, J. Komasa, K. S. E. Eikema, K. Pachucki and W. Ubachs, Phys. Chem. Chem. Phys., 2013, 110, 193601 CAS.
- P. Ayotte, G. H. Weddle and M. A. Johnson, J. Chem. Phys., 1999, 110, 7129 CrossRef CAS.
- O. Rodriguez and J. M. Lisy, J. Phys. Chem. Lett., 2011, 2, 1444 CrossRef CAS.
- J.-C. Jiang, Y.-S. Wang, H.-C. Chang, S. H. Lin, Y. T. Lee, G. Niedner-Schatteburg and H.-C. Chang, J. Am. Chem. Soc., 2000, 122, 1398 CrossRef CAS.
- L. J. M. Kempkes, J. Martens, G. Berden, K. J. Houthuijs and J. Oomens, Faraday Discuss., 2019, 217, 434 RSC.
- M. Mayer and K. R. Asmis, J. Phys. Chem. A, 2021, 125, 2801 CrossRef CAS PubMed.
- H. H. Loeffler and B. M. Rode, J. Chem. Phys., 2002, 117, 110 CrossRef CAS.
- C. Baldauf, K. Pagel, S. Warnke, G. von Helden, B. Koksch, V. Blum and M. Scheffler, Chem. – Eur. J., 2013, 19, 11224 CrossRef CAS PubMed.
- K. D. Collins, Biophys. J., 1997, 72, 65 CrossRef CAS PubMed.
- Z. Jing, C. Liu, S. Y. Cheng, R. Qi, B. D. Walker, J.-P. Piquemal and P. Ren, Annu. Rev. Biophys., 2019, 48, 371 CrossRef CAS PubMed.
- K. S. Amin, X. Hu, D. R. Salahub, C. Baldauf, C. Lim and S. Noskov, J. Chem. Phys., 2020, 153, 144102 CrossRef CAS PubMed.
- M. Ropo, M. Schneider, C. Baldauf and V. Blum, Sci. Data, 2016, 3, 160009 CrossRef CAS PubMed.
- J. T. O’Brien and E. R. Williams, J. Am. Chem. Soc., 2012, 134, 10228 CrossRef PubMed.
- N. Heine and K. R. Asmis, Int. Rev. Phys. Chem., 2015, 34, 1 Search PubMed.
- M. Brümmer, C. Kaposta, G. Santambrogio and K. R. Asmis, J. Chem. Phys., 2003, 119, 12700 CrossRef.
- N. Heine, M. R. Fagiani, M. Rossi, T. Wende, G. Berden, V. Blum and K. R. Asmis, J. Am. Chem. Soc., 2013, 135, 8266 CrossRef CAS PubMed.
- B. M. Elliott, R. A. Relph, J. R. Roscioli, J. C. Bopp, G. H. Gardenier, T. L. Guasco and M. A. Johnson, J. Chem. Phys., 2008, 129, 94303 CrossRef PubMed.
- W. R. Bosenberg and D. R. Guyer, J. Opt. Soc. Am. B, 1993, 10, 1716 CrossRef CAS.
- N. Heine and K. R. Asmis, Int. Rev. Phys. Chem., 2016, 35, 507 Search PubMed.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 275 CrossRef CAS.
- F. Weigend, F. Furche and R. Ahlrichs, J. Chem. Phys., 2003, 119, 12753 CrossRef CAS.
- J. Bloino and V. Barone, J. Chem. Phys., 2012, 136, 124108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao and N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 34106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 73005 CrossRef PubMed.
- A. Tkatchenko, R. A. DiStasio, R. Car and M. Scheffler, Phys. Rev. Lett., 2012, 108, 236402 CrossRef PubMed.
- A. Tkatchenko, A. Ambrosetti and R. A. DiStasio, J. Chem. Phys., 2013, 138, 74106 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8(8), e1327 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175 CrossRef CAS.
- V. Kapil, M. Rossi, O. Marsalek, R. Petraglia, Y. Litman, T. Spura, B. Cheng, A. Cuzzocrea, R. H. Meißner, D. M. Wilkins, B. A. Helfrecht, P. Juda, S. P. Bienvenue, W. Fang, J. Kessler, I. Poltavsky, S. Vandenbrande, J. Wieme, C. Corminboeuf, T. D. Kühne, D. E. Manolopoulos, T. E. Markland, J. O. Richardson, A. Tkatchenko, G. A. Tribello, V. van Speybroeck and M. Ceriotti, Comput. Phys. Commun., 2019, 236, 214 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 14101 CrossRef PubMed.
- C. Baldauf and M. Rossi, J. Phys.: Condens. Matter, 2015, 27, 493002 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03431j |
| This journal is © The Royal Society of Chemistry 2022 |