
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding fragility and engineering activation stability in two-dimensional covalent organic frameworks†
Dongyang
Zhu‡
 a,
Jun-Jie
Zhang‡
b,
Xiaowei
Wu‡
cd,
Qianqian
Yan
b,
Fangxin
Liu
a,
Yifan
Zhu
b,
Xiaodong
Gao
e,
Muhammad M.
Rahman
b,
Boris I.
Yakobson
bf,
Pulickel M.
Ajayan
b and
Rafael
Verduzco
*ab
a,
Jun-Jie
Zhang‡
b,
Xiaowei
Wu‡
cd,
Qianqian
Yan
b,
Fangxin
Liu
a,
Yifan
Zhu
b,
Xiaodong
Gao
e,
Muhammad M.
Rahman
b,
Boris I.
Yakobson
bf,
Pulickel M.
Ajayan
b and
Rafael
Verduzco
*ab
aDepartment of Chemical and Biomolecular Engineering, Rice University, 6100 Main Street, MS-362, Houston, Texas 77005, USA. E-mail: rafaelv@rice.edu
bDepartment of Materials Science and NanoEngineering, Rice University, 6100 Main Street, MS-325, Houston, Texas 77005, USA
cCAS Key Laboratory of Design and Assembly of Functional Nanostructures, Fujian Provincial Key Laboratory of Nanomaterials, Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences, Fuzhou 350002, China
dXiamen Key Laboratory of Rare Earth Photoelectric Functional Materials, Xiamen Institute of Rare Earth Materials (XMIREM), Haixi Institutes, Chinese Academy of Sciences, Xiamen 361021, China
eDepartment of Earth, Environmental, and Planetary Sciences, Rice University, 6100 Main Street, MS-126, Houston, Texas 77005, USA
fDepartment of Chemistry, Rice University, MS-60, 6100 Main Street, Houston, Texas 77005, USA
First published on 22nd July 2022
Abstract
The sensitivity of covalent organic frameworks (COFs) to pore collapse during activation processes is generally termed activation stability, and activation stability is important for achieving and maintaining COF crystallinity and porosity which are relevant to a variety of applications. However, current understanding of COF stability during activation is insufficient, and prior studies have focused primarily on thermal stability or on the activation stability of other porous materials, such as metal–organic frameworks (MOFs). In this work, we demonstrate and implement a versatile experimental approach to quantify activation stability of COFs and use this to establish a number of relationships between their pore size, the type of pore substituents, pore architecture, and structural robustness. Additionally, density functional theory calculations reveal the impact on both inter-and intra-layer interactions, which govern activation stability, and we demonstrate that activation stability can be systematically tuned using a multivariate synthesis approach involving mixtures of functionalized and unfunctionalized COF building blocks. Our findings provide novel fundamental insights into the activation stability of COFs and offer guidance for the design of more robust COFs.
1 Introduction
Covalent organic frameworks (COFs) are an emerging class of reticular materials with nanoscale pores, long-range ordering, and crystallinity.1–7 The high degree of crystallinity and porosity makes them attractive for various applications, including gas adsorption and storage,8–10 metal-free heterogeneous catalysis,11–16 membrane separations,17,18 and energy storage.19–24 In order to produce dry, crystalline, and porous COFs for applications, an activation procedure is required to remove impurities such as unreacted monomers, oligomers, and/or guest solvent molecules. The simplest and most readily accessible activation approach is through solvent washing followed by vacuum drying. However, solvent evaporation can produce significant capillary forces on the nanoscale pores, in some cases leading to structural distortion, such as degradation of the pores or mismatch between layers.25–27 This process is generally termed “pore collapse” and can disrupt or destroy the crystallinity and porosity of COFs, limiting their utility for use in various applications.Foundational studies with metal organic frameworks (MOFs), a related class of porous materials, provided insight into the pore collapse of MOFs during activation.28,29 These studies demonstrated that solvent surface tension plays an important role in maintaining MOF porosity and crystallinity, and subsequent work has established best practices for the preparation and activation of MOFs.30 While this work was useful for demonstrating factors that may influence pore collapse in all porous materials, pore collapse in COFs has not been studied in such significant detail. Additionally, there are important differences between MOFs and COFs that may influence pore collapse. MOFs have metal coordination bonds, while COFs have covalent bonds. These different bond types will likely lead to different bond strengths and structural changes when disturbed by capillary forces. Additionally, COFs are typically two-dimensional materials, while MOFs are three dimensional. As a result, we expect differences in structural distortion and pore collapse during activation of MOFs and COFs.
Similar to MOFs, the sensitivity of COFs to pore collapse during the solvent activation process, namely the activation stability, depends on their chemistries, structures, and architectures. For some 2D COFs termed fragile COFs, the crystallinity and porosity can be readily disrupted during activation with polar solvents. Other COFs, termed robust COFs, are less sensitive to activation procedures and can maintain crystallinity and porosity under a variety of solvent conditions and activation procedures.25–27 However, the factors governing COF fragility and robustness are unclear. Medina and coworkers hypothesized that fragility in 2D COFs was governed primarily by the strength of interlayer interactions and that loss of crystallinity reflected disrupted alignment or stacking of COF molecular layers.26 They reported that after degradation, crystallinity and porosity in some COFs could be recovered through supercritical CO2 activation. However, other studies have observed irreversible loss of crystallinity and porosity in COFs,27 which suggests more significant structural degradation after solvent removal. Zhao and coworkers observed both reversible and irreversible interlayer shifting induced by different solvent evaporations depending on strength of adjacent COF layer interaction.31 Although these prior studies provided some insightful analyses, we still lack a clear and systematic understanding of fragility and robustness in 2D COFs. In particular, we do not have an understanding of how to design COFs that are fragile or robust, and how the COF chemistries, structures and architectures influence fragility or robustness. We also lack a method for probing fragility and robustness in a series of COF samples that enables comparison and understanding trends in molecular structure and chemistry. Insights into these questions can provide useful information not only for the design of COFs for specific applications but also for understanding the fracture and degradation of COFs under different activation conditions.
Recent work probed the thermal degradation of COFs.32,33 These studies were focused on understanding structural and chemical changes in COFs at elevated temperatures and revealed two distinct degradation mechanisms. The first degradation involves structural distortion or a phase transition from an eclipsed to staggered conformation and is reflected in a loss or reduction of crystallinity, and at higher temperatures the COF will degrade chemically. These studies also established empirical rules for degradation stability and demonstrated that stability was improved with decreasing pore size and with smaller pore substituents. Other related studies have developed engineering approaches to construct more thermally and chemically stable COFs, but these prior studies have not investigated stability during solvent evaporation or COF activation. For example, Smaldone and coworkers designed COFs with amide functional groups that produced interlayer hydrogen bonding interactions and enhanced the thermal and chemical stability of the resulting COF.34 Lotsch and coworkers introduced side chains in large pore COFs to improve thermal stability.33 Other approaches include the introduction of fluorine atoms35 and intralayer hydrogen bonding interactions36 to produce COFs with improved chemical stability. However, these studies did not specifically focus on stability during solvent drying or activation. For many applications such as organic solvent adsorption or liquid-phase catalysis, COFs have to be dried and regenerated multiple times to enable reuse. Loss of crystallinity and porosity during the solvent removal process may significantly decrease their recycle performance. Although our prior study and others suggest that low surface tension solvents such as perfluorohexane, hexane and supercritical CO2 can help maintain high crystallinity and porosity in fragile COFs,25–27 the molecular design of COFs to provide intrinsic activation stability has not been investigated. Therefore, molecular engineering of stable COFs that can resist pore collapse or structural distortion during the solvent removal procedure is critically needed for applications that involve solvent evacuation and material reuse.
Herein, we report a systematic study on the activation stability during solvent evaporation process in 2D COFs varying in pore size, functionality, and architecture. We previously found that the surface tension of the solvent used in COF activation played an important role in maintaining or degrading COF crystallinity and porosity.25 Fragile COFs exhibited complete loss of crystallinity and porosity when activated using a solvent with a surface tension higher than a threshold value. Depending on the COF studied, the threshold value ranged from 22.7 to 37.1 mN m−1. Conversely, robust COFs could maintain crystallinity and porosity even after activation with high surface tension solvents. We also showed that the structural integrity of 2D COFs could be measured as a function of solvent surface tension (Fig. 1A). Using this approach, we evaluated the activation stability in a series of 2D COFs varying in pore size, pore substituents, and framework topologies (Fig. 1B). We demonstrate that activation stability decreases with increasing pore sizes, and bulky or polar substituents on COF frameworks significantly enhance their robustness during activation, in contrast to some prior studies on thermal stability of COFs.32 The trends hold true for two different pore topologies (hexagonal and rhombic), suggesting that these trends may be universal. Density functional theory (DFT) calculations were used to elucidate the strength of interlayer interactions and the energy surface as functions of distortion dihedral angles of molecular models, and they show that both interlayer interactions and the probability of intralayer structural distortions contribute to COF robustness. Finally, by using a multivariate approach, the simplest molecular engineering method, by mixing building blocks with or without pore substituents, we show that we can finely tune COF fragility and engineer materials with a desired robustness (Fig. 1C). This work provides insight into chemical, structural, and architectural details that govern COF pore collapse or stability during solvent evaporation process, offers a practical approach to study fragility in 2D COFs, and demonstrates a simple and straightforward approach to construct COFs with tunable robustness. We anticipate that this work will prove useful for the design of robust COFs for a wide range of applications and enable future studies focused on understanding structural degradation during activation of COFs.
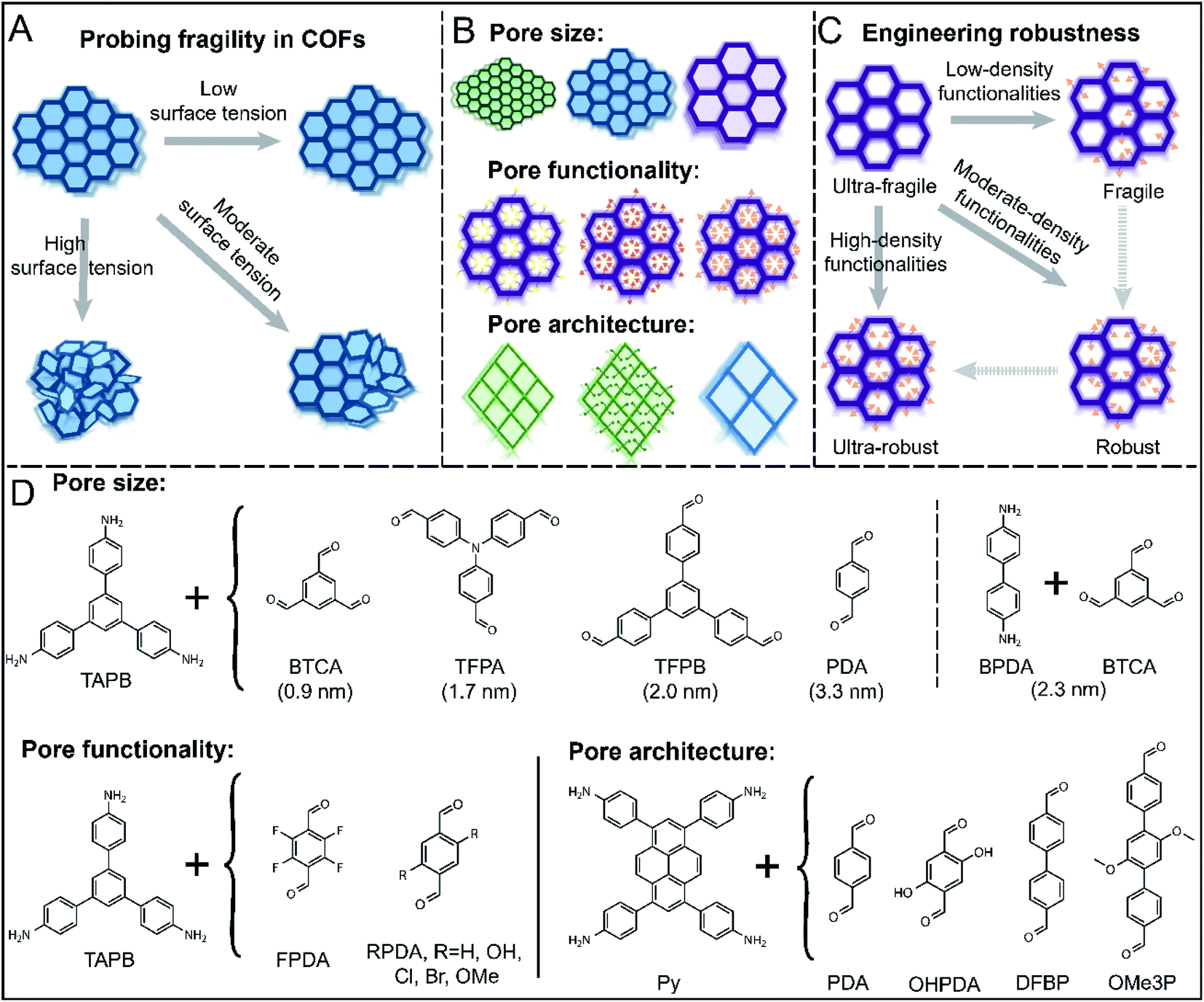 | ||
| Fig. 1 Overview of pore collapse in COFs of varying pore sizes, pore functionalities, and topologies. Schematics for (A) pore collapse after activation with low, moderate, and high surface-tension solvents, (B) series of 2D COFs studied varying in pore size and pore functionality and of rhombic or hexagonal topology, and (C) systematic molecular engineering of robustness of COFs through introduction of increasing number of side-chain functionalities. (D) COF chemistries studied in this work. TAPB: 1,3,5-tris(4-aminophenyl)benzene, BTCA: benzene-1,3,5-tricarbaldehyde, TFPA: tris(4-formylphenyl)amine, TFPB: 1,3,5-tri(4-formylphenyl)benzene, PDA: terephthaldehyde, BPDA: [1,1′-biphenyl]-4,4′-diamine, FPDA: tetrafluoroterephthaldehyde, OHPDA: 2,5-dihydroxy-1,4-benzenedicarboxaldehyde, ClPDA: 2,5-dichloroterephthalic dicarboxaldehyde, BrPDA: 2,5-dibromoterephthalaldehyde, OMePDA: 2,5-dimethoxyterephthalaldehyde, Py: 1,3,6,8-tetrakis(4-aminophenyl)pyrene, DFPB: 4,4′-diformylbiphenyl, OMe3P: 2′,5′-dimethoxy-[1,1':4′,1′′-terphenyl]-4,4′′-dicarbaldehyde. | ||
2 Results and discussion
Our objectives were to understand the relationship between structural integrity and pore size, pore functionality, and pore architecture in 2D COFs during the solvent evaporation process (Fig. 1A). We synthesized a series of COFs varying in pore sizes, functionalities, and topologies and characterized their activation stability in the presence of low, moderate, or high surface tension solvents (Fig. 1A–C). Specifically, we used perfluorohexane, methanol, and tetrahydrofuran, which have surface tensions of 11.9, 22.7, and 26.4 mN m−1. Higher solvent surface tensions produce greater capillary forces during drying, and by screening the stability of COFs against different solvents, we could compare COF robustness. Other factors, such as hydrogen bonding interactions may also be important, but have not been reported to play a role in activation. The most robust COFs will retain crystallinity and porosity after washing with highly polar solvents and drying under vacuum, while fragile COFs will readily lose structural integrity after treatments with even nonpolar solvents with low surface tensions (Fig. 1A). The level of crystallinity can be observed by powder X-ray diffraction (PXRD) analyses, and the porosity can be quantified through gas adsorption measurements.To examine the influence of pore size, we initially designed a series of imine COFs with building blocks of varying pore sizes ranging from 0.9 nm to 3.3 nm (Fig. 1D). All COFs in this series had a hexagonal topology, the most common 2D COF topology studied to date. To study the effect of pore functionality, we investigated a series of COFs with a COF-TAPB-PDA backbone and varying in the size and polarity of substituents: F, OH, Cl, Br, and methoxy (OMe). Finally, to study the effect of pore architecture, we built a set of COFs with a rhombic shape and varying in pore size and pore functionality. All COFs were synthesized using solvothermal conditions, and detailed synthetic procedures are provided in the ESI†.
To compare the robustness for various COFs studied, we performed a vacuum activation process using solvents of different surface tensions. Briefly, each COF was first washed thoroughly using THF and acetone to remove unreacted monomers and oligomers. Next, any residual acetone and THF was removed by washing and solvent exchanging with perfluorohexane (PFH, low surface tension, 11.9 mN m−1), methanol (MeOH, moderate surface tension, 22.7 mN m−1), or tetrahydrofuran (THF, high surface tension, 26.4 mN m−1), respectively. The samples were then dried under vacuum in a heating oven (80 °C, pressure = 1 Torr or 0.13 kPa) and then characterized using a combination of PXRD and nitrogen sorption measurements to measure their crystallinity and porosity. This enabled us to understand the factors that control the porosity and crystallinity along with the trends in fragility across the series of COF samples studied.
2.1 Effect of pore size
The pore size is a well-defined and measurable feature of COFs, and it has been shown to play an important role in separation, ion conduction, and many adsorption-related phenomena.19,37–41 We hypothesized that increasing the pore size would result in more fragile COFs. To test this hypothesis, we synthesized five different imine-linked COFs TAPB-BTCA (0.9 nm), TAPB-TFPA (1.7 nm), TAPB-TFPB (2.0 nm), PI-2 (2.3 nm, constructed by BPDA and BTCA) and TAPB-PDA (3.3 nm) COFs (see Fig. 1D). Four of these COFs contained the same TAPB building block, and the fifth (PI-2) had structurally similar building blocks. No COF in this series contained pore functionalities, and all possessed a 2D hexagonal hca/hcb topology. The most significant difference across the series was the pore size, and we observed clear trends in fragility across this set of samples. COFs with the smallest pore size (i.e. TAPB-BTCA COF, 0.9 nm pore size) were very robust, and no loss of porosity or crystallinity occurred after vacuum activation (Fig. 2A). The PXRD patterns exhibited sharp diffraction peaks, and the nitrogen sorption curves were similar and indicative of highly porous materials after activation with PFH, MeOH and THF (Fig. 2, B and C). The nitrogen sorption curves increased sharply at low relative pressures (P/P0 < 0.01), indicative of rich microporous structures. The Brunauer–Emmett–Teller (BET) surface areas of these samples were similar: 1022, 1001 and 955 m2 g−1 for samples activated using PFH, MeOH and THF, respectively. Finally, the pore size distributions calculated using quenched solid density functional theory (QSDFT) were uniform and centered around 0.9 nm (Fig. S1†), which were very close to that in prior studies of these materials.42 Furthermore, the incremental pore volume (y-axis of pore size distribution plots, Fig. S1†) showed similar values, indicating these samples shared similar pore volumes.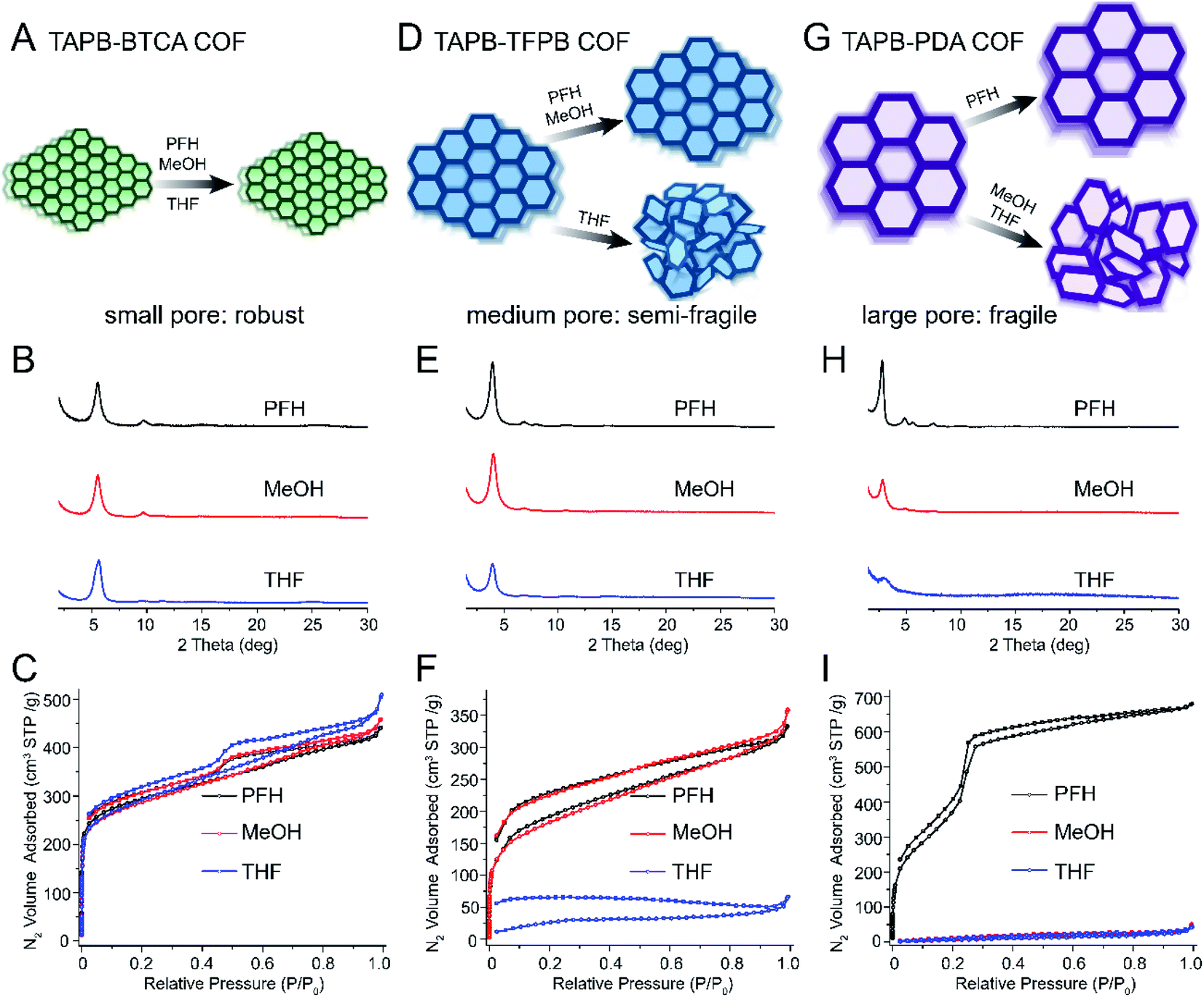 | ||
| Fig. 2 Schematics, PXRD, and nitrogen sorption isotherms demonstrating increased fragility with increased pore size. (A) Schematics showing that small-pore COFs (TAPB-BTCA) retain crystallinity for all activation solvents. (B) PXRD and (C) nitrogen sorption isotherms of small-pore COFs after activation using PFH, MeOH and THF. (D) Schematics showing COFs with medium pore size (TAPB-TFPB) retain crystallinity after activation using PFH, MeOH but not after THF activation. (E) PXRD and (F) nitrogen sorption isotherms of medium-pore COFs after activation using PFH, MeOH and THF. (G) Schematics showing COFs with large pore size (TAPB-PDA) only retain crystallinity after PFH activation, but pore collapse occurs after MeOH and THF activation. (H) PXRD and (I) nitrogen sorption isotherms of large-pore COFs after activation using PFH, MeOH and THF. Note: the structures of all monomers presented here are shown in Fig. 1. | ||
With increasing pore size, we observed increasing fragility. TAPB-TFPB COFs (2.0 nm pore size) retained excellent crystallinity and porosity after activation with PFH or MeOH (Fig. 2 D–F), but a clear loss of porosity was observed after activation with THF (high surface tension). PXRD analysis revealed that the sample retained some crystallinity after activation by all solvents, but the intensity of the primary diffraction peak decreased significantly after activation with THF (Fig. 2E). The nitrogen sorption curve for TAPB-TFPB after activation with THF did not exhibit a sharp increase at low relative pressures (Fig. 2F), and the BET surface area after activation by THF (108 m2 g−1) was significantly lower compared with the sample activated by PFH (687 m2 g−1) or MeOH activation (651 m2 g−1). Next, TAPB-PDA COFs, with a pore size of 3.3 nm, the largest in the series, were very fragile and lost crystallinity and porosity after activation with MeOH or THF (Fig. 2G–I). TAPB-PDA COFs exhibited excellent crystallinity and porosity after PFH activation (Fig. 2H and I) and had a very high BET surface area of 1919 m2 g−1. However, the porous structure was completely destroyed after MeOH and THF activation, and the resulting materials exhibited a broad primary diffraction peak indicative of short-range ordering (Fig. 2H). The nitrogen sorption isotherms (Fig. 2I) and pore size distributions (Fig. S5†) further confirmed loss of structural integrity. COFs after activation using MeOH and THF displayed extremely low nitrogen sorption capacity and low surface areas (45 and 30 m2 g−1 after MeOH and THF activation, respectively, Table S2†). Their incremental pore volume (Fig. S5†) was also close to zero and pore size became disordered, indicating the complete loss of porous structures.
The results for TAPB-TFPA COF (1.7 nm pores) and PI-2 COFs (2.3 nm pores) are presented in the ESI (Fig. S7–S14).† These COFs followed the trend of increasing fragility with increasing pore size. TAPB-TFPA exhibited moderate fragility between that of TAPB-BTCA and TAPB-TFPB. TAPB-TFPA retained crystallinity after activation with all three solvents, but we measured a significant loss of porosity after activation with THF. This sample is therefore more fragile than TAPB-BTCA, but more robust than TAPB-TFPB, which exhibited very poor porosity and crystallinity after THF activation.
As a method to visualize trends in the structural integrity of these COFs as a function of pore size, we plotted the ratio of BET surface areas after PFH and THF activation (SBET(THF)/SBET(PFH)) and those after MeOH and THF activation ((SBET(THF)/SBET(MeOH)) (Fig. S15†). These ratios represent the fragility or robustness of each material, with a value of 1 most robust and 0 the most fragile. This plot reveals a clear trend with increasing pore size, showing increasing fragility (or decreasing activation stability) with increasing pore size. This provides a simple quantitative measure for robustness that can be used to compare different COFs.
Our results therefore demonstrate that fragility in the series of COFs increases with pore size. While prior work has identified fragile and robust COFs,25–27,31 this is the first example of an empirical trend of activation stability in a series of COFs varying in pore size. To be clear, the pore size is related to the density of intermolecular interactions, which have been noted previously to be important to thermal stability.32–34 In our series of materials, it may be that the strength of these intermolecular interactions governs activation stability. The pore size can also affect the evaporation rate, which may play a role in activation stability, but was not studied here. The roles of the strength of interlayer interactions are further studied through molecular simulations and discussed in Section 2.4.
2.2 Effect of pore functionality
Pore functionalities or substituents can be introduced by modifying COF building blocks, and this strategy has been widely explored for various applications including catalysis, ion transport, adsorption and separations.43–47 Prior studies have also shown that pore functionalities increase structural stability in certain cases,25–27,31,33 but this effect has not previously been studied in a systematic series. We hypothesized that pore functionalities would improve activation stability, and that larger and more polar substituents would have a stronger effect. To test this hypothesis, we prepared five COFs all containing the same framework architecture and varying in pore functionality: F, OH, Cl, Br, and OMe. Each COF was treated by vacuum activation after washing with PFH, MeOH, or THF and characterized by PXRD and nitrogen sorption measurements. The results for TAPB-FPDA, TAPB-OHPDA, and TAPB-BrPDA are shown in Fig. 3 and ESI,† and those for TAPB-ClPDA and TAPB-OMePDA are provided in the ESI†.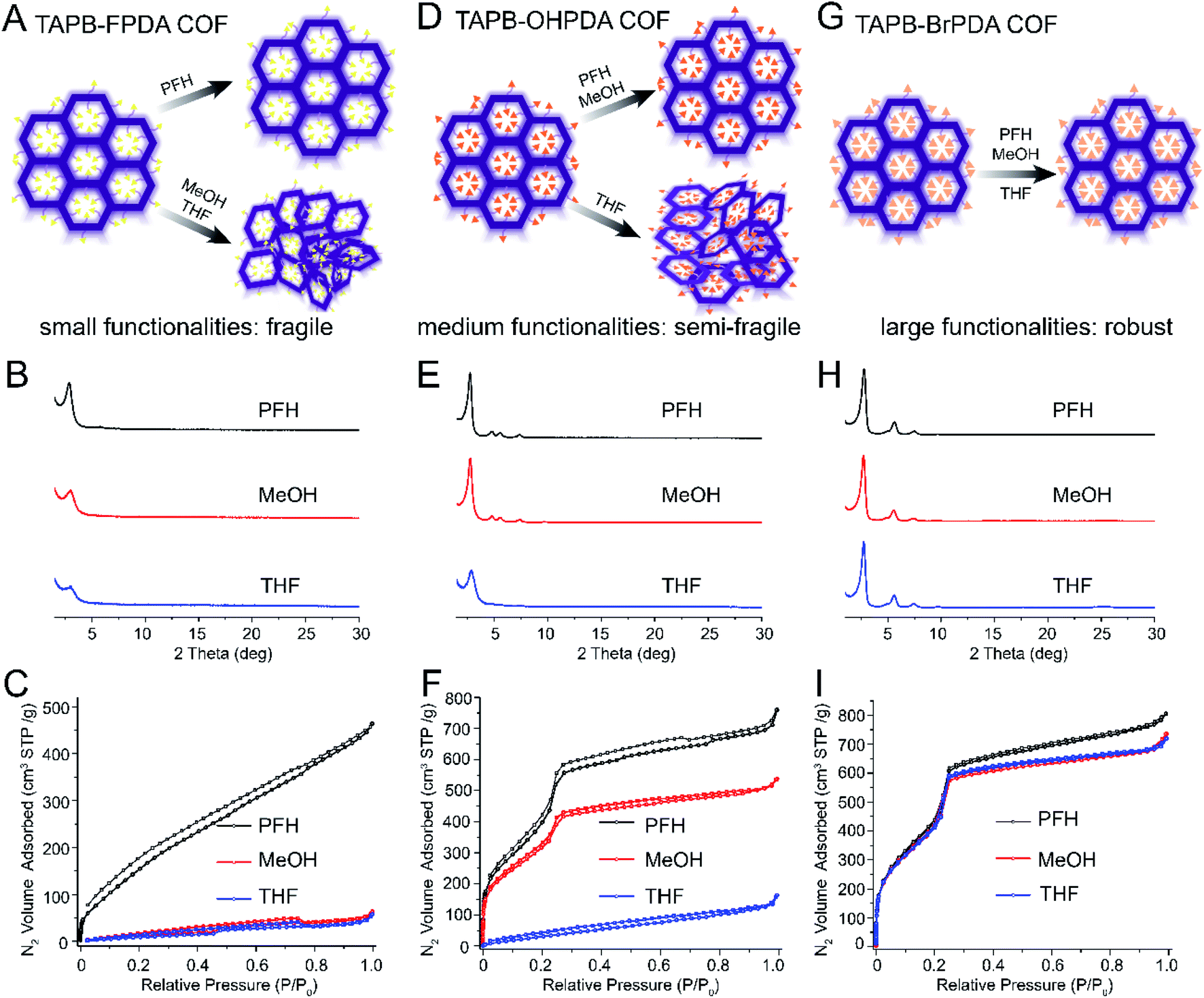 | ||
| Fig. 3 Schematics, PXRD, and nitrogen sorption isotherms demonstrating increased robustness with increasing size of pore functionality. (A) Schematic showing that COFs with small pore functionalities lose crystallinity after MeOH and THF activation. (B) PXRD and (C) nitrogen sorption isotherms of TAPB-FPDA COF after activation using PFH, MeOH and THF. (D) Schematics showing that COFs with medium-sized functional groups are more robust and retain crystallinity and porosity after activation with PFH or MeOH activation but not after THF activation. (E) PXRD and (F) nitrogen sorption isotherms of TAPB-OHPDA COFs (G) Schematics showing COFs with large pore functionalities retain crystallinity and porosity after activation with PFH, MeOH, or THF. (H) PXRD and (I) nitrogen sorption isotherms of TAPB-BrPDA COFs after activation using PFH, MeOH and THF. Note: the structures of all monomers presented here are shown in Fig. 1. | ||
The COF with the smallest substituent (TAPB-FPDA) was fragile and lost crystallinity and porosity after activation with either MeOH or THF (Fig. 3A–C). Similar to TAPB-PDA COF (Fig. 2G–I), after activation, the PXRDs of TAPB-FPDA COFs (Fig. 3B) had broad peaks indicative of only short-range ordering, and the BET surface areas were very low (75 and 55 m2 g−1 for MeOH and THF, respectively). The BET traces reflect generally poor porosity of these samples even when activation with PFH, and more porous samples of TAPB-FPDA could not be obtained despite several attempts. However, the BET surface area after PFH activation (653 m2 g−1) was still much larger than those after activation using MeOH and THF.
Next, we analyzed COFs with hydroxyl-functionalized pores (Fig. 3D). TAPB-OHPDA COFs exhibited excellent crystallinity and high nitrogen sorption capacity after activation with MeOH (Fig. 3E and F), in clear contrast to TAPB-FPDA COFs. Nitrogen isotherms exhibited a sharp increase at low relative pressures indicative of microporosity, and we measured narrow pore size distributions for these samples (Fig. S18†). The BET surface area for the MeOH activated sample reached a value as high as 1190 m2 g−1, which was only slightly lower than that after PFH activation (1452 m2 g−1). However, TAPB-OHPDA COFs lost crystallinity and porosity after activation with THF (Fig. 3D–F). We measured a significantly lower BET surface area (155 m2 g−1) for TAPB-OHPDA COFs after THF activation compared with the samples activated by either MeOH or PFH. Pore size distributions of TAPB-OHPDA COFs after THF activation displayed a much broader and disordered range compared with those samples after PFH and MeOH activation (Fig. S18†).
Finally, COFs with Cl, Br, or OMe substituents were very robust. As shown in Fig. 3G–I and S20–S29,† all three COFs exhibited PXRD patterns with a sharp primary diffraction peak at 2.8° and second order peaks above 4°. No peak attenuation was observed for these samples when activated using high surface tension solvents. Nitrogen sorption isotherms for TAPB-BrPDA COFs or TAPB-ClPDA COFs after PFH, MeOH and THF activations were nearly coincident in the P/P0 range from 0 to 0.4 (Fig. 3I and S21†), while the isotherms for TAPB-OMePDA COFs are almost identical over the entire P/P0 range tested (Fig. S27†). BET surface areas are very high for each sample even after THF activation (Table S3†).
We also calculated BET surface area ratios (SBET(THF)/SBET(PFH) and SBET(MeOH)/SBET(PFH)) ratios for this series of COFs, and they increased with the size of side functionalities (Fig. S30†). These results show a clear trend in fragility of COFs sharing the same backbone but different types of substituents. COFs with Cl, Br, or OMe pore functionalities are more robust, which may be due to the fact that larger side functionalities may prevent shifting between layers of 2D COFs due to stronger interlayer interactions. Other factors, such as the substituent polarity can also play a role, and the series of samples studied here do not provide clear insight into whether size, polarity, or other factors govern activation stability. However, these results are consistent with studies of the thermal stability of COFs by Lotsch and coworkers.33 They found that pore substituents increased the strength of interlayer interactions and improved COF thermal stability. Interestingly, a prior report by Dichtel and coworkers reported the opposite trend in the thermal stability of COFs.32 They found that pore substituents reduced the thermal stability and resulted in loss of crystallinity with heating at lower temperatures compared to non-substituted COFs.
2.3 Effect of pore architecture
Most 2D COFs studied to date have a hexagonal topology, but a variety of 2D COFs with rhombic or tetragonal topologies have also been reported. We were interested in whether the trends in pore size and pore functionality we observed in hexagonal 2D COFs would also apply to COFs with a different pore architecture. We focused on a series of COFs with a rhombic topology synthesized using a pyrene-based amine building block as a node (Py, Fig. 1D) and four different aldehyde linkers varying in length and functionality.Rhombic COFs with a small pore size (Py-1P, 1.5 nm) were robust to all activation solvents (Fig. 4, A–C). While some loss of porosity was observed with increasing activation solvent surface tension, Py-1P retained high BET surface areas after solvent activation with PFH, MeOH and THF (Fig. 4B, C and Table S4†). The samples also exhibited a sharp increase in the nitrogen sorption isotherms in the low relative pressure range, indicating that these samples retained their microporous structure (Fig. 4C). All samples displayed narrow pore size distributions with a sharp peak near 1.5 nm (Fig. S31†). These findings are consistent with the robustness observed in hexagonal COFs with small pore sizes.
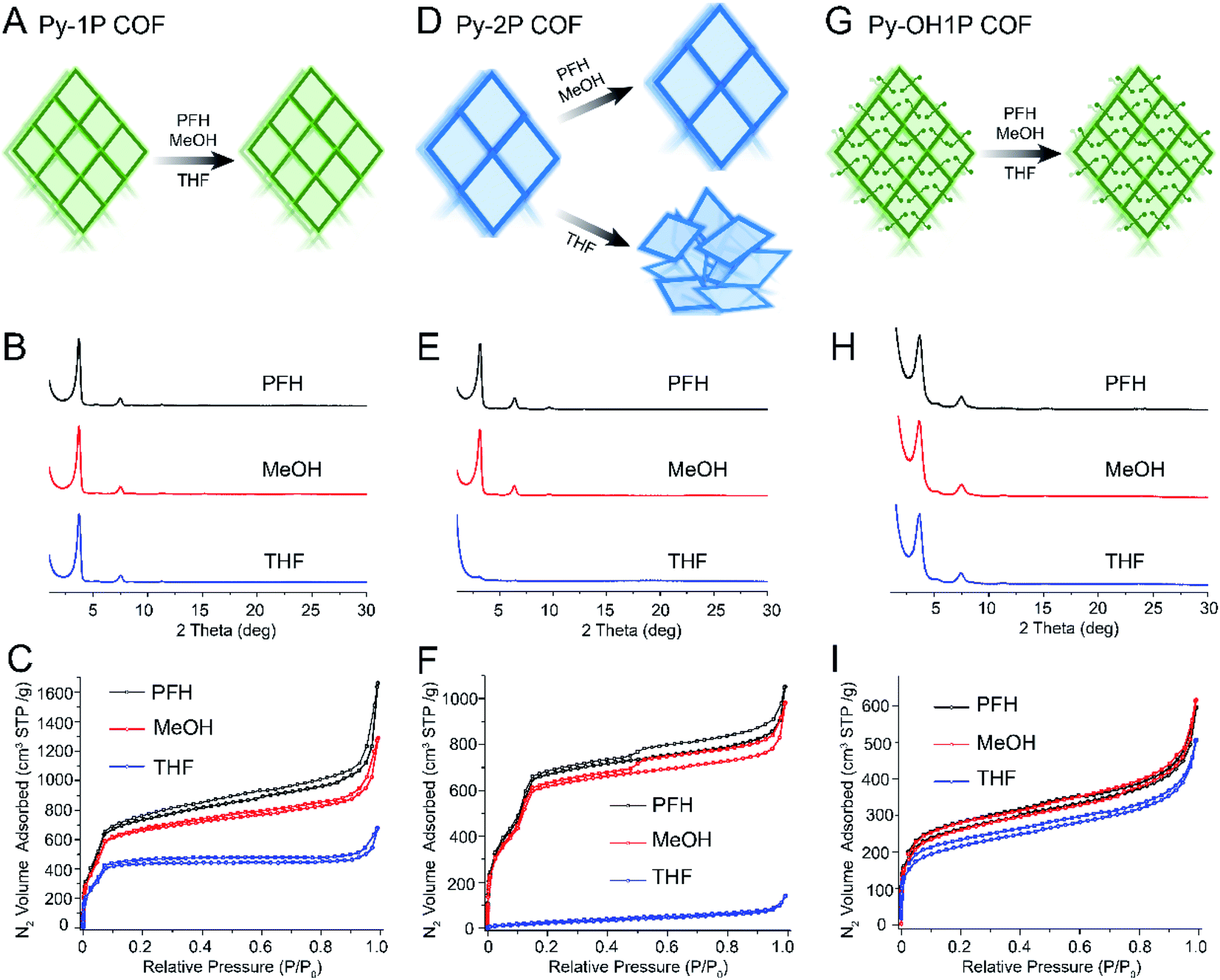 | ||
| Fig. 4 Schematics, PXRD, and nitrogen sorption isotherms demonstrating trends of fragility in rhombic topology COFs varying in pore size and functionality. (A) Schematic showing that rhombic COFs with small pore size retain crystallinity and porosity after activation using PFH, MeOH and THF. (B) PXRD and (C) nitrogen sorption isotherms of Py-1P COF after activation using PFH, MeOH and THF. (D) Schematics showing that rhombic COFs with moderate pore size retain crystallinity after activation using PFH and MeOH, but pore collapse occurs after THF activation. (E) PXRD and (F) nitrogen sorption isotherms of Py-2P COF after activation using PFH, MeOH and THF. (G) Schematics showing that rhombic COFs with small pore size and moderately-sized pore functionalities retain crystallinity after PFH and MeOH activation but not after THF activation. (H) PXRD and (I) nitrogen sorption isotherms of Py-OH1P COF after activation using PFH, MeOH and THF. Note: the structures of all monomers presented here are shown in Fig. 1. Py-1P COF, Py-2P COF, and Py-OH1P COF are composed of Py and PDA, Py and DFBP, and Py and OHPDA, respectively. | ||
Next, we studied rhombic COFs with larger pore sizes but without substituents (Fig. 4D–F). Py-2P COFs have a larger pore size of 2.6 nm and exhibited excellent crystallinities and high BET surface areas after activation with PFH or MeOH (3312 and 3121 m2 g−1, Fig. 4E and F). However, crystallinity and porosity were completely disrupted after activation with THF. The samples exhibited non-porous features with very low BET surface areas (91 m2 g−1, Fig. 4F), and PXRD patterns revealed only a very weak and broad primary diffraction peak (Fig. 4E). These results demonstrate that Py-2P is more fragile compared with Py-1P, consistent with the expected trend of fragility with pore size.
To understand the effect of pore substituents, we synthesized a rhombic COF with a framework identical to Py-1P but containing a hydroxyl functionality, termed Py-OH1P (Fig. 4G–I). The resulting COFs exhibited improved structural robustness compared with that of Py-1P. A loss of surface area was measured only after activation by THF, and the BET surface areas were 901, 938, and 763 m2 g−1 for Py-OH1P after activation with PFH, MeOH, and THF, respectively (Fig. 4G–I). We also prepared a rhombic COF with a large pore size (3.0 nm) and large pore functionalities (–OMe) (Fig. 1D and S37–S42†). It maintained crystallinity and porosity when activated using PFH or MeOH, but collapsed when activated by THF (Fig. S39 and S40†). Nitrogen sorption isotherms for the THF activated sample reflected a complete loss of porosity, and the pore size distributions also become disordered and broader (Fig. S41†).
These results demonstrate that similar trends in pore size and functionality apply to rhombic COFs as for hexagonal COFs. As was observed for the series of hexagonal COFs, increasing the pore size decreases robustness while introducing pore substituents improves robustness. However, our data also demonstrated that pore architecture does impact fragility. For example, while hexagonal COFs with a pore size of 3.3 nm and OMe substituent (TAPB-OMePDA) were very robust, rhombic COFs with a smaller pore size (3.0 nm) and OMe substituent were not as robust. Therefore, pore architecture has an impact on robustness, but both topologies studied display the same trends in pore size and pore substituents.
2.4 DFT calculations
We conducted DFT calculations to understand how pore size and functionalities influence COF activation stability. COFs have been previously reported to switch between crystalline and non-crystalline morphologies due to reversibility of weak linkages or interlayer interactions,31 and therefore we used DFT calculations to quantify interlayer binding energies for COFs of different pore size and functionality. We calculated the interlayer binding energy (Eb) of COFs, where Eb was defined as the difference between the energy of a bilayer and that of two monolayers, Eb = Ebilayer − 2Emonolayer. A larger value of Eb reflects stronger interlayer interactions, which serve to stabilize interlayer crystallinity. The DFT calculations were conducted based on the crystal model for each COF (Fig. S55†). As shown in Fig. 5A, Eb decreased with the increase of pore size, presumably due to reduced interlayer interactions per unit area. This is consistent with our experimental results which revealed increased fragility with pore size. Next, we calculated Eb for COFs with pore substituents by relying on molecular models for –H, –OH and –OMe substituents (i.e. TAPB-PDA, TAPB-OHPDA COF and TAPB-OMePDA COF), as shown in Fig. S56.† These calculations revealed that Eb increased with the increased size of side functionalities, possibly due to the dispersion interactions between the side chains (Fig. 5B). These results are consistent with our experimental observations that showed that pore functionalities can dramatically enhance COF activation stability and suggest that interlayer interactions play an important role in stabilizing 2D COFs.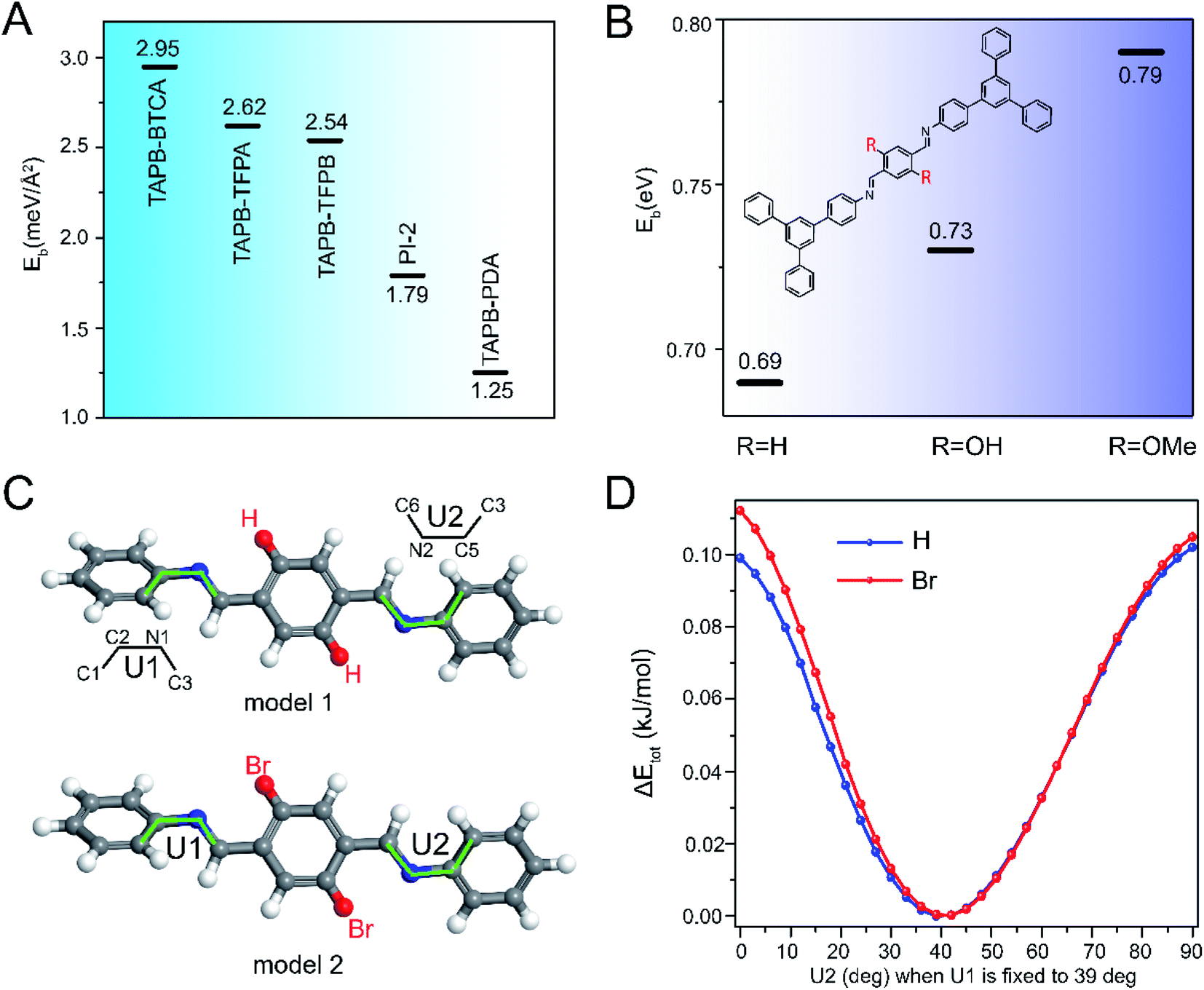 | ||
| Fig. 5 DFT Analysis of interlayer interactions and twisting and bending deformations in 2D COFs. Calculated interlayer binding energy Eb for COFs with (A) different pore sizes and (B) pore substituents. (C) TAPB-PDA-TAPB unit (model 1) and TAPB-BrPDA-TAPB unit (model 2) for the distortion calculations. (D) Distortion energy as a function of dihedral angles U2 when U1 is fixed to 39°. | ||
We also performed DFT calculations to understand the potential effect of distortions due to stretching and twisting of network bonds within a single COF layer. Using simplified molecular models, we calculated the energy barrier for twisting within a layer, as reflected in the values of dihedral angles U1 and U2 (see molecular models in Fig. 5C). We studied conformational changes for COFs with H and Br side functionalities (i.e. TAPB-PDA and TAPB-BrPDA COF). The moieties linked at both sides of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– bonds have two types of dihedral angles U and Z in the molecular models (Fig. 5C and Fig. S57A†), but we found that the distortion energy along Z dihedral angles was significantly higher than that along the U direction, indicating that distortions in the Z-direction were suppressed (Fig. S57,† see details in ESI†). Therefore, when calculating the molecular models (model 1 and model 2 in Fig. 5C) with two –CN– bonds, we focused on distortions along U1 and U2. The DFT calculations revealed that COFs with either H or Br pore functionality both had a lowest energy conformation at the dihedral angle U1 = U2 = 40° (Fig. S58†). However, the presence of Br substituents increased steric repulsion in the twisted conformation, and therefore Br substituents increased the energy for rotation about U1 and U2 (Fig. 5D and S58†). This suggests that substituents may also stabilize the intra-layer structure and reduce the amplitude of deformations associated with twisting and rotation of 2D COF sheets.
N– bonds have two types of dihedral angles U and Z in the molecular models (Fig. 5C and Fig. S57A†), but we found that the distortion energy along Z dihedral angles was significantly higher than that along the U direction, indicating that distortions in the Z-direction were suppressed (Fig. S57,† see details in ESI†). Therefore, when calculating the molecular models (model 1 and model 2 in Fig. 5C) with two –CN– bonds, we focused on distortions along U1 and U2. The DFT calculations revealed that COFs with either H or Br pore functionality both had a lowest energy conformation at the dihedral angle U1 = U2 = 40° (Fig. S58†). However, the presence of Br substituents increased steric repulsion in the twisted conformation, and therefore Br substituents increased the energy for rotation about U1 and U2 (Fig. 5D and S58†). This suggests that substituents may also stabilize the intra-layer structure and reduce the amplitude of deformations associated with twisting and rotation of 2D COF sheets.
2.5 Engineering of robustness in 2D COFs
Our studies of hexagonal and rhombic 2D COFs clearly demonstrated that pore substituents improved the structural stability of COFs. The trend in fragility as a function of pore functionality suggested that robustness could be systematically engineered. We sought to demonstrate this by synthesizing a series of COFs using a single node (TAPB) and a combination of two linear linkers, one of which (2,5-bis(octyloxy)terephthalaldehyde, C8PDA) contained octyloxy functionality while the other (PDA) remained unfunctionalized (Fig. 6A). This approach enabled us to systematically control the quantity of octyloxy substituent in the final COF by varying the feed ratio of C8PDA and PDA. For simplicity, these COFs will be referred to as TAPB-PDAx-C8PDAy, where x and y represent the feed ratio of PDA and C8PDA, respectively. The presence and quantities of octyloxy chains in the final COFs were monitored by FTIR analysis (see highlights in Fig. S59†). Consistent with increasing presence of C8PDA repeat unit in the final COF, the intensity of vibration peaks near 2900 cm−1 and 2800 cm−1 increased relative to the peak intensity at 550 cm−1 with increasing amount of C8PDA.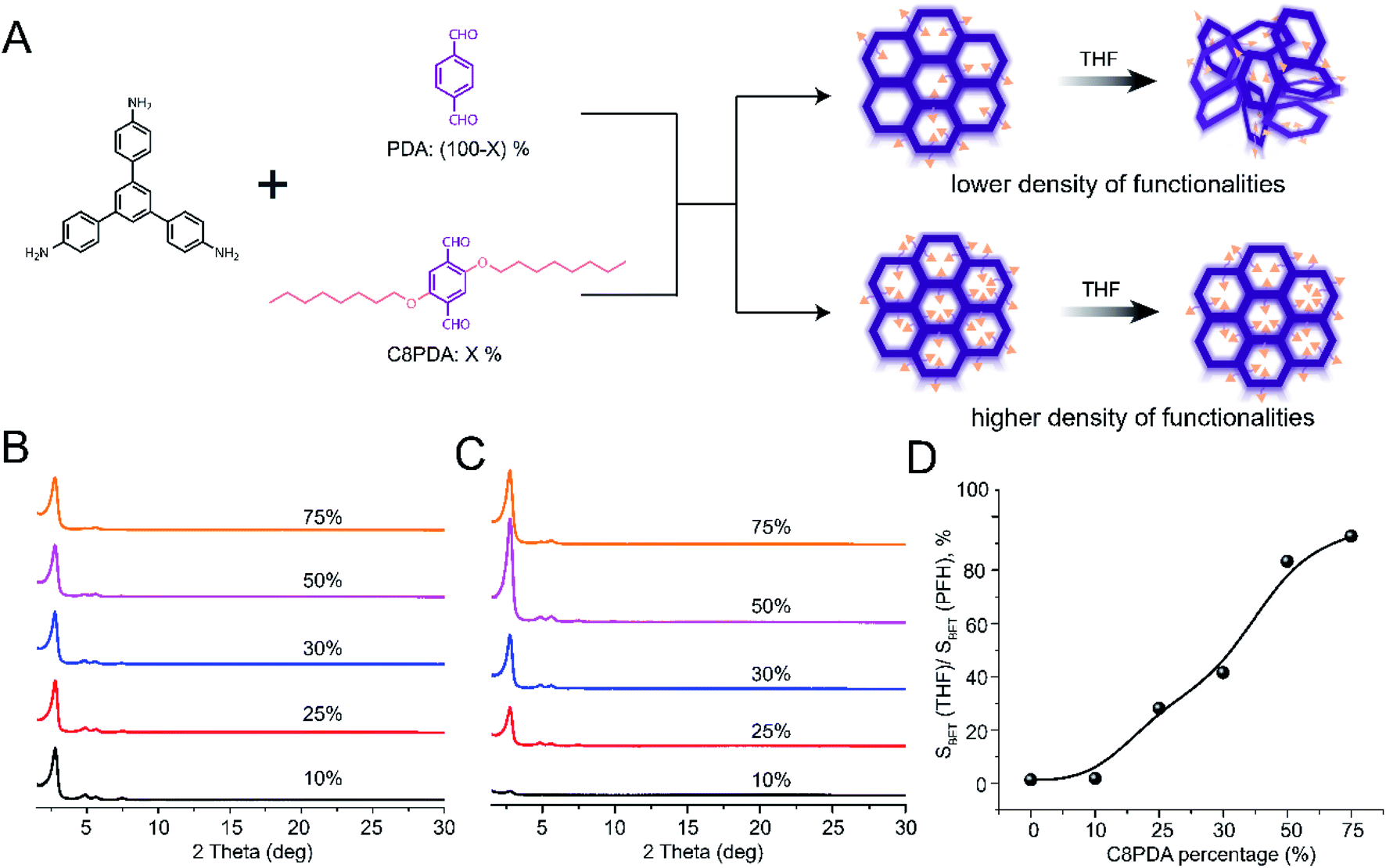 | ||
| Fig. 6 Engineering robustness of 2D COFs. (A) Reaction scheme and schematic for tailoring COF robustness through systematic variations in pore functionality. The schematic shows that increasing the number of pore functionalities improves robustness. (Band C) PXRD spectra of multicomponent COFs synthesized with different feed ratio of C8PDA monomer after (B) PFH activation and (C) THF activation. (D) BET surface area of COFs activated by THF relative to the surface area measured after PFH activation. COFs with a higher octyloxy pore functionality content are more robust, as measured by the percentage of BET surface area retained after THF activation relative to that after PFH activation. | ||
We then analyzed the crystallinities and porosities of TAPB-PDAx-C8PDAy. All samples exhibited excellent crystallinities (Fig. 6B) and porosities (Fig. S60†) after PFH activation. However, a lower BET surface area was measured for samples with high loadings of C8PDA, and the sample with 75% C8PDA had the lowest BET surface area of the entire series (see Fig. S6†). This is consistent with the observation reported in prior studies that large pore substituents reduce the accessible pore volume, resulting in a decrease in the BET surface area.48,49 This is also consistent with other studies of multivariant COFs,50 in which the pore size changed continuously and became smaller with higher loadings of C8PDA feed ratios (Fig. S61†), due to occupation of the pore channels by long octyloxy side chains. The decreased BET surface areas after PFH activation (Fig. S60†) and the decreased pore size (Fig. S61†) therefore reflect the presence of both PDA and C8PDA in the multivariate COFs.
To understand the impact of pore functionalities on activation stability, we analyzed each TAPB-PDAx-C8PDAy COF after THF activation through PXRD and nitrogen sorption measurements. These measurements revealed a clear trend in the activation stability of TAPB-PDAx-C8PDAy with the feed ratio of C8PDA (Fig. 6C, S62, S63 and Table S6†). TAPB-PDA90-C8PDA10 were amorphous after THF activations, while samples with higher C8PDA (x > 10) contents exhibited a primary diffraction peak, the intensity of which increased significantly as the C8PDA content went up. We further found that a 50% feed ratio of C8PDA resulted in a multivariate COF with highest crystallinity and BET surface area after THF activation, while the 75% feed ratio led to a multivariate COF with slightly lower BET surface area after THF activation. This shows that robustness increases while maintaining a high accessible pore volume and surface area up to a threshold value of octyloxy side chains. However, a larger number of octyloxy side chains results in decreased accessible pore volume while also improving the stability of the COF.
To quantitatively analyze trends in robustness, we turned to nitrogen sorption measurements. For each TAPB-PDAx-C8PDAy COF, we measured the surface area after THF activation (SBET(THF), Fig. S62 and Table S6†) and compared it with the surface area measured after PFH activation (SBET(PFH), Fig. S60 and Table S6†). By calculating the percentage of surface area retained after THF activation relative to that measured after PFH activation, we can get a quantitative measure of robustness for each COF. As shown in Fig. 6D, we observed a steady increase in this ratio of surface areas as a function of C8PDA content from 10 to 75%. The percentage of surface area retained increased from 0% at 10% C8PDA to approximately 90% for 75% C8PDA. This provides strong evidence that the pore substituents improve COF activation stability, and that the fragility of COFs can be suppressed by quantitative tuning of pore substituents. We performed a similar set of experiments using methoxy (OMe) functionality (ESI, Fig. S64–S72†). Similar results were disclosed except that a larger quantity of OMe substituents (at least 50%) was needed to produce a measurable impact on COF robustness.
We note that the reactivities of the PDA and C8PDA may be different, and as a result the C8 pore substituents may not be homogeneously distributed. Other factors such as steric interaction may prevent excessive crowding and clustering of the C8 chains, and dynamic rearrangements during synthesis likely play a role in the distribution of C8 chains. Nevertheless, the clear trend in activation stability as a function of C8 content indicates that this is an effective approach to tune COF activation stability.
3 Conclusion
In summary, we systematically investigated the relationship between activation stability or structural robustness, COF molecular chemistry, and architecture including pore size, pore functionalities, and pore architecture. We quantified the activation stability of a large series of COFs through solvent activation with solvents varying in surface tension. This process enabled us to quantitatively analyze the fragility of COFs through analysis of the crystallinity and surface area after activation. We found that increasing pore size produced increasingly fragile COFs, while introducing pore functionalities produce increasingly robust COFs. These trends applied to both hexagonal and rhombic topologies, and DFT calculations demonstrated that fragility was related to both inter- and intra-layer interactions. Finally, we demonstrated that we could systematically tune COF robustness using a multivariate approach involving building blocks with and without side-functionalities. This work is relevant to a wide range of practical applications of COFs and provides experimental and theoretical insight into framework integrity in 2D COFs, demonstrates a simple molecular-engineering approach to reinforce COF materials, and gives specific guidelines for the design of robust COFs that maintain crystallinity and porosity during activation.4 Experimental section
4.1 COF synthesis
The typical synthesis procedure for TAPB-PDA COF is described here. In detail, 0.16 mmol 1,3,5-tris(4-aminophenyl)benzene (TAPB) and 0.24 mmol terephthaldehyde (PDA) were weighed and placed in a Pyrex tube, then a solvent mixture of 3.2 mL dioxane and 0.8 mL mesitylene was added. To uniformly disperse or dissolve the monomers, the Pyrex tube was sonicated for 5 minutes and followed by addition of 0.4 mL 6 M acetic acid as the catalyst. Finally, the Pyrex tube was flame sealed and placed in an oven to react at 120 °C for 3 days. To separate the dry COF powders, the produced precipitates were thoroughly washed using THF and acetone through centrifugation. Then, the wet powder samples were averagely separated into three aliquots. In the last washing step, the three aliquots were washed using perfluorohexane, methanol and THF respectively. After filtration, all samples were placed in a vacuum oven at 80 °C to dry overnight, and the as-prepared samples were measured for PXRD, BET and FTIR analysis. All the other COFs follow the same procedure except for the reaction solvents used, which are listed in Table S1.†4.2 Instrumentations and characterizations
PXRD data were detected on a Rigaku SmartLab XRD with 2θ ranging from 1° to 30° with 0.02° increment in a continuous mode. Powder samples without prior grinding were directly placed on zero background sample holders and leveled flat using a glass microscope slide. Fourier-transform infrared spectroscopy (FTIR) of all solid samples was tested using a ThermoNicolet iS10 FT-IR spectrometer with a diamond ATR attachment. The spectra were tested using 64 scans with a resolution of 4. The testing range was set from 4000 to 500 cm−1. The spectra shown are uncorrected. Nitrogen sorption measurements were conducted on Quantachrome Autosorb-iQ-MP/Kr BET Surface Analyzer. All samples were washed using different solvents (PFH, MeOH and THF) and dried under vacuum at 80 °C overnight in a laboratory heating oven. All samples were tested directly without grinding. BET surface areas for all samples were calculated using BET adsorption models (P/P0 ranging from 0.05–0.2) included in the instrument software (ASiQwin version 5.2). Pore size distributions were calculated using the quenched solid density functional theory (QSDFT) model included in the instrument software, which is mostly used for COF pore size calculations and matches the COF models best.4.3 DFT calculation
The crystal model was performed using the plane wave basis Vienna ab initio simulation package (VASP) code.1,2 The generalized gradient approximation in the Perdew–Burke–Ernzerhof (GGA-PBE) formulation is used with a cutoff energy of 600 eV. The vacuum space of ∼20 Å is intercalated into interlamination to eliminate the interaction between layers. A 6 × 6 2D grid uniform k-points is applied for Density-functional theatrical (DFT) calculations. The van der Waals (vdW) corrections were employed (DFT-D3) to calculate the interlayer distance in bulk phases.The molecular model was carried out with the Gaussian 16 code based on DFT.3 The Becke exchange functional (B) and the Lee–Yang–Parr (LYP) correlation functional within a generalized gradient approximation (GGA) was used to describe the interaction between the ionic cores and electrons.4,5 The hybrid basic set b3lyp/6-311 + g(d,p) was employed to optimize all structures and calculate total energy.
Data availability
All data are available in the main text or the ESI.†Author contributions
D. Z. and R. V designed research; D. Z., J. J. Z, Q. Y., F. L., Y. Z and X. G. performed research; D. Z. J. J. Z, X. W, B.·I.·Y. and R. V. analyzed data; and D. Z., J. J. Z., X. W. B. I. Y., and R. V. wrote the original draft of the paper. D. Z., J. J. Z., X. W. B. I. Y., M. M. R, P. A, and R. V. revised and edited the paper. #D. Z., J. J. Z, and X. W. contributed equally to this work.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge financial support from the Army Research Laboratory (W911NF-18-2-0062) and the Welch Foundation for Chemical Research (C-2124). Computational modeling (J. J. Z. and B. I. Y.) was supported by the U.S. Department of Energy, BES (DE-SC0012547). The authors also acknowledge Shared Equipment Authority at Rice University for access and utilization of characterization instruments.References
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Porous, Crystalline, Covalent Organic Frameworks, Science, 2005, 310(5751), 1166–1170, DOI:10.1126/science.1120411.
- S.-Y. Ding and W. Wang, Covalent Organic Frameworks (COFs): From Design to Applications, Chem. Soc. Rev., 2012, 42(2), 548–568, 10.1039/C2CS35072F.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Chemically Stable Polyarylether-Based Covalent Organic Frameworks, Nat. Chem., 2019, 11(6), 587–594, DOI:10.1038/s41557-019-0238-5.
- R.-R. Liang, S.-Y. Jiang, R.-H. A. and X. Zhao, Two-Dimensional Covalent Organic Frameworks with Hierarchical Porosity, Chem. Soc. Rev., 2020, 49(12), 3920–3951, 10.1039/D0CS00049C.
- J. Zhao, J. Ren, G. Zhang, Z. Zhao, S. Liu, W. Zhang and L. Chen, Donor-Acceptor Type Covalent Organic Frameworks, Chem.– Eur. J., 2021, 27(42), 10781–10797, DOI:10.1002/chem.202101135.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, New Synthetic Strategies toward Covalent Organic Frameworks, Chem. Soc. Rev., 2020, 49(10), 2852–2868, 10.1039/D0CS00199F.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent Organic Frameworks: Design, Synthesis, and Functions, Chem. Rev., 2020, 120(16), 8814–8933, DOI:10.1021/acs.chemrev.9b00550.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, Covalent Organic Frameworks as Exceptional Hydrogen Storage Materials, J. Am. Chem. Soc., 2008, 130(35), 11580–11581, DOI:10.1021/ja803247y.
- H. Furukawa and O. M. Yaghi, Storage of Hydrogen, Methane, and Carbon Dioxide in Highly Porous Covalent Organic Frameworks for Clean Energy Applications, J. Am. Chem. Soc., 2009, 131(25), 8875–8883, DOI:10.1021/ja9015765.
- L. Zhu and Y.-B. Zhang, Crystallization of Covalent Organic Frameworks for Gas Storage Applications, Molecules, 2017, 22(7), 1149, DOI:10.3390/molecules22071149.
- H. Xu, J. Gao and D. Jiang, Stable, Crystalline, Porous, Covalent Organic Frameworks as a Platform for Chiral Organocatalysts, Nat. Chem., 2015, 7(11), 905–912, DOI:10.1038/nchem.2352.
- R. Chen, Y. Wang, Y. Ma, A. Mal, X.-Y. Gao, L. Gao, L. Qiao, X.-B. Li, L.-Z. Wu and C. Wang, Rational Design of Isostructural 2D Porphyrin-Based Covalent Organic Frameworks for Tunable Photocatalytic Hydrogen Evolution, Nat. Commun., 2021, 12(1), 1354, DOI:10.1038/s41467-021-21527-3.
- Y. Liu, W. Zhou, W. L. Teo, K. Wang, L. Zhang, Y. Zeng and Y. Zhao, Covalent-Organic-Framework-Based Composite Materials, Chem, 2020, 6(12), 3172–3202, DOI:10.1016/j.chempr.2020.08.021.
- S. Chen, C. Zhu, W. Xian, X. Liu, X. Liu, Q. Zhang, S. Ma and Q. Sun, Imparting Ion Selectivity to Covalent Organic Framework Membranes Using de Novo Assembly for Blue Energy Harvesting, J. Am. Chem. Soc., 2021, 143(25), 9415–9422, DOI:10.1021/jacs.1c02090.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Covalent Organic Frameworks Linked by Amine Bonding for Concerted Electrochemical Reduction of CO2, Chem, 2018, 4(7), 1696–1709, DOI:10.1016/j.chempr.2018.05.003.
- M. Liu, L. Guo, S. Jin and B. Tan, Covalent Triazine Frameworks: Synthesis and Applications, J. Mater. Chem. A, 2019, 7(10), 5153–5172, 10.1039/C8TA12442F.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Covalent Organic Frameworks for Separation Applications, Chem. Soc. Rev., 2020, 49(3), 708–735, 10.1039/C9CS00827F.
- P. Zhang, Z. Wang, P. Cheng, Y. Chen and Z. Zhang, Design and Application of Ionic Covalent Organic Frameworks, Coord. Chem. Rev., 2021, 438, 213873, DOI:10.1016/j.ccr.2021.213873.
- D. Zhu, G. Xu, M. Barnes, Y. Li, C.-P. Tseng, Z. Zhang, J.-J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Covalent Organic Frameworks for Batteries, Adv. Funct. Mater., 2021, 31(32), 2100505, DOI:10.1002/adfm.202100505.
- X. Li, Q. Hou, W. Huang, H.-S. Xu, X. Wang, W. Yu, R. Li, K. Zhang, L. Wang, Z. Chen, K. Xie and K. P. Loh, Solution-Processable Covalent Organic Framework Electrolytes for All-Solid-State Li–Organic Batteries, ACS Energy Lett., 2020, 5(11), 3498–3506, DOI:10.1021/acsenergylett.0c01889.
- X. Li, H. Wang, Z. Chen, H.-S. Xu, W. Yu, C. Liu, X. Wang, K. Zhang, K. Xie and K. P. Loh, Covalent-Organic-Framework-Based Li–CO2 Batteries, Adv. Mater., 2019, 31(48), 1905879, DOI:10.1002/adma.201905879.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, High-Lithium-Affinity Chemically Exfoliated 2D Covalent Organic Frameworks, Adv. Mater., 2019, 31(29), 1901640, DOI:10.1002/adma.201901640.
- Y. Zhang, R.-L. Zhong, M. Lu, J.-H. Wang, C. Jiang, G.-K. Gao, L.-Z. Dong, Y. Chen, S.-L. Li and Y.-Q. Lan, Single Metal Site and Versatile Transfer Channel Merged into Covalent Organic Frameworks Facilitate High-Performance Li-CO2 Batteries, ACS Cent. Sci., 2021, 7(1), 175–182, DOI:10.1021/acscentsci.0c01390.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Covalent–Organic Frameworks: Advanced Organic Electrode Materials for Rechargeable Batteries, Adv. Energy Mater., 2020, 10(19), 1904199, DOI:10.1002/aenm.201904199.
- D. Zhu and R. Verduzco, Ultralow Surface Tension Solvents Enable Facile COF Activation with Reduced Pore Collapse, ACS Appl. Mater. Interfaces, 2020, 12(29), 33121–33127, DOI:10.1021/acsami.0c09173.
- T. Sick, J. M. Rotter, S. Reuter, S. Kandambeth, N. N. Bach, M. Döblinger, J. Merz, T. Clark, T. B. Marder, T. Bein and D. D. Medina, Switching on and off Interlayer Correlations and Porosity in 2D Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(32), 12570–12581, DOI:10.1021/jacs.9b02800.
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Rapid Synthesis of High Surface Area Imine-Linked 2D Covalent Organic Frameworks by Avoiding Pore Collapse During Isolation, Adv. Mater., 2020, 32(2), 1905776, DOI:10.1002/adma.201905776.
- A. P. Nelson, O. K. Farha, K. L. Mulfort and J. T. Hupp, Supercritical Processing as a Route to High Internal Surface Areas and Permanent Microporosity in Metal–Organic Framework Materials, J. Am. Chem. Soc., 2009, 131(2), 458–460, DOI:10.1021/ja808853q.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Are Zr6-Based MOFs Water Stable? Linker Hydrolysis vs. Capillary-Force-Driven Channel Collapse, Chem. Commun., 2014, 50(64), 8944–8946, 10.1039/C4CC02401J.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Best Practices for the Synthesis, Activation, and Characterization of Metal–Organic Frameworks, Chem. Mater., 2017, 29(1), 26–39, DOI:10.1021/acs.chemmater.6b02626.
- C. Kang, Z. Zhang, V. Wee, A. K. Usadi, D. C. Calabro, L. S. Baugh, S. Wang, Y. Wang and D. Zhao, Interlayer Shifting in Two-Dimensional Covalent Organic Frameworks, J. Am. Chem. Soc., 2020, 142(30), 12995–13002, DOI:10.1021/jacs.0c03691.
- A. M. Evans, M. R. Ryder, W. Ji, M. J. Strauss, A. R. Corcos, E. Vitaku, N. C. Flanders, R. P. Bisbey and W. R. Dichtel, Trends in the Thermal Stability of Two-Dimensional Covalent Organic Frameworks, Faraday Discuss., 2021, 225, 226–240, 10.1039/D0FD00054J.
- S. T. Emmerling, R. Schuldt, S. Bette, L. Yao, R. E. Dinnebier, J. Kästner and B. V. Lotsch, Interlayer Interactions as Design Tool for Large-Pore COFs, J. Am. Chem. Soc., 2021, 143(38), 15711–15722, DOI:10.1021/jacs.1c06518.
- S. B. Alahakoon, K. Tan, H. Pandey, S. D. Diwakara, G. T. McCandless, D. I. Grinffiel, A. Durand-Silva, T. Thonhauser and R. A. Smaldone, 2D-Covalent Organic Frameworks with Interlayer Hydrogen Bonding Oriented through Designed Nonplanarity, J. Am. Chem. Soc., 2020, 142(30), 12987–12994, DOI:10.1021/jacs.0c03409.
- X. Chen, M. Addicoat, S. Irle, A. Nagai and D. Jiang, Control of Crystallinity and Porosity of Covalent Organic Frameworks by Managing Interlayer Interactions Based on Self-Complementary π-Electronic Force, J. Am. Chem. Soc., 2013, 135(2), 546–549, DOI:10.1021/ja3100319.
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, Locking Covalent Organic Frameworks with Hydrogen Bonds: General and Remarkable Effects on Crystalline Structure, Physical Properties, and Photochemical Activity, J. Am. Chem. Soc., 2015, 137(9), 3241–3247, DOI:10.1021/ja509602c.
- G. He, R. Zhang and Z. Jiang, Engineering Covalent Organic Framework Membranes, Acc. Mater. Res., 2021, 2(8), 630–643, DOI:10.1021/accountsmr.1c00083.
- A. R. Corcos, G. A. Levato, Z. Jiang, A. M. Evans, A. G. Livingston, B. J. Mariñas and W. R. Dichtel, Reducing the Pore Size of Covalent Organic Frameworks in Thin-Film Composite Membranes Enhances Solute Rejection, ACS Mater. Lett., 2019, 1(4), 440–446, DOI:10.1021/acsmaterialslett.9b00272.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Bulk COFs and COF Nanosheets for Electrochemical Energy Storage and Conversion, Chem. Soc. Rev., 2020, 49(11), 3565–3604, 10.1039/D0CS00017E.
- M. Zhang, R. Zheng, Y. Ma, R. Chen, X. Sun and X. Sun, N-Rich Covalent Organic Frameworks with Different Pore Size for High-Pressure CO2 Adsorption, Microporous Mesoporous Mater., 2019, 285, 70–79, DOI:10.1016/j.micromeso.2019.04.021.
- Z. Li, T. He, Y. Gong and D. Jiang, Covalent Organic Frameworks: Pore Design and Interface Engineering, Acc. Chem. Res., 2020, 53(8), 1672–1685, DOI:10.1021/acs.accounts.0c00386.
- X. Li, Q. Gao, J. Aneesh, H.-S. Xu, Z. Chen, W. Tang, C. Liu, X. Shi, K. V. Adarsh, Y. Lu and K. P. Loh, Molecular Engineering of Bandgaps in Covalent Organic Frameworks, Chem. Mater., 2018, 30(16), 5743–5749, DOI:10.1021/acs.chemmater.8b02560.
- B. P. Biswal, S. Kandambeth, S. Chandra, D. B. Shinde, S. Bera, S. Karak, B. Garai, U. K. Kharul and R. Banerjee, Pore Surface Engineering in Porous, Chemically Stable Covalent Organic Frameworks for Water Adsorption, J. Mater. Chem. A, 2015, 3(47), 23664–23669, 10.1039/C5TA07998E.
- N. Huang, R. Krishna and D. Jiang, Tailor-Made Pore Surface Engineering in Covalent Organic Frameworks: Systematic Functionalization for Performance Screening, J. Am. Chem. Soc., 2015, 137(22), 7079–7082, DOI:10.1021/jacs.5b04300.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Pore Surface Engineering in Covalent Organic Frameworks, Nat. Commun., 2011, 2(1), 536, DOI:10.1038/ncomms1542.
- H. Vardhan, A. Nafady, A. M. Al-Enizi and S. Ma, Pore Surface Engineering of Covalent Organic Frameworks: Structural Diversity and Applications, Nanoscale, 2019, 11(45), 21679–21708, 10.1039/C9NR07525A.
- Y. Yang, M. Faheem, L. Wang, Q. Meng, H. Sha, N. Yang, Y. Yuan and G. Zhu, Surface Pore Engineering of Covalent Organic Frameworks for Ammonia Capture through Synergistic Multivariate and Open Metal Site Approaches, ACS Cent. Sci., 2018, 4(6), 748–754, DOI:10.1021/acscentsci.8b00232.
- X. Wu, Y. Hong, B. Xu, Y. Nishiyama, W. Jiang, J. Zhu, G. Zhang, S. Kitagawa and S. Horike, Perfluoroalkyl-Functionalized Covalent Organic Frameworks with Superhydrophobicity for Anhydrous Proton Conduction, J. Am. Chem. Soc., 2020, 142(33), 14357–14364, DOI:10.1021/jacs.0c06474.
- G. Zhang, Y. Hong, Y. Nishiyama, S. Bai, S. Kitagawa and S. Horike, Accumulation of Glassy Poly(Ethylene Oxide) Anchored in a Covalent Organic Framework as a Solid-State Li+ Electrolyte, J. Am. Chem. Soc., 2019, 141(3), 1227–1234, DOI:10.1021/jacs.8b07670.
- R. L. Li, A. Yang, N. C. Flanders, M. T. Yeung, D. T. Sheppard and W. R. Dichtel, Two-Dimensional Covalent Organic Framework Solid Solutions, J. Am. Chem. Soc., 2021, 143(18), 7081–7087, DOI:10.1021/jacs.1c01683.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2191120. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03489a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |