
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed intramolecular enantioselective C(sp3)–H insertion of donor/donor carbenes†
Wendeng
Li‡
a,
He
Zhang‡
a,
Kai
Chen
 b,
Huanfeng
Jiang
a,
Jianwei
Sun
c and
Shifa
Zhu
*a
b,
Huanfeng
Jiang
a,
Jianwei
Sun
c and
Shifa
Zhu
*a
aKey Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou, 510640, P. R. China. E-mail: zhusf@scut.edu.cn
bCollege of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, P. R. China
cDepartment of Chemistry, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, P. R. China
First published on 30th September 2022
Abstract
Herein, the first palladium-catalyzed intramolecular enantioselective C(sp3)–H insertion reaction of donor–donor carbenes has been successfully achieved. This facile protocol enables the rapid construction of a collection of enantioenriched decorated indolines with two contiguous stereocenters in a single step. Both enynones and diazo compounds are efficient donor–donor carbene precursors for this reaction. By an adjustment of ligands and protecting groups of the substrates, the palladium–carbene intermediates from diazo compounds afford sparse trans-indolines with excellent enantioselectivities, while carbenes from enynones deliver cis-indolines exclusively. Based on the control reactions and Hammett analysis, a stepwise Mannich-type pathway through a short-lived and compact zwitterionic intermediate is proposed.
The asymmetric C–H insertion of metal carbene is one of the most powerful methods for the construction of chiral molecules through carbon–carbon bond formation.1,2 In the past several decades, many transition metal complexes, especially dirhodium(II) complexes,2,3 have emerged as effective catalysts for the enantioselective carbene C–H insertion reactions.
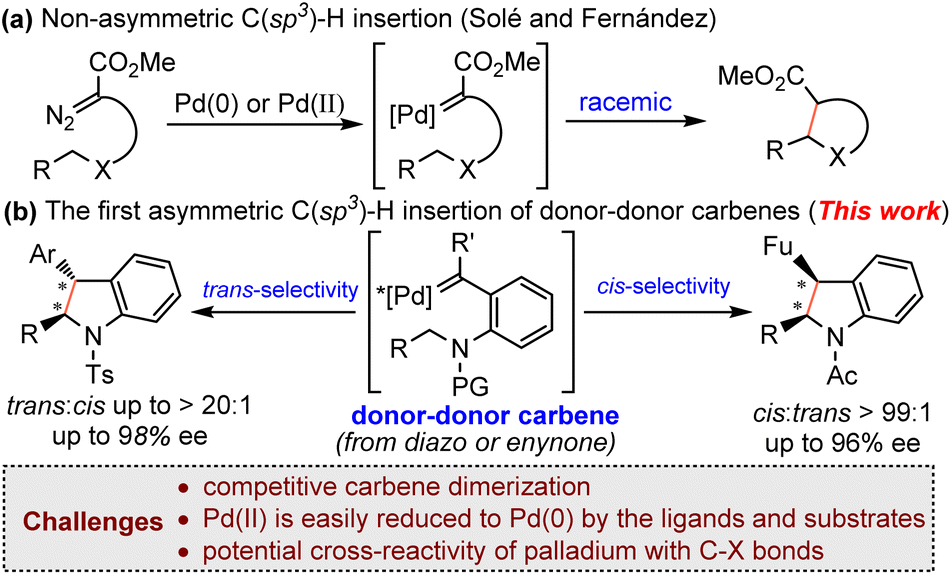
Palladium is versatile and indispensable in the formation of C–C and C–heteroatom bonds in cross-coupling chemistry.4 Palladium-catalyzed carbene transfer reactions, especially cross-couplings via the migratory insertion process,5,6 have been extensively investigated in the past two decades. However, palladium catalyzed carbene-involving asymmetric transformations are still in their infant stage.7 Taking C–H bond insertion as an example, there are only very limited examples of asymmetric carbenoid C–H insertion reactions in the presence of a palladium catalyst. In 2015 and 2018, Zhou and coworkers reported palladium-catalyzed asymmetric formal carbene insertion into C(sp2)–H of electron-rich indole and pyrrole derivatives through a Friedel-Crafts-like process with axially chiral bipyridine ligands using aryl diazoesters as the carbene precursor.7e,h In 2016, Solé and Fernández reported the first palladium-catalyzed non-asymmetric carbene insertion into an intramolecular C(sp3)–H bond using diazocarbonyl compounds.8 It was found that the reaction could be catalyzed by both Pd(0) and Pd(II) (Scheme 1a). DFT calculations revealed that palladium carbene-involving C(sp3)–H insertion reactions had quite different reaction mechanisms from the related Rh(II)- and Cu-catalyzed reactions.9 Most notably, the mechanisms of Pd-catalytic systems were affected by the metal valence and substrate structures. For example, Pd(0) and Pd(II) have totally different mechanisms.8,10 To the best of our knowledge, the palladium-catalyzed enantioselective carbene insertion of C(sp3)–H bonds still remains unknown. This is probably due to the unique mechanism of palladium carbene chemistry.
 | ||
| Scheme 1 Pd-catalyzed carbene insertion of a C–H bond. | ||
Donor-type metal carbenes (donor carbene and donor–donor carbene),11 owing to the presence of a donor-substituent for stabilization of the carbene carbon center, are typically less reactive (compared with acceptor-type metal carbenes) and have attracted increasing attention. Recently, Shaw’s group12 and our group13 have demonstrated the utility of donor-type carbenes in an intramolecular asymmetric C(sp3)–H insertion reaction in the presence of rhodium and ruthenium catalysts, and diazo compounds, enynones and azaenynes could be used as carbene precursors. As part of our continuing efforts to develop asymmetric carbene transformations,13,14 along with the unique nature of palladium carbene chemistry, we speculate that the less reactive donor-type carbene intermediate might provide a good opportunity to realize the palladium-catalyzed enantioselective insertion of a C(sp3)–H bond. However, there are several challenging issues associated with this project: (1) donor-type carbenes are known to easily undergo undesired intermolecular dimerization, resulting in the formation of carbene dimers;11b (2) Pd(II) is easily reduced to Pd(0) by the ligands or substrates,4b,15 which will bring additional complexities in controlling the enantioselectivity; (3) the potential cross-reactivity of palladium with the C–X bond of the reaction components will make the reaction more complicated.5b Herein, we report a Pd(II)-catalyzed asymmetric donor–donor carbene insertion into the C(sp3)–H bond using diazo compounds and enynones (Scheme 1b). This reaction represents the first example of palladium-catalyzed enantioselective C(sp3)–H bond insertion of donor-type carbenes. Both the diastereo- and enantioselectivity could be well-controlled by tuning the catalytic system.
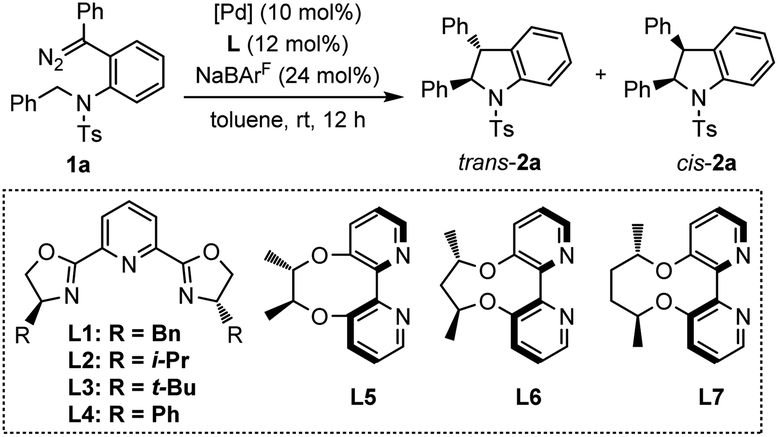
At the beginning of this investigation, diazo compound 1a tethered to N-benzyltosylamide was chosen as the model substrate to screen the asymmetric intramolecular C(sp3)–H insertion reaction conditions. As shown in Table 1, different chiral PyBox ligands L1–L4 were initially tested in the presence of Pd(PhCN)2Cl2 as the catalyst precursor with NaBArF as an additive in toluene (Table 1, entries 1–4). However, the reactions afforded the cis-indoline 2a as the major product with low enantioselectivity. Subsequently, electron-rich axially chiral 2,2′-bipyridines L5–L7 were then evaluated, which were proven to be effective ligands in promoting the asymmetric C(sp2)–H functionalization of indoles and pyrroles in Zhou's system.7e,h Surprisingly, rare trans-indoline162a (trans/cis = 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) could be observed as a slightly dominant product with 93% ee when employing L6 as a ligand (entry 6). The cis/trans-selectivity reversal under this reaction condition may arise from the interference of the Ts group with chiral cavity. With a smaller or larger dihedral angle upon chelation with palladium, L5 or L7 delivered trans-indoline with diminished enantioselectivity (entry 5 and entry 7). Therefore, we chose L6 as the ligand for further investigation of trans-indoline. The reaction was conducted in CHCl3 with poor trans-selectivity and enantioselectivity (entry 8). Slightly better trans-selectivity was observed in DCE but the enantioselectivity decreased to 88% (entry 9). Solvent screening revealed that PhCF3 was optimal with a moderate trans-selectivity (trans/cis = 1.6:1) and a maintained enantioselectivity of 93% ee (entry 10). In addition, the nature of the palladium sources had great impact on the reactivity and selectivity. When using PdCl2, the reaction proceeded to afford the trans-indoline 2a in 46% yield with a 1.7:1 trans/cis ratio and 85% ee (entry 11). PdCl2(cod), containing a 1,5-cyclooctadiene ligand, exhibited a moderate enantioselectivity of 79% ee (entry 12). Pd(PPh3)2Cl2, which had more electron-rich triphenylphosphine ligands, strongly eroded the reactivity and enantioselectivity, with only 17% yield and 6% ee (entry 13). To our delight, excellent trans-selectivity (trans/cis = 5.2:1) and enantioselectivity (95% ee) were obtained when Pd(CH3CN)2Cl2 was used (entry 14). The chloride abstraction additive NaBArF was proven necessary for enantioselective control as the ee value dropped dramatically to 4% without NaBArF (entry 15). Moreover, palladium(0) complexes were almost ineffective in catalyzing this reaction (see the ESI for details†). In the optimal combination system with Pd(CH3CN)2Cl2 and PhCF3, the same type of ligands L5 and L7 were retested. L5 exhibited similar results to L6 but with a slight improvement of trans-selectivity (trans/cis = 7.3:1) and enantioselectivity (95% ee) (entry 16), while L7 presented slightly decreased selectivities (trans/cis = 2.8:1, 91% ee) (entry 17).
:1) could be observed as a slightly dominant product with 93% ee when employing L6 as a ligand (entry 6). The cis/trans-selectivity reversal under this reaction condition may arise from the interference of the Ts group with chiral cavity. With a smaller or larger dihedral angle upon chelation with palladium, L5 or L7 delivered trans-indoline with diminished enantioselectivity (entry 5 and entry 7). Therefore, we chose L6 as the ligand for further investigation of trans-indoline. The reaction was conducted in CHCl3 with poor trans-selectivity and enantioselectivity (entry 8). Slightly better trans-selectivity was observed in DCE but the enantioselectivity decreased to 88% (entry 9). Solvent screening revealed that PhCF3 was optimal with a moderate trans-selectivity (trans/cis = 1.6:1) and a maintained enantioselectivity of 93% ee (entry 10). In addition, the nature of the palladium sources had great impact on the reactivity and selectivity. When using PdCl2, the reaction proceeded to afford the trans-indoline 2a in 46% yield with a 1.7:1 trans/cis ratio and 85% ee (entry 11). PdCl2(cod), containing a 1,5-cyclooctadiene ligand, exhibited a moderate enantioselectivity of 79% ee (entry 12). Pd(PPh3)2Cl2, which had more electron-rich triphenylphosphine ligands, strongly eroded the reactivity and enantioselectivity, with only 17% yield and 6% ee (entry 13). To our delight, excellent trans-selectivity (trans/cis = 5.2:1) and enantioselectivity (95% ee) were obtained when Pd(CH3CN)2Cl2 was used (entry 14). The chloride abstraction additive NaBArF was proven necessary for enantioselective control as the ee value dropped dramatically to 4% without NaBArF (entry 15). Moreover, palladium(0) complexes were almost ineffective in catalyzing this reaction (see the ESI for details†). In the optimal combination system with Pd(CH3CN)2Cl2 and PhCF3, the same type of ligands L5 and L7 were retested. L5 exhibited similar results to L6 but with a slight improvement of trans-selectivity (trans/cis = 7.3:1) and enantioselectivity (95% ee) (entry 16), while L7 presented slightly decreased selectivities (trans/cis = 2.8:1, 91% ee) (entry 17).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | L | 2a yield |
2a
trans:cis |
ee (trans) | ee (cis) |
| a 1a was prepared in situ through the oxidization of the corresponding hydrazone by MnO2 (8.0 eq.); 1a (0.1 mmol), [1a] = 0.033 M; the yield was the isolated yield; the dr value (trans/cis) was determined by using the 1H NMR spectrum of the crude reaction mixture. The ee values of trans-2a and cis-2a were determined by HPLC using a chiral stationary phase. b The reaction was conducted in CHCl3. c The reaction was conducted in DCE. d The reaction was conducted in PhCF3. e The reaction was conducted in PhCF3 for four days without NaBArF. | ||||||
| 1 | Pd(PhCN)2Cl2 | L1 | 66% | 1:15.7 |
2% | 10% |
| 2 | Pd(PhCN)2Cl2 | L2 | 44% | 1:7.3 |
1% | 2% |
| 3 | Pd(PhCN)2Cl2 | L3 | 58% | 1:7.3 |
15% | 4% |
| 4 | Pd(PhCN)2Cl2 | L4 | 67% | 1:9.0 |
3% | 11% |
| 5 | Pd(PhCN)2Cl2 | L5 | 54% | 1:5.7 |
28% | 0% |
| 6 | Pd(PhCN)2Cl2 | L6 | 66% | 1.2:1 |
93% | 8% |
| 7 | Pd(PhCN)2Cl2 | L7 | 41% | 1:3.8 |
63% | 7% |
| 8b | Pd(PhCN)2Cl2 | L6 | 69% | 1:11.5 |
5% | 20% |
| 9c | Pd(PhCN)2Cl2 | L6 | 71% | 1.7:1 |
88% | 2% |
| 10d | Pd(PhCN)2Cl2 | L6 | 63% | 1.6:1 |
93% | 9% |
| 11d | PdCl2 | L6 | 46% | 1.7:1 |
85% | 10% |
| 12d | PdCl2(cod) | L6 | 49% | 1:2.3 |
79% | 44% |
| 13d | Pd(PPh3)2Cl2 | L6 | 17% | 1:2.7 |
6% | 42% |
| 14 | Pd(CH 3 CN) 2 Cl 2 | L6 | 73% |
5.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
94% | 21% |
| 15e | Pd(CH3CN)2Cl2 | L6 | 19% | 1:2.1 |
4% | 2% |
| 16 | Pd(CH 3 CN) 2 Cl 2 | L5 | 65% |
7.3:1
|
95% | 12% |
| 17d | Pd(CH3CN)2Cl2 | L7 | 57% | 2.8:1 |
91% | 19% |
Having identified the optimal reaction conditions (Table 1, entry 14 and entry 16), we then turned our attention to investigate the substrate scope with different diaryl diazo compounds. As shown in Scheme 2, the substrate scope of this asymmetric C(sp3)–H insertion reaction was found to be quite general, especially the substrates with N-benzyl groups. For example, the benzyl moieties with both electron-rich and electron-deficient aryl groups were compatible, furnishing the desired trans-indolines in 42–91% yields with 89–98% ee (2a–t). Different substituents, such as MeO, MeS, CF3O, MeO2C, vinyl, F, Cl, Br and even I, could be introduced at different positions of the benzyl moiety. The reaction enantioselectivities were highly robust for the insertion of benzylic C–H, regardless of the electronic or steric properties, with the ee values typically higher than 90%. However, the diastereoselectivities were a little bit more sensitive to the properties of substituents, with the dr ranging from 1.7:1 to 7.3:1. The N-alkyl substrate, which represents a more challenging substrate, could also be subjected to the catalytic reaction conditions, affording the desired products 2u–v in much better diastereoselectivities (15.7:1 and 24:1 dr) but with diminished enantioselectivities (60% and 86% ee). Furthermore, the varying of groups R2 had a great effect on the reaction performance. When R2 was aryl, trans-indolines with excellent enantioselectivities could be obtained (2w, 2x). However, the alkyl and ester ones (2y, 2z, and 2ad) were ineffective in this protocol (see the ESI for details†). To further demonstrate the practicality of this methodology, a gram-scale reaction was conducted, delivering the desired product 2a in 79% yield, slightly lower diastereoselectivity (3.1:1 dr) and maintained enantioselectivity (94% ee). In addition, the N-Ts group of trans-indoline 2a could be easily removed in the Mg/MeOH system in good yield, and both the diastereo- and enantioselectivity remained unchanged.
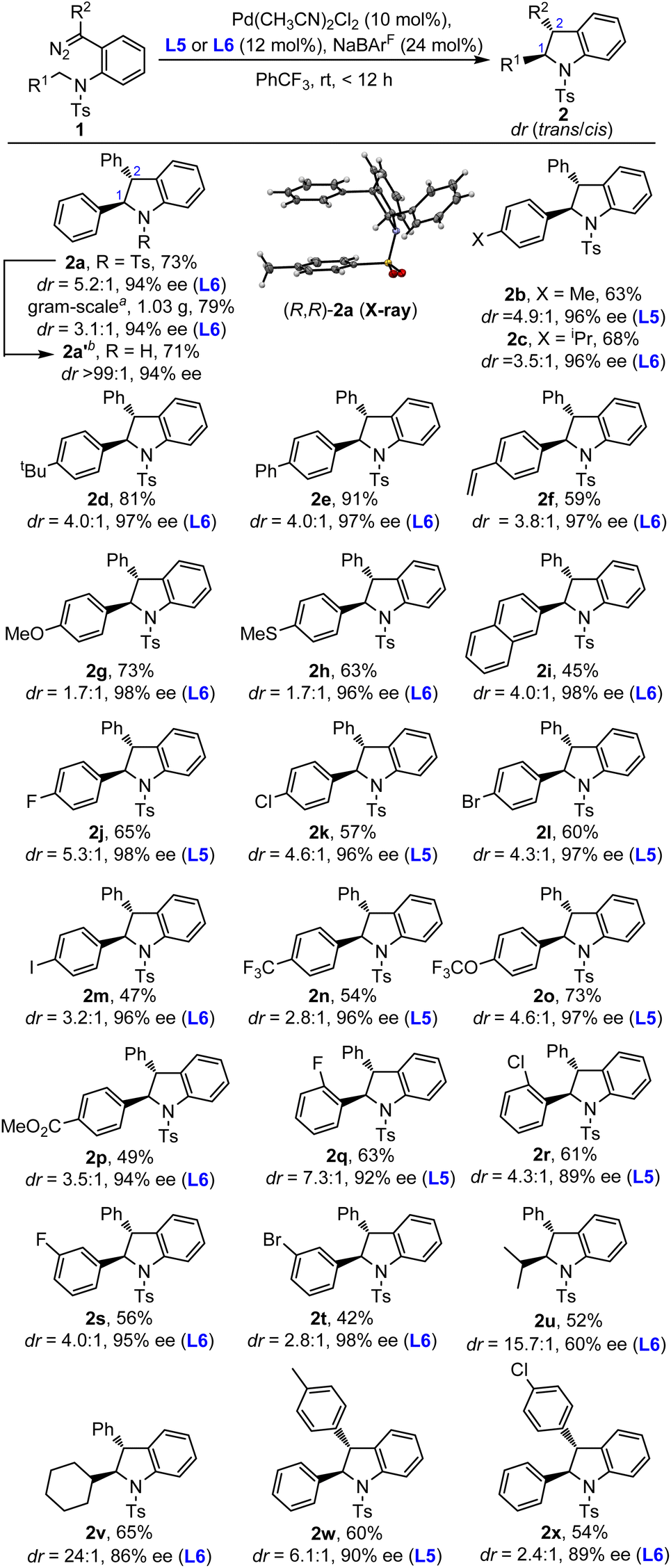 | ||
| Scheme 2 Substrate scope of diazo compounds. Reaction conditions: 1 was prepared through the oxidization of the hydrazones by MnO2 (8.0 eq.) and filtered followed by concentration. Then the reactions were run with 1 (0.1 mmol, 0.033 M), Pd(CH3CN)2Cl2 (10 mol%), L5 or L6 (12 mol%) and NaBArF (24 mol%) in PhCF3 under a N2 atmosphere for 12 h until completion by TLC. aThe reaction was run with 1a (3 mmol, 1.40 g, 0.15 M), Pd(CH3CN)2Cl2 (9 mol%), L6 (10.8 mol%) and NaBArF (21.6 mol%) in PhCF3 (15 mL) for 15 h. btrans-2a (0.13 mmol), Mg powder (14.0 eq.), methanol (5 mL), 50 °C, sonicate, 2 h. | ||
In addition to diazo compounds, this palladium-catalytic system could also be extended to an enynone system under modified conditions (see the ESI for more details†). Unlike the above diazo-based system catalyzed by Pd(CH3CN)2Cl2/bipyridine, in which trans-indolines were obtained, the enynone-based system enabled by Pd(PhCN)2Cl2/PyBox afforded the corresponding indolines 4 with the cis-isomers dominating. As shown in Scheme 3, enynones with different substituted benzylacetamide side chains were subjected to the standard conditions, leading to the desired indolines 4a–f in 76–85% yields with 80–95% ee. The reactions were a little sensitive to the electronic properties of the substituted group R1. The electron-rich benzyl groups gave the products in relatively lower ee (4c, 80% ee). However, the electron-deficient benzyl groups furnished the products 4d–f in better enantioselectivities (91–95% ee). Furthermore, this system could be extended to N-benzylic enynones with different R2 groups, delivering the desired products 4g–h in 63–85% yield and 92–96% ee. However, in the case of N-alkyl enynones 3i, only trace oxidation of carbene was observed with no detectable insertion product even at 120 °C (see the ESI for details†). The absolute configuration of the major enantiomer product was determined by single crystal X-ray diffraction of compound 4h, which showed a cis-(2R, 3S) configuration. It is noted that the indolines 4 were obtained in higher than 99:1 dr in all cases, possibly due to the favorable π–π interaction between phenyl and furan substituents.
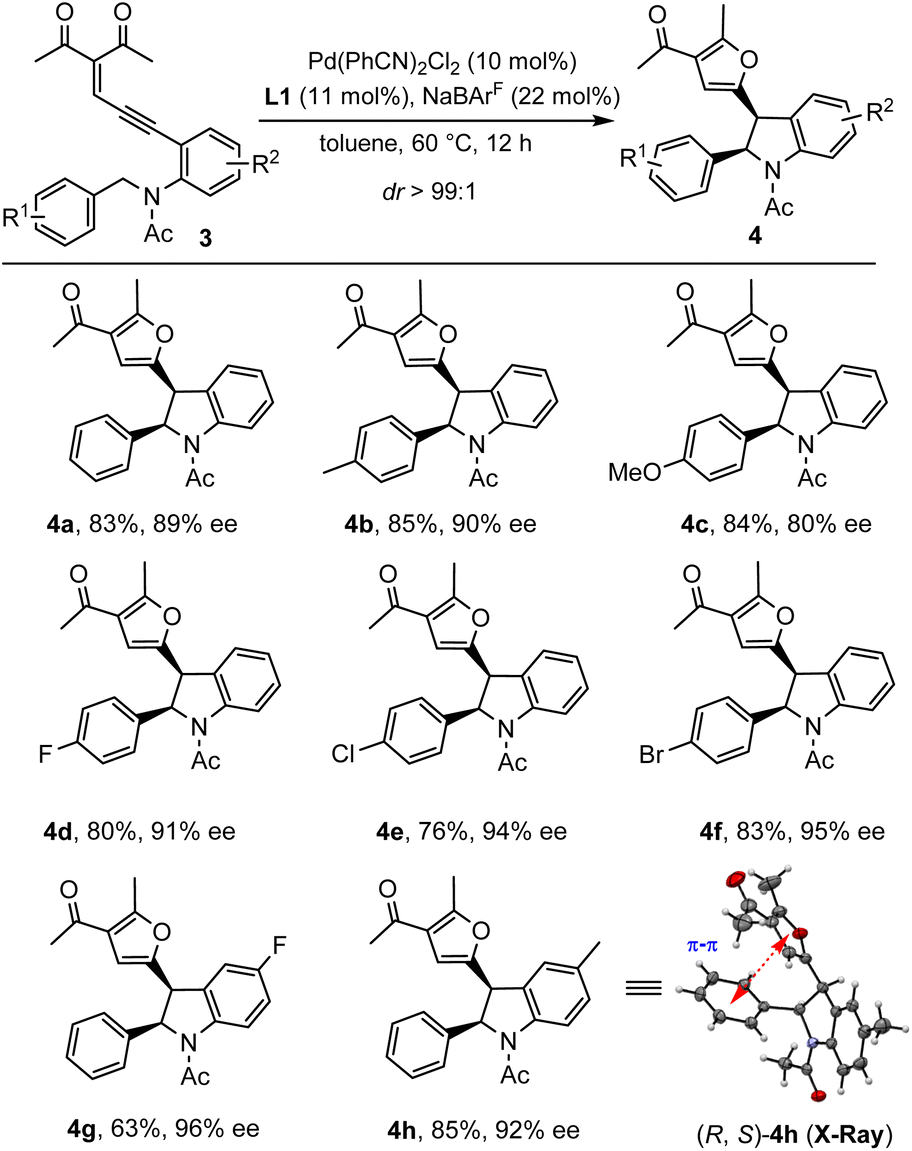 | ||
| Scheme 3 Substrate scope of enynones. Reaction conditions: 3 (0.2 mmol), Pd(PhCN)2Cl2 (10 mol%), L1 (11 mol%) and NaBArF (22 mol%) in 1 mL toluene under a N2 atmosphere at 60 °C for 12 h. | ||
According to the previous mechanistic studies performed by Solé and Fernández, there are two possible mechanisms for the Pd(II)-catalyzed C(sp3)–H insertion of an acceptor-type carbene system: (i) the concerted metalation-deprotonation (CMD) process assisted by carbonate or acetate;8a (ii) stepwise Mannich-type reaction through a zwitterionic intermediate.8b As the reactions in our system were conducted under neutral conditions without the base of carbonate or acetate, the base-assisted CMD mechanism is not likely to be operative. Another stepwise Mannich-type reaction through a zwitterionic intermediate seems more likely. To elucidate the reaction mechanism of our system, several control reactions were conducted. First, when diazo compound d2-1a (99% D) with deuterated methylene was employed as the substrate under the standard reaction conditions, deuterium was completely transferred to the carbene carbon of d2-2a without measurable scrambling of the isotope label (Scheme 4a). Such results indicated that the H-shift occurred intramolecularly without interference from the external solvent or reagents. Second, the kinetic isotope effect (KIE) experiment of monodeuterated diazo compound d-1a was also performed (see the ESI for details†). The result suggested that the C–H bond cleavage process might not be involved in a rate-determining step. To get more insight into the C(sp3)–H insertion process, chiral diazo compound (R)-5a (97% ee) with a tertiary carbon stereocenter was then examined (Scheme 4b). Treatment of (R)-5a with a palladium catalyst under the standard reaction conditions but with the racemic ligand L6 provided the cis-indoline 6a as the major product in 68% yield and 94:6 dr without loss of chiral integrity (Scheme 4b, entry 1). Besides, the chiral ligand (Sa, R, R)-L6 produced the stereoretentive indoline 6a in 63% yield and 95:5 dr (entry 2). Interestingly, a decreased yield of 36% and enantioselectivity of 92% ee were observed when another stereoisomer of the ligand (Ra, S, S)-L6 was used (entry 3), which indicated a mismatch in stereochemical preference between the substrate and Pd-(Ra, S, S)-L6 catalyst. What is most unexpected is that the product stereoselectivities further dropped to 87% ee and 68:32 dr when the catalytic system was cooled down to −10 °C (entry 4). The erosion of chiral integrity during the C–H insertion process indicated that the reaction did not proceed through a concerted process. But the slightly decreased enantioselectivity implied that there might exist a short-lived and compact zwitterionic intermediate after a stereoselective hydride shift which is dominated by the substrate and palladium catalyst.8b We envisioned that the chirality erosion of C1 might be caused by the trivial rotation around the Caryl–N bond of the zwitterionic intermediate.
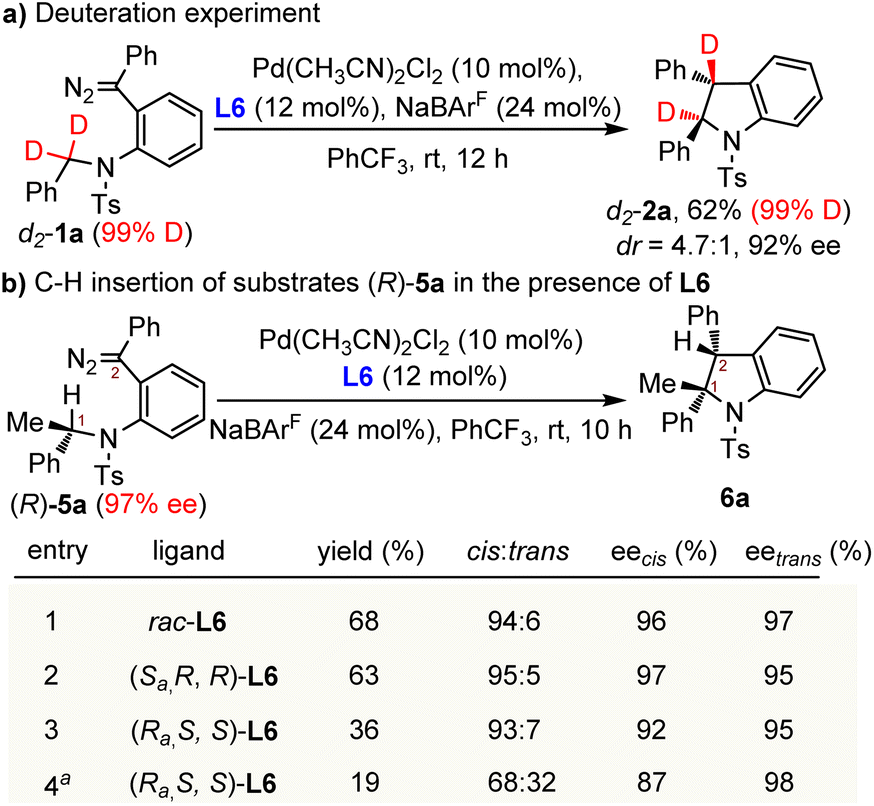 | ||
| Scheme 4 Mechanistic investigations. aThe reaction was conducted at −10 °C. | ||
To prove the existence of this zwitterionic intermediate, a Hammett analysis was also conducted with diazo compounds 1 bearing different para-substituted benzyl amides. As illustrated in Fig. 1, the small magnitude of the ρ value of −0.29 (R2 = 0.8338) suggests a slight positive charge buildup at the benzylic carbon atom of the C–H insertion step. Collectively, the stepwise Mannich-type mechanism through a zwitterionic intermediate is likely to be operative for this system.
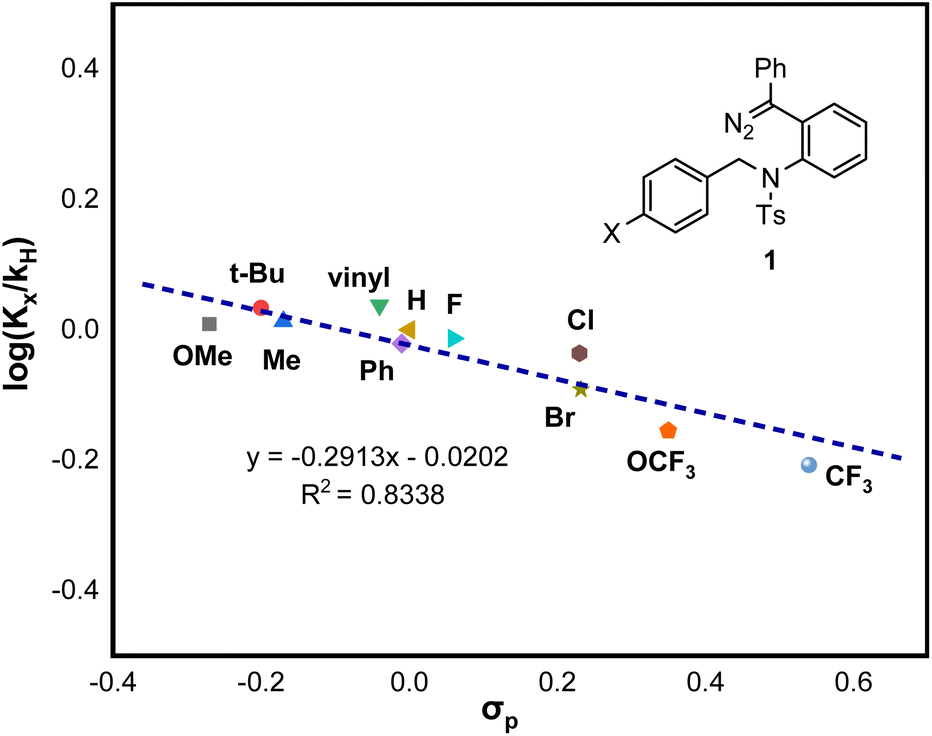 | ||
| Fig. 1 Hammett analysis. | ||
In light of the opposite diastereoselectivities of C–H insertion behavior for diazo compounds and enynones, we suspected that the difference of N-protected groups might lead to the divergence of products. To verify this hypothesis, Ac/Ms/Bn-substituted diazo compound 1aa/1ab/1ac was then prepared and subjected to the standard conditions (Scheme 5). As expected, the Ac-substituted diazo 1aa exclusively produced the cis-indoline 2aa in 54% yield. Ms-substituted diazo 1ab delivered the trans-indoline 2ab with a 4.3:1 trans:cis ratio similar to that of 2a. However, as for Bn-substituted diazo 1ac, only traces of the C(sp3)–H insertion product were observed, probably due to the coordination between the amine and palladium complex. These results indicated that the N-protected groups of diazo substrates dictated the reaction diastereoselectivities.
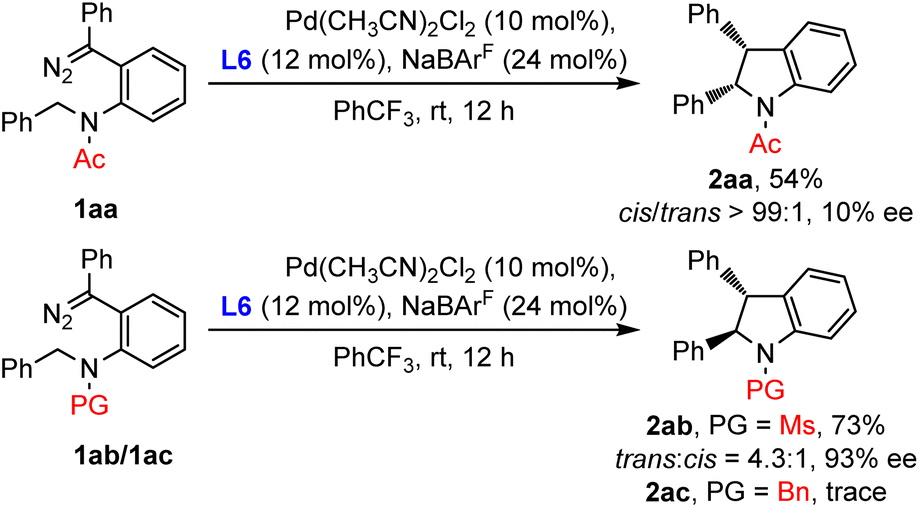 | ||
| Scheme 5 Investigation into diastereoselectivity. | ||
Based on the above mechanistic experiments, a plausible mechanism is proposed in Scheme 6. Taking diazo compounds 1a/1aa for example, the reaction starts with the coordination of the diazo and Pd catalyst, followed by extrusion of nitrogen to generate palladium carbene II. The intermediate II subsequently undergoes a rapid insertion into the C(sp3)–H bond through a short-lived zwitterionic intermediate III to form the products 2a/2aa with the Pd catalyst being regenerated. Based on the structure of ligand L6 reported by Zhou’s group7e and the rhodium-diphenylcarbenes reported by Fürstner, Davies and co-workers,17 two models with different N-protected groups are also depicted. The π–π interaction between two phenyl rings could be found in either Ac or Ts-substituted palladium carbene II. For N-Ac IIa, the acetyl group tends to be planar and the rigid structure of acetamide restricts the phenyl on C1 to approaching methyl on acetamide. Combined with the favorable π–π interaction between two phenyl rings, cis-indoline is produced exclusively. However, as for N-Ts IIb, the sulfonyl group is tetrahedral and could rotate to a proper conformation. The introduction of the Ts group would probably change the spatial distribution and thus reduce steric repulsion or structure strain between the substrate and chiral catalyst. Therefore, a more favorable trans configuration would be adopted even though the π–π interaction between two phenyl rings has been weakened. In addition, a high enantioselectivity for trans-indoline is attained probably owing to one of the pyridine rings of the axially chiral 2,2′-bipyridine ligand stretching to the reaction center to some extent. It is noteworthy that a stereoretentive ring closure occurs quickly due to the rotation restriction of the iminium ion, and thus the erosion of chirality on C1 by this effect contributes little.
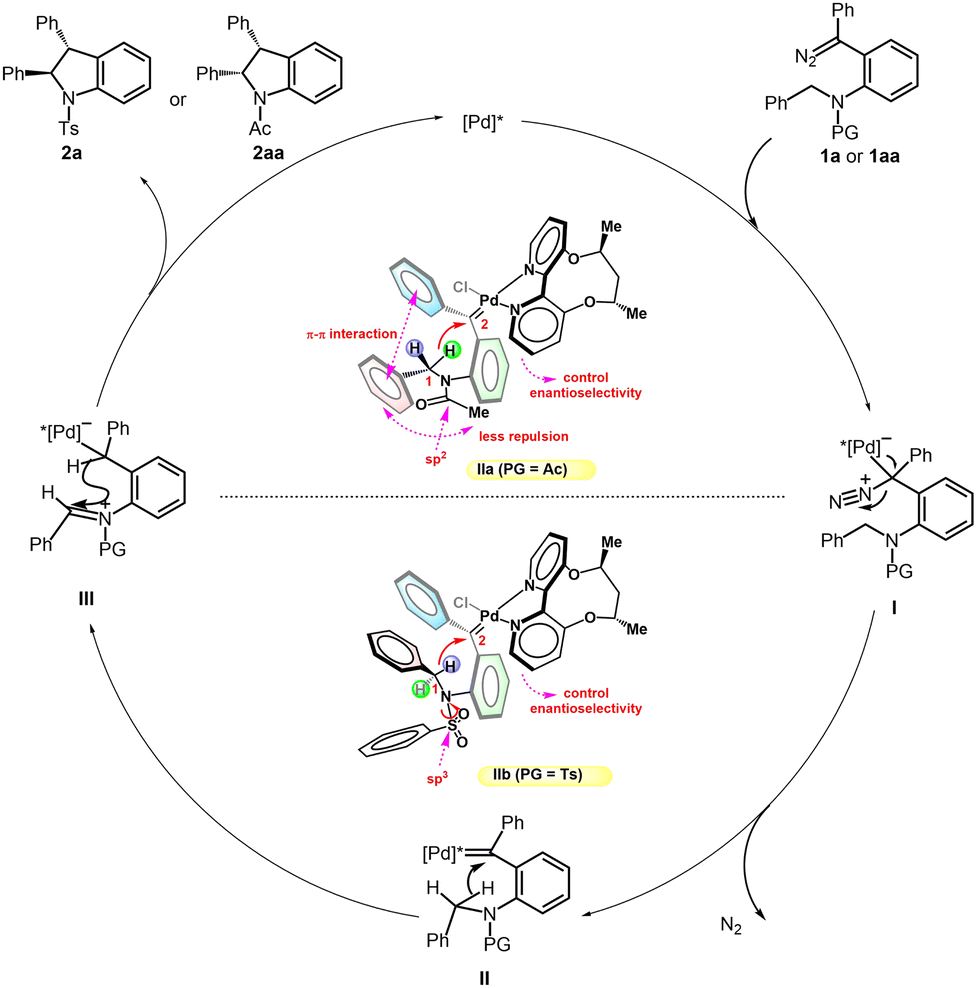 | ||
| Scheme 6 Plausible reaction mechanism. | ||
Conclusions
In summary, we have described an effective protocol for the first palladium-catalyzed diastereodivergent asymmetric intra-molecular C(sp3)–H insertion reaction of donor–donor carbenes. Both diazo compounds and enynones can be used as carbene precursors. The reaction shows good functional group tolerance and exhibits excellent diastereo- and enantioselectivity. Opposite configurations of indolines can be accessed by adopting different protecting groups, demonstrating a complementary approach to divergent synthesis. The mechanistic experiments suggest that a stepwise Mannich-type mechanism through a zwitterionic intermediate is more operative for our system. These findings may open new vistas for palladium carbene-involving asymmetric synthesis, especially asymmetric C(sp3)–H insertions.Data availability
All the data including experimental procedures, NMR, IR, HRMS, and HPLC spectra and crystallographic data of 2a, 4h, (2R, 3S)-6a are recorded in the ESI†.Author contributions
Conceptualization, funding acquisition, resources and supervision were done or provided by S. Zhu. W. Li and H. Zhang performed all the experiments. K. Chen performed the optimizations of models on reaction diastereoselectivity and enantioselectivity. H. Jiang and J. Sun provided their help with useful discussions and suggestions. W. Li and S. Zhu contributed to the conception of the experiments, discussion of the results and preparation of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate financial support from the NSFC (21871096, 22071062, and 22271096) and 111 Project (no. B20003). In addition, Prof. Yonggui Zhou of the Dalian Institute of Chemical Physics (DICP) was acknowledged for his generous supply of the first batch of chiral ligands L6 and L7.Notes and references
- For books on asymmetric C–H insertion, see: (a) S.-L. You, Asymmetric Functionalization of C–H Bonds, RSC, Cambridge, U.K., 2015, pp. 1–66 RSC; (b) F. Colobert and J. WencelDelord, C–H Activation for Asymmetric Synthesis, Wiley-VCH, Weinheim, 2019, pp. 1–49 CrossRef; (c) J. Wang, C.-M. Che and M. P. Doyle, Transition Metal–Catalyzed Carbene Transformations, Wiley-VCH, Weinheim, 2022, pp. 1–24 CrossRef.
- For reviews on asymmetric C–H insertion, see: (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2903 CrossRef CAS PubMed; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed; (c) Y.-P. Li, Z.-Q. Li and S.-F. Zhu, Tetrahedron Lett., 2018, 59, 2307–2316 CrossRef CAS; (d) H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS PubMed; (e) Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. Madron du Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed; (f) Y. Liu, T. You, H.-X. Wang, Z. Tang, C.-Y. Zhou and C.-M. Che, Chem. Soc. Rev., 2020, 49, 5310–5358 RSC; (g) Y. He, Z. Huang, K. Wu, J. Ma, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2022, 51, 2759–2852 RSC; (h) B. Hong, L. Shi, L. Li, S. Zhan and Z. Gu, Green Synth. Catal., 2022, 3, 137–149 CrossRef.
- For recent selected examples on asymmetric C–H insertion catalyzed by rhodium, see: (a) X. Xu, Y. Deng, D. N. Yim, P. Y. Zavalij and M. P. Doyle, Chem. Sci., 2015, 6, 2196–2201 RSC; (b) K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. Davies, Nature, 2016, 533, 230–234 CrossRef CAS PubMed; (c) K. Liao, T. C. Pickel, V. Boyarskikh, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2017, 551, 609–613 CrossRef CAS PubMed; (d) K. Liao, Y. F. Yang, Y. Li, J. N. Sanders, K. N. Houk, D. G. Musaev and H. M. L. Davies, Nat. Chem., 2018, 10, 1048–1055 CrossRef CAS PubMed; (e) J. Fu, Z. Ren, J. Bacsa, D. G. Musaev and H. M. L. Davies, Nature, 2018, 564, 395–399 CrossRef CAS PubMed; (f) W. Liu, Z. Ren, A. T. Bosse, K. Liao, E. L. Goldstein, J. Bacsa, D. G. Musaev, B. M. Stoltz and H. M. L. Davies, J. Am. Chem. Soc., 2018, 140, 12247–12255 CrossRef CAS PubMed; (g) Z. J. Garlets, B. D. Wertz, W. Liu, E. A. Voight and H. M. L. Davies, Chem, 2020, 6, 304–313 CrossRef CAS PubMed; (h) Z. J. Garlets, J. N. Sanders, H. Malik, C. Gampe, K. N. Houk and H. M. L. Davies, Nat. Catal., 2020, 3, 351–357 CrossRef CAS; (i) K. Dong, X. Fan, C. Pei, Y. Zheng, S. Chang, J. Cai, L. Qiu, Z.-X. Yu and X. Xu, Nat. Commun., 2020, 11, 2363 CrossRef CAS PubMed; (j) Z. Li, Y. Chen, C. Wang, G. Xu, Y. Shao, X. Zhang, S. Tang and J. Sun, Angew. Chem., Int. Ed., 2021, 60, 25714–25718 CrossRef CAS PubMed.
- (a) J. Tsuji, Palladium Reagents and Catalysts: New Perspectives for the 21st Century, Wiley-VCH, Chichester, 2005 Search PubMed; (b) T. J. Colacot, New Trends in Cross-Coupling: Theory and Applications, RSC, Cambridge, U.K., 2015 Search PubMed.
- For reviews on palladium-catalyzed cross-couplings through carbene migratory insertion, see: (a) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed; (b) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed; (c) K. Wang and J. Wang, Synlett, 2019, 30, 542–551 CrossRef CAS; (d) Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592–10605 CrossRef CAS PubMed.
- For selected examples on palladium-catalyzed cross-couplings through carbene migratory insertion, see: (a) K. L. Greenman, D. S. Carter and D. L. Van Vranken, Tetrahedron, 2001, 57, 5219–5225 CrossRef CAS; (b) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587–5590 CrossRef CAS PubMed; (c) R. Kudirka, S. K. Devine, C. S. Adams and D. L. Van Vranken, Angew. Chem., Int. Ed., 2009, 48, 3677–3680 CrossRef CAS PubMed; (d) Y. Gao, G. Wu, Q. Zhou and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 2716–2720 CrossRef CAS PubMed; (e) Z. Liu, S. Cao, J. Wu, G. Zanoni, P. Sivaguru and X. Bi, ACS Catal., 2020, 10, 12881–12887 CrossRef CAS; (f) Y. Ping, R. Wang, Q. Wang, T. Chang, J. Huo, M. Lei and J. Wang, J. Am. Chem. Soc., 2021, 143, 9769–9780 CrossRef CAS PubMed; (g) X. Ning, Y. Chen, F. Hu and Y. Xia, Org. Lett., 2021, 23, 8348–8352 CrossRef CAS PubMed; (h) S. Li, M. Li, S.-S. Li and J. Wang, Chem. Commun., 2022, 58, 399–402 RSC.
- For examples on palladium carbene-involving asymmetric transformations, see: (a) A. Khanna, C. Maung, K. R. Johnson, T. T. Luong and D. L. Van Vranken, Org. Lett., 2012, 14, 3233–3235 CrossRef CAS PubMed; (b) D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356–13360 CrossRef CAS PubMed; (c) Y. Zhu, X. Liu, S. Dong, Y. Zhou, W. Li, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2014, 53, 1636–1640 CrossRef CAS PubMed; (d) X.-L. Xie, S.-F. Zhu, J.-X. Guo, Y. Cai and Q.-L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 2978–2981 CrossRef CAS PubMed; (e) X. Gao, B. Wu, W.-X. Huang, M.-W. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2015, 54, 11956–11960 CrossRef CAS PubMed; (f) J. Feng, B. Li, Y. He and Z. Gu, Angew. Chem., Int. Ed., 2016, 55, 2186–2190 CrossRef CAS PubMed; (g) V. Arredondo, S. C. Hiew, E. S. Gutman, I. D. U. A. Premachandra and D. L. Van Vranken, Angew. Chem., Int. Ed., 2017, 56, 4156–4159 CrossRef CAS PubMed; (h) H.-Q. Shen, C. Liu, J. Zhou and Y.-G. Zhou, Org. Chem. Front., 2018, 5, 611–614 RSC; (i) H.-Q. Shen, H.-P. Xie, L. Sun and Y.-G. Zhou, Organometallics, 2019, 38, 3902–3905 CrossRef CAS; (j) J. Huo, K. Zhong, Y. Xue, M. Lyu, Y. Ping, Z. Liu, Y. Lan and J. Wang, J. Am. Chem. Soc., 2021, 143, 12968–12973 CrossRef CAS PubMed; (k) J. Huo, K. Zhong, Y. Xue, M. Lyu, Y. Ping, W. Ouyang, Z. Liu, Y. Lan and J. Wang, Chem. - Eur. J., 2022, 28, e202200191 CAS; (l) B. Yang, B. K. Cao, G. Zhao, J. Yang and J. Zhang, J. Am. Chem. Soc., 2022, 144, 15468–15474 CrossRef CAS PubMed.
- (a) D. Solé, F. Mariani, M. L. Bennasar and I. Fernández, Angew. Chem., Int. Ed., 2016, 55, 6467–6470 CrossRef PubMed; (b) D. Solé, F. Pérez-Janer, M. L. Bennasar and I. Fernández, Eur. J. Org. Chem., 2018, 2018, 4446–4455 CrossRef.
- E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181–7192 CrossRef CAS PubMed.
- (a) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015–1026 CrossRef; (b) D. G. Musaev, T. M. Figg and A. L. Kaledin, Chem. Soc. Rev., 2014, 43, 5009–5031 RSC.
- (a) M. Mato, C. García-Morales and A. M. Echavarren, ChemCatChem, 2018, 11, 53–72 CrossRef; (b) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC; (c) B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed; (d) R. Vicente, Chem. Rev., 2021, 121, 162–226 CrossRef CAS PubMed; (e) M. Mato, A. Franchino, C. García-Morales and A. M. Echavarren, Chem. Rev., 2021, 121, 8613–8684 CrossRef CAS PubMed.
- (a) C. Soldi, K. N. Lamb, R. A. Squitieri, M. González-López, M. J. Di Maso and J. T. Shaw, J. Am. Chem. Soc., 2014, 136, 15142–15145 CrossRef CAS PubMed; (b) K. N. Lamb, R. A. Squitieri, S. R. Chintala, A. J. Kwong, E. I. Balmond, C. Soldi, O. Dmitrenko, M. Castiñeira Reis, R. Chung, J. B. Addison, J. C. Fettinger, J. E. Hein, D. J. Tantillo, J. M. Fox and J. T. Shaw, Chem. - Eur. J., 2017, 23, 11843–11855 CrossRef CAS PubMed; (c) L. W. Souza, R. A. Squitieri, C. A. Dimirjian, B. M. Hodur, L. A. Nickerson, C. N. Penrod, J. Cordova, J. C. Fettinger and J. T. Shaw, Angew. Chem., Int. Ed., 2018, 57, 15213–15216 CrossRef CAS PubMed; (d) L. A. Nickerson, B. D. Bergstrom, M. Gao, Y. S. Shiue, C. J. Laconsay, M. R. Culberson, W. A. Knauss, J. C. Fettinger, D. J. Tantillo and J. T. Shaw, Chem. Sci., 2020, 11, 494–498 RSC; (e) S. N. Dishman, C. J. Laconsay, J. C. Fettinger, D. J. Tantillo and J. T. Shaw, Chem. Sci., 2022, 13, 1030–1036 RSC; (f) B. D. Bergstrom, A. T. Merrill, J. C. Fettinger, D. J. Tantillo and J. T. Shaw, Angew. Chem., Int. Ed., 2022, 61, e2022030 CrossRef PubMed.
- (a) D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2016, 55, 8452–8456 CrossRef CAS PubMed; (b) D. Zhu, L. Chen, H. Zhang, Z. Ma, H. Jiang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 12405–12409 CrossRef CAS PubMed; (c) S. Qiu, X. Gao and S. Zhu, Chem. Sci., 2021, 12, 13730–13736 RSC.
- (a) R. Wu, K. Chen, J. Ma, Z.-X. Yu and S. Zhu, Sci. China: Chem., 2020, 63, 1230–1239 CrossRef CAS; (b) Z. Liu, L. Chen, D. Zhu and S. Zhu, Org. Lett., 2021, 23, 1275–1279 CrossRef CAS PubMed; (c) R. Wu, J. Lu, T. Cao, J. Ma, K. Chen and S. Zhu, J. Am. Chem. Soc., 2021, 143, 14916–14925 CrossRef CAS PubMed; (d) D. Zhu, T. Cao, K. Chen and S. Zhu, Chem. Sci., 2022, 13, 1992–2000 RSC.
- Y. V. Tomilov, V. A. Dokitchev, U. M. Dzhemilev and O. M. Nefedov, Russ. Chem. Rev., 1993, 62, 799–838 CrossRef.
- M. Santi, S. T. R. Müller, A. A. Folgueiras-Amador, A. Uttry, P. Hellier and T. Wirth, Eur. J. Org. Chem., 2017, 2017, 1889–1893 CrossRef CAS.
- (a) C. Werlé, R. Goddard, P. Philipps, C. Farès and A. Fürstner, J. Am. Chem. Soc., 2016, 138, 3797–3805 CrossRef PubMed; (b) M. Lee, Z. Ren, D. G. Musaev and H. M. L. Davies, ACS Catal., 2020, 10, 6240–6247 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2142261, 1908561 and 2142262. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03524c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |