
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembling diuranium complexes in different states of charge with a bridging redox-active ligand†
Dieuwertje K.
Modder‡
a,
Mikhail S.
Batov‡
 a,
Thayalan
Rajeshkumar
b,
Andrzej
Sienkiewicz
cd,
Ivica
Zivkovic
c,
Rosario
Scopelliti
a,
Laurent
Maron
b and
Marinella
Mazzanti
*a
a,
Thayalan
Rajeshkumar
b,
Andrzej
Sienkiewicz
cd,
Ivica
Zivkovic
c,
Rosario
Scopelliti
a,
Laurent
Maron
b and
Marinella
Mazzanti
*a
aGroup of Coordination Chemistry, Institut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne. Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratoire de Physique et Chimie des Nano-objets, Institut National des Sciences Appliquées, Cedex 431077 ToulouseFrance
cLaboratory for Quantum Magnetism, Institute of Physics, École Polytechnique Fédérale de Lausanne (EPFL), 1015 LausanneSwitzerland
dADSresonances Sàrl, Route de Genève 60B1028 PréverengesSwitzerland
First published on 31st August 2022
Abstract
Radical-bridged diuranium complexes are desirable for their potential high exchange coupling and single molecule magnet (SMM) behavior, but remain rare. Here we report for the first time radical-bridged diuranium(IV) and diuranium(III) complexes. Reaction of [U{N(SiMe3)2}3] with 2,2′-bipyrimidine (bpym) resulted in the formation of the bpym-bridged diuranium(IV) complex [{((Me3Si)2N)3UIV}2(μ-bpym2−)], 1. Reduction with 1 equiv. KC8 reduces the complex, affording [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-bpym)], 2, which is best described as a radical-bridged UIII–bpym˙−–UIII complex. Further reduction of 1 with 2 equiv. KC8, affords [K(2.2.2-cryptand)]2[{((Me3Si)2N)3UIII}2(μ-bpym2−)], 3. Addition of AgBPh4 to complex 1 resulted in the oxidation of the ligand, yielding the radical-bridged complex [{((Me3Si)2N)3UIV}2(μ-bpym˙−)][BPh4], 4. X-ray crystallography, electrochemistry, susceptibility data, EPR and DFT/CASSCF calculations are in line with their assignments. In complexes 2 and 4 the presence of the radical-bridge leads to slow magnetic relaxation.
Introduction
F-element complexes led recently to a paradigm shift in molecular magnetism.1 In particular multimetallic f-element complexes with strong superexchange-type metal–metal interactions mediated by radical bridging ligands are currently excellent candidates for the development of single-molecule magnets (SMMs) operating at temperatures compatible with practical applications.1a,d,2 In this context, the large radial extension of 5f orbitals should allow for stronger exchange coupling in polymetallic uranium complexes making them ideal candidates for the development of exchange-coupled SMMs.3 However, uranium complexes showing SMM behavior are mainly limited to mononuclear complexes4 while examples of uranium-based exchange-coupled SMMs remain extremely rare and all except one5 consist of 5f-3d systems.3f,6 This is due to both the difficulty of designing rational syntheses of polymetallic complexes of uranium and to the poor understanding of the parameters leading to magnetic exchange in polynuclear uranium complexes. In contrast, some unambiguous examples of magnetic coupling between uranium centers have been reported, but none of these systems showed SMM properties so far.7Redox-active ligands and in particular N-heterocyclic ligands such as 2,2′-bipyrimidine (bpym) were identified more than 10 years ago as suitable bridging ligands capable of promoting magnetic communication between two lanthanide(III) centers leading to single molecule magnet behaviour.2c,8 Seminal studies by Schelter and Kiplinger showed that the redox-active cyano-substituted terpyridine ligands could be used to assemble multimetallic 5f-4f and 5f-5f complexes exhibiting metal–metal communication,9 but further development of exchange-coupled uranium SMMs based on redox-active ligands has lagged behind. Particularly desirable in this context is the design of radical-bridged diuranium(III) complexes. Uranium(III) complexes have been reported to promote the reductive coupling of polyazines such as pyrazine and 2,4,6-tris(2-pyridyl)-1,3,5-triazine,10 but reduction of pyridine, 2,2′-bipyridine (2,2′-bpy) and 4,4′-bipyridine (4,4′-bpy) was not observed for U(III) complexes supported by cyclopentadienyl or hydrotris(3,5-dimethylpyrazolyl) borate ligands.10a,11 Direct reduction of bipyridine by a U(III) complex was only reported recently by Meyer and coworkers for the tris–aryloxide complex [((Ad,tBuArO)3tacn)U] complex.12 A few examples of mononuclear uranium complexes of bipyridine radicals11a,11c,13a–g are known, including a unique U(III)–bipy˙− compound that showed zero field slow magnetic relaxation behavior.11c However, examples of dinuclear complexes of uranium containing a redox-active ligand in different states of charge are very rare.7k,14 Recently, our group and the Schelter group showed that the reaction of the amide supported U(III) and U(II) complexes [U{N(SiMe3)2}3]n−, with redox active ligands (N,N-dimethylbenzamide, benzophenone and bipyridine) leads either to mononuclear U(IV)-ligand radical complexes or dinuclear complexes bridged by dianionic ligands but multiple states of charge were not identified for the bridging ligand.15 Here we show that the [U{N(SiMe3)2}3] complex provides a convenient precursor to access 2,2′-bipyrimidine-bridged dinuclear complexes for which different states of charge can be stabilised. Structural, spectroscopic and computational studies indicate that dinuclear radical-bridged UIV-bpym˙−-UIV and UIII-bpym˙−-UIII complexes have been obtained for the first time. Despite the absence of an obvious metal–metal coupling, a slow magnetic relaxation is observed in both complexes.
Results and discussion
Syntheses
Since we recently showed that the bulky amide N(SiMe3)2 ligand allows the binding of 2,2′-bpy to a [U{N(SiMe3)2}3]+ fragment,16 the amide supported uranium(III) complex [U{N(SiMe3)2}3] was chosen as a convenient precursor to investigate the possibility of accessing bpym-bridged dinuclear complexes.Upon addition of a THF solution of bpym to [U{N(SiMe3)2}3], the color changed from purple to light green and crystals of the diuranium bpym-bridged complex [{((Me3Si)2N)3U}2(μ-bpym)], 1 (Scheme 1), were obtained in 62% yield. The 1H-NMR spectrum of 1 measured at room temperature showed broad signals at −7.7 and −23.3 ppm, as well as a sharp signal at 33.9 ppm (Fig. S2†), assigned to the N(SiMe3)2 ligands and the protons of the bpym ligand, respectively. Upon increasing the temperature to 60 °C coalescence of the two broad peaks was observed, in agreement with a higher fluxionality of the amide ligands.
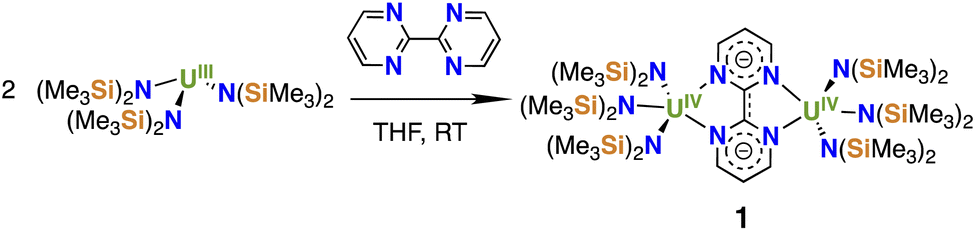 | ||
| Scheme 1 Synthesis of [{((Me3Si)2N)3U}2(μ-bpym)], 1. | ||
With complex 1 on hand reduction and oxidation reactions were pursued with the objective of accessing radical-bridged diuranium complexes. Addition of 1 equiv. KC8, in the presence of 1 equiv. 2.2.2-cryptand (Scheme 2) to a solution of 1 in THF, resulted in a color change from light green to brown. Crystals of the complex [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-bpym)], 2, were isolated in 73% yield by slow diffusion of hexane into a concentrated THF solution. The 1H-NMR spectrum of 2 at room temperature shows one broad signal at −8.9 and a sharp signal at 13.1 ppm (Fig. S4†) corresponding to the N(SiMe3)2 ligands and the protons of the bpym ligand, respectively. Upon decreasing the temperature, the broad signal splits into two broad signals, which can be observed both at 0 °C and at −40 °C indicating a decreased fluxionality of the amide ligands.
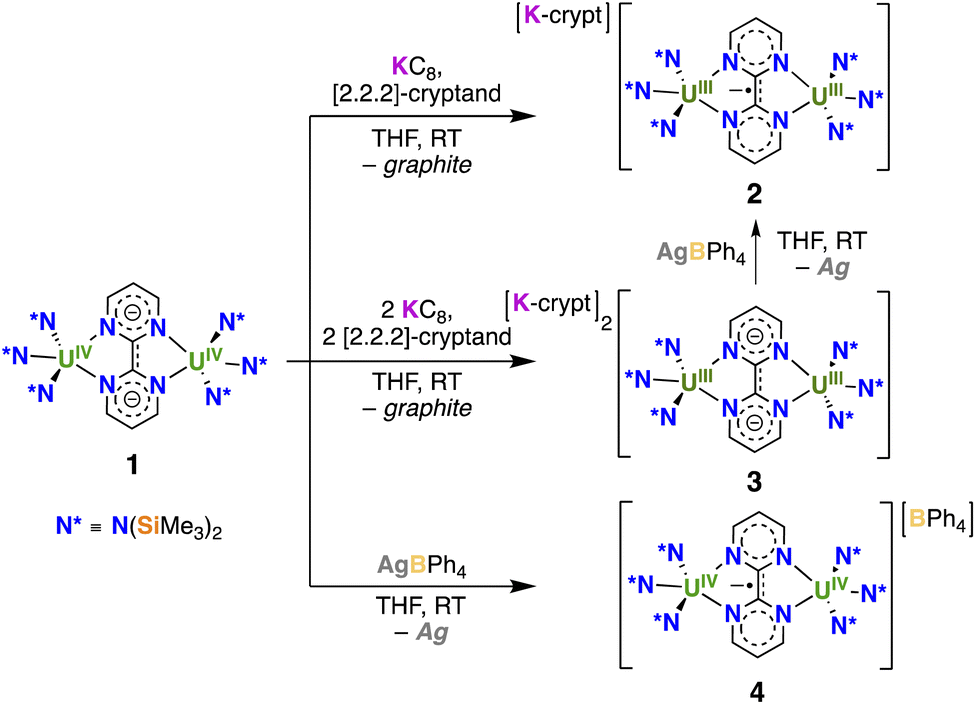 | ||
| Scheme 2 Synthesis of [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-bpym)], 2, [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-bpym)], 3, and [{((Me3Si)2N)3U}2 (μ-bpym)][BPh4], 4. | ||
Addition of 2 equiv. KC8, in the presence of 2 equiv. 2.2.2-cryptand (Scheme 2) to a solution of 1 in THF at room temperature, afforded, after crystallization from Et2O at −40 °C, single crystals of [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-bpym)], 3, in 57% yield. Complex 3 was also obtained from the reaction of the previously reported masked U(II) complex [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-O)]16,17 with bpym at −80 °C as indicated by 1H-NMR spectroscopy (Fig. S9†), but it requires working at low temperatures and its isolation is more difficult due to the formation of the terminal oxo complex [K(2.2.2-cryptand)][U(O){N(SiMe3)2}3] as byproduct. Oxidation of 3 with 1 equiv. AgBPh4 in THF solution resulted in the formation of 2 as indicated by 1H-NMR spectroscopy (Fig. S10†).
Further reduction of 3 with excess KC8 (up to 10 equiv. KC8) only resulted in partial decomposition of the complex, releasing KN(SiMe3)2 as identified by 1H-NMR spectroscopy (Fig. S11†). The 1H-NMR spectrum of 3 measured at room temperature shows a broad signal at −7.3 and a sharp signal at 12.8 ppm (Fig. S6†). Upon decreasing the temperature to 0 °C, the former broadens.
The reaction of 1 with AgBPh4 caused a color change from green to brown. Slow diffusion of hexane into the resulting THF solution yielded dark brown crystals, characterised by X-ray spectroscopy as [{((Me3Si)2N)3U}2(μ-bpym)][BPh4], 4 (Scheme 2), in 62% yield. Complex 4 shows very broad NMR signals (Fig. S8†).
In these complexes the assignment of the bridging ligand's charge and of the U centers' oxidation states is not straightforward. In order to assign the oxidation state of the uranium centers and the charge of the bpym ligand (neutral, a radical anion, or a dianion) in complexes 1-4 we performed structural studies, cyclic voltammetry, EPR, magnetometry and computational DFT studies.
Solid-state structures
The molecular structures of complexes 1–4 (Fig. 1 and S12–15†) all consist of two [((Me3Si)2N)3U] units bridged by a bpym ligand and related by an inversion center. In all complexes the two uranium centers are five-coordinate with a distorted trigonal bipyramidal geometry. Each uranium center is bound by three N(SiMe3)2 ligands and two adjacent nitrogen atoms from the bpym ligand. Complex 1 is neutral, complexes 2 and 3 consist of negatively charged uranium complexes with one and two [K(2.2.2-cryptand)] counter cations, respectively, while complex 4 consists of a positively charged uranium complex with a BPh4 counter anion. For the latter, the asymmetric unit cell consists of two independent halves of the molecule.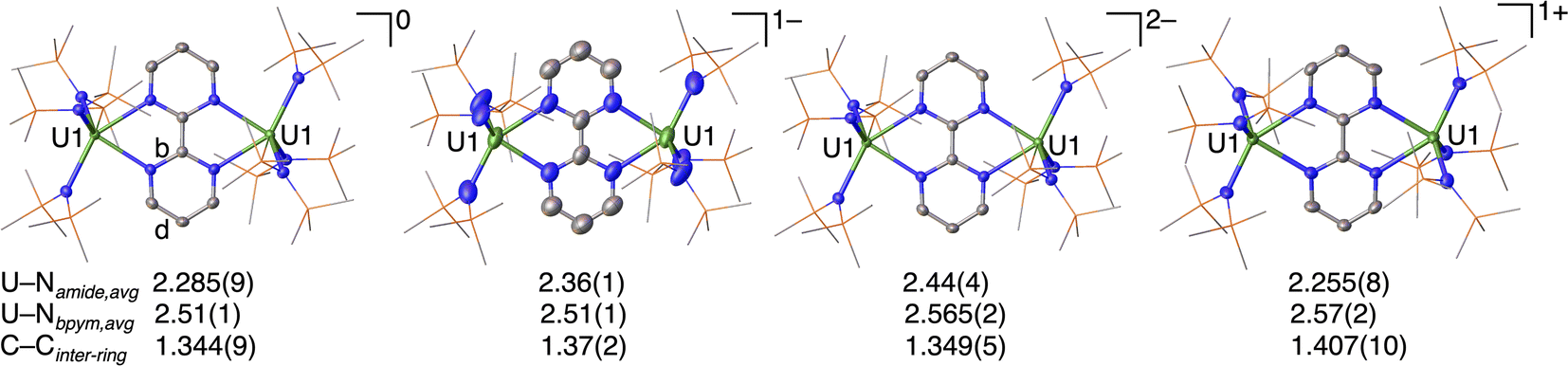 | ||
| Fig. 1 Molecular structures of [{((Me3Si)2N)3U}2(μ-bpym)], 1, [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-bpym)], 2, [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-bpym)], 3, and [{((Me3Si)2N)3U}2(μ-bpym)][BPh4], 4, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. For labeled structures and bond lengths see ESI.† U atoms are shown in green, N in blue, Si in orange and C in grey. | ||
Bpym is a redox-active ligand that can exist in three different oxidation states (0 to −2) and the inter-ring C–C bond (C–C bond between the two pyrimidyl groups) distance is the parameter most indicative of the degree of reduction. Notably, a value of 1.501(1) Å was reported for the inter-ring C–C bond distance in free bpym, while the inter-ring C–C bond distance of 1.359(1) Å found in the complex [(Cp*2Yb)2(μ-bpym)] was interpreted in terms of the presence of bpym2−.18 For comparison we also pursued the isolation of the one-electron reduced bpym ligand. Reduction with one equivalent of KC8 in the presence of 2.2.2-cryptand yielded a few crystals, characterised by X-ray spectroscopy as a mixture of the neutral and one-electron reduced bpym ligand, 5 (Fig. S16†). The C–Cinter-ring bond length of 1.506(3) Å for free bpym in the mixed crystal structure is in agreement with the reported value.18 An inter-ring C–C bond length of 1.423(4) Å was measured for [K(2.2.2-cryptand][bpym] that can be used as a reference for the compounds presented here.
The C–Cinter-ring bond lengths in 1 (1.344(9) Å) and 3 (1.349(5) Å) are in line with the presence of a double bond, indicating the two-electron reduction of the bpym ligand, similarly to what was observed by Andersen and colleagues in the bpym-bridged diytterbium complex (C1–C1′ 1.359(1) Å).18 The loss of aromaticity is further supported by the uneven distribution (Table S2, bonds b and d are elongated†) of bond lengths within the rings compared to free bpym.
In contrast, the C–Cinter-ring bond length in 4 (1.407(10) Å) is significantly longer than in 1 and 3, but shorter than in the free bpym ligand (1.506(3) Å), and close to the inter-ring C–C bond length of the one-electron reduced bpym ligand (1.423(4) Å). These features suggest that, based on structural data, 4, should be formulated as a diuranium(IV) complex bridged by a bpym˙− ligand. The inter-ring C–C bond length in 4 falls within the range previously reported for transition metal and lanthanide bpym˙− complexes (1.396(9)–1.432(18) Å).8a,19 Furthermore, the elongation of the bond lengths b and d (Table S2†) in the rings is less pronounced than in the bpym2− complexes 1 and 3.
In 2, the C–Cinter-ring bond length (1.37(2) Å), is similar to that found in 4. However, the error in the bond lengths of 2 is relatively high, due both to crystal quality and to the specific crystallographic symmetry, preventing a definitive assignment based on the structural data.
The mean value of the U1–Namide bond length increases progressively from 1 (2.285(9) Å), to 2 (2.36(1) Å) and 3 (2.44(4) Å). A similar increase of the U–N bond length is found from the U(IV)/U(IV) [{((Me3Si)2N)3U}2(μ-O)] (2.29(1) Å), to the U(III)/U(III) [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-O)], (2.430(5) Å) oxo-bridged complexes.16,20 The observed increase in U1–Namide length from 1 to 3, is consistent with the presence of U(IV)/U(IV) and U(III)/U(III) complexes, respectively. In the U(III)/U(IV) [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-O)] complex the U–N amide distances suggest a localised valence with mean values of U(IV)–N at 2.335(6) Å and U(III)–N at 2.381(6) Å. The mean value of the U–N bond lengths (2.36(1) Å) in 2 and the equivalence of the two uranium centers seems to indicate that 2 is better described as a U(III)–bpym˙−–U(III) complex but an alternative formulation as a U(III)–bpym2−–U(IV) with a delocalised valence cannot be completely ruled out. In 4, on the other hand, the mean value of the U1–Namide bond lengths (2.255(8) Å) is similar to that found in 1, providing further evidence for the assignment of 4 as a diuranium(IV) complex bridged by a (bpym)˙− ligand. Nevertheless, solid-state structures cannot always unambiguously assign oxidation states. Therefore, additional characterization techniques were used to corroborate assignments.
Electrochemistry
The observed chemical reductions and oxidation were further investigated with cyclic voltammetry for all complexes. The voltammogram of complex 1 (Fig. 2) shows two chemically reversible reduction waves II and I at E1/2 = −1.94 V and E1/2 = −2.55 V, corresponding to the reductions to complexes 2 and 3, respectively. Furthermore, one chemically reversible oxidation wave III was observed at E1/2 = −0.95 V, in line with the oxidation of the bpym ligand forming complex 4. The voltammograms of the complexes 2, 3, and 4 (Fig. 2) confirm this assignment and have similar half wave potentials, with the average potentials of the waves at E1/2 = −0.94 V, E1/2 = −1.92 V, and E1/2 = −2.54 V. Finally, no reduction wave could be observed after U(III)/U(III). This is in line with the chemical reactivity, which indicated no further reduction of complex 3 is possible. The obtained voltammograms were independent of the scan direction. The cyclic voltammogram of free bpym ligand (Fig. S17 and S22†) shows two chemically reversible waves at E1/2 = −2.47 V and E1/2 = −3.04 V, indicating that the reduction of the bpym ligand is facilitated by binding to uranium.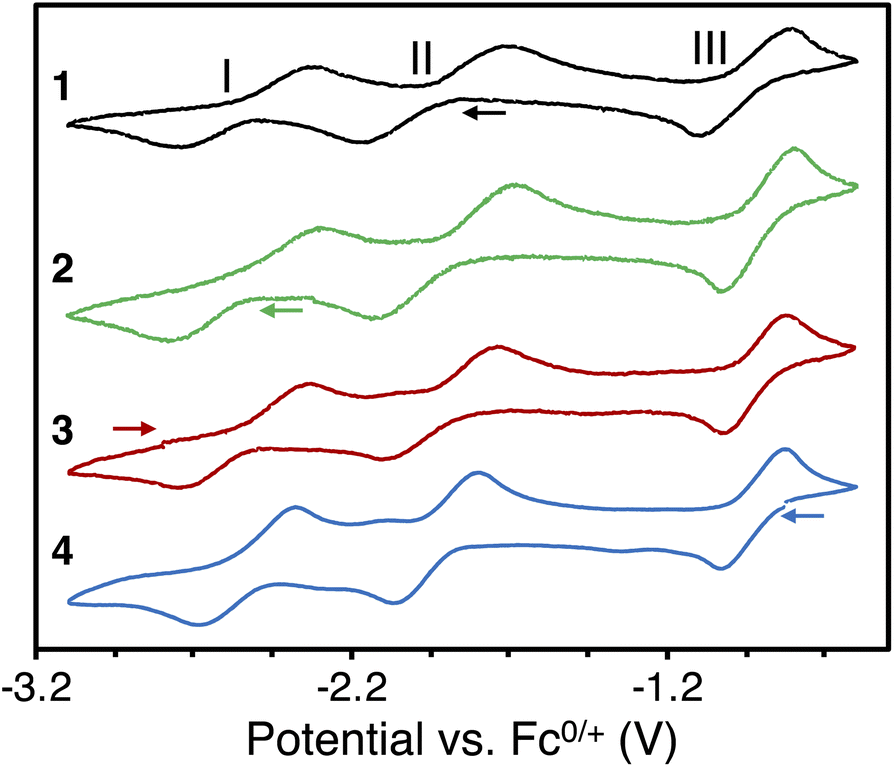 | ||
| Fig. 2 Cyclic voltammograms of [{((Me3Si)2N)3U}2(μ-bpym)], 1 (black), [K(2.2.2-cryptand)][{((Me3Si)2N)3U}2(μ-bpym)], 2 (green), [K(2.2.2-cryptand)]2[{((Me3Si)2N)3U}2(μ-bpym)], 3 (red), and [{((Me3Si)2N)3U}2(μ-bpym)][BPh4], 4 (blue), in THF/0.06 M [NBu4][BPh4] at a 50 mV s−1 scan rate versus Fc/Fc+ using a Pt0 disk as a working electrode (arrows indicate scan direction). | ||
The reductions to complex 2 (wave II) and complex 3 (wave I) show a large separation, both for the reduction and the oxidation waves. Theoretically, the reduction or oxidation of two equivalent non-interacting redox centers gives a separation of 36 mV. Larger separations, such as 240 mV for a uranium(IV) 1,4-phenylenediketimide-bridged complex, have been attributed to metal–metal communication.9b The separation of the reduction waves I and II varies between 560 and 640 mV and the separation of the oxidation waves between 570 and 650 mV (Table S4†). Such large separation could be assigned to strong metal–metal communication or, more likely, could indicate that the two waves correspond to different processes: one involving the metal reduction and one involving the ligand reduction. Notably, wave II can be assigned to the reduction of U(IV) to U(III), paired with an electron transfer from the bpym ligand to the other U(IV) center, resulting in a symmetric U(III)–bpym˙−–U(III) complex, while wave I corresponds to the reduction of the U(III)-bound bpym˙−ligand (the reduction of the U-bound bpym˙− to U-bound bpym2− is shifted towards more positive potentials compared to the reduction of the free bpym ligand (Fig. S17†) as anticipated upon binding to the metal). This assignment is consistent with the assignment of complex 2 as a U(III)–bpym˙−–U(III) species (see below magnetic and computational studies).
Magnetism and EPR spectroscopy
Susceptibility data for complexes 1–4 were recorded by performing a temperature scan in the 2–300 K range under an applied magnetic field of 0.1 T (Fig. 3 and 4). The χ versus T plots for 2, 3 and 4 show a continuous increase towards low temperatures, characteristic of magnetically isolated uranium centers, but the presence of weak magnetic coupling cannot be ruled out.21 The magnetic susceptibility curve of 1 also shows a continuous increase but starts to plateau around 12 K until rapidly rising below 5 K (Fig. S27†), possibly hinting at a weak magnetic interaction between the uranium centers, which is obscured by a paramagnetic impurity. However, it has been shown that such plateaus are also observed for isolated U(IV) ions in a singlet state at low temperature and are associated with the presence of non-negligible single-ion crystal field effects within the system.22 In contrast, the nitride bridged diuranium(IV) complex supported by N(SiMe3)2 ligands showed a clear antiferromagnetic coupling.23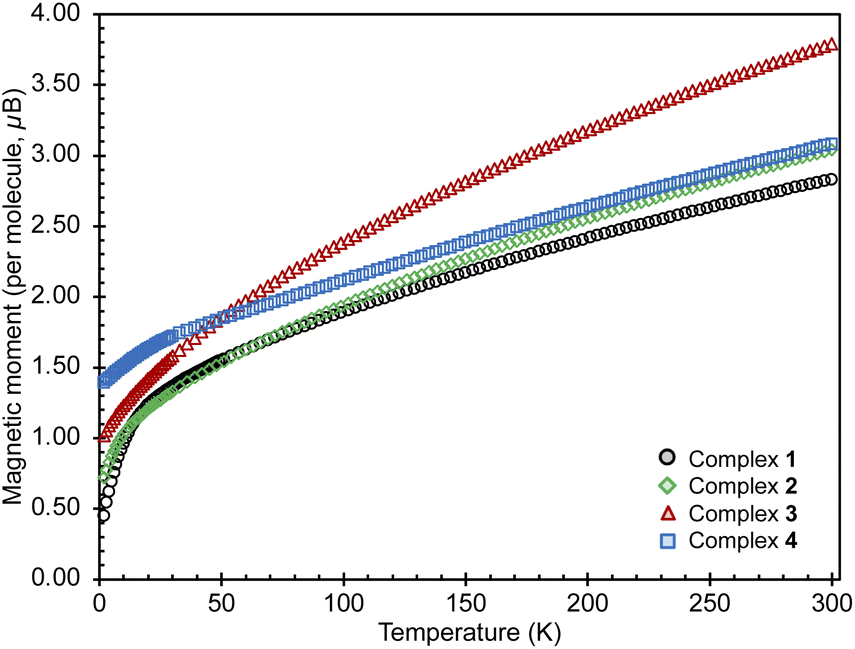 | ||
| Fig. 3 Magnetic moments for complexes 1–4, measured under an applied field of 0.1 T (1, black; 2, green; 3, red; 4, blue). | ||
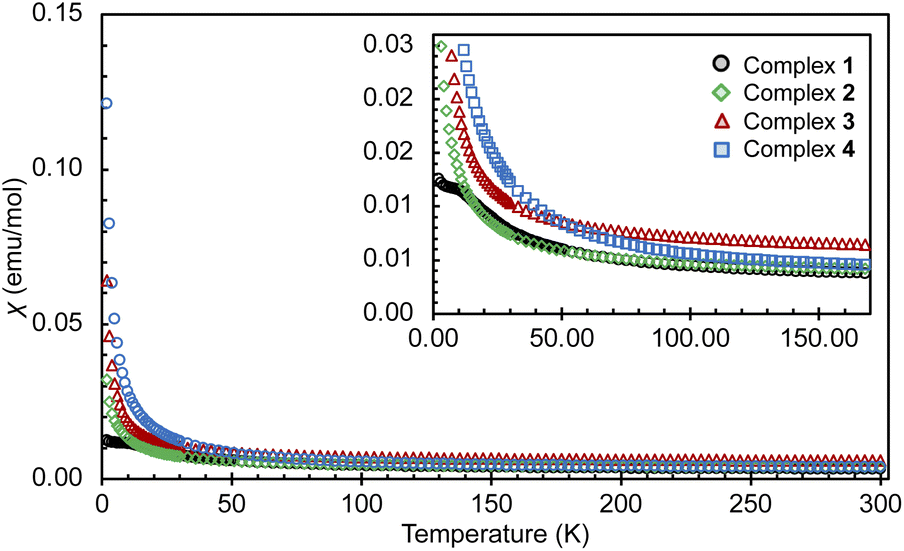 | ||
| Fig. 4 Overlay of χ versus T plots for complexes 1–4, measured under an applied field of 0.1 T up to 300 K and with a zoom up to 150 K (1, black; 2, green; 3, red; 4, blue). | ||
The χMT vs. T plots (Fig. S23 and S24†) for complexes 1–4 do not show any evidence of a maximum that could be associated with strong ferromagnetic coupling, but the possibility of weak magnetic interactions cannot be completely be ruled out. For complex 1 the χMT vs. T curve approaches zero when cooled to 2 K, thus confirming the presence of a non-magnetic 5f2 non-Kramers (3H4) ground state.
The range of magnetic moments of U(III), U(IV) and U(V) can overlap for various compounds, making it complicated to assign an oxidation state solely based on this.21a Nevertheless, in our systems the ligands are identical for all complexes, thus enabling a direct comparison.
The magnetic moments of 1 of 0.32 μB and 1.92 μB per uranium center at 2 K and 300 K (per complex: 0.45 μB at 2 K and 2.83 μB at 300 K), respectively, are in the range of those previously reported for bimetallic uranium(IV) complexes (2 K: 0.2–1.1 μB; 300 K: 1.65–5.17 μB).7f,g,7l,14b,15,21a,22,24 The values of the magnetic moment of 3 are higher compared to those measured for 1, being 0.67 μB and 2.74 μB per uranium center at 2 K and at 300 K, respectively (per complex: 1.01 μB at 2 K and 3.79 μB at 300 K). These values are within the range reported for bimetallic U(III) complexes (2 K: 0.24–0.74 μB; 300 K: 1.5–3.74 μB).7n,21a
The values of the magnetic moment of 2 at 2 K (0.72 μB per complex and 0.51 μB per uranium center) and at 300 K (3.04 μB per complex and 2.15 μB per uranium center) are slightly higher than those found for 1 but lower than those found for 3. Upon cooling the magnetic moment drops monotonically with a faster decrease below 20 K. The value of the magnetic moment at 2 K may denote an antiferromagnetic interaction between the moment of the radical bpym˙— and the U(III) in a UIII–bpym˙−–UIII complex. Complexes associating uranium(III) and radical ligands are rare9c,11a,11c,25 but a similar magnetic behavior was reported for two mononuclear uranium(III) complexes with a radical bipyridine or terpyridine ligand.11c,13,13a Alternatively the low magnetic moment could be interpreted in terms of the presence of a mixed valent complex UIII-bpym2—UIV (comparable magnetic data were previously reported for a nitride-bridged dinuclear U(III)/U(IV) complex [{((Me3Si)2N)2U(THF)}2(μ-N)]24d).
Upon examination of the magnetic behavior of 4 no evidence is seen of any features that could be attributed to magnetic coupling between two U(IV) centers, tentatively expected from a radical-bridged f-block compound. However, the rather large magnetic moments of 1.39 μB at 2 K and 3.08 μB at 300 K per complex are likely to result from the contribution of an unpaired electron present in the complex. The magnetic moment of 4 at 300 K is considerably higher than that of 1, in line with the presence of an extra unpaired electron and excluding the possibility of the presence of a UV–bpym2−–UIV complex. Furthermore, the high magnetic moment at 2 K, normally uncharacteristic for a U(IV) and not observed for complex 1, was also observed for monouranium(IV)-radical complexes, such as [((AdArO)3tacn)UIV(CO˙−)] (1.51 μB; (AdArOH)3tacn = 1,4,7-tris(3-adamantyl-5-tert-butyl-2-hydroxybenzyl)1,4,7-triazacyclononane),26, [((t−BuArO)3tacn)UIV(OC˙−t-BuPh2)] (1.61 μB at 5 K)27,MesPDIMe˙−UIVI3(THF) (1.5 μB; MesPDIMe = 2,6-(2,4,6-Me3-C6H2–N=CMe)2C5H3),28 the charge-separated amide radical, [U{OC˙(Ph)(NMe2)}(N(SiMe3)2)3]15 and for the radical bridged U(IV)/U(IV) complex [(MesPDIMe)˙3−UIVI]2 (1.03 μB).29 In these systems, the high values of the low temperature magnetic moments were attributed to magnetic contribution from the unpaired electron of the radical ligand [U(IV)L˙−] alone or combined to contributions from the [U(III)L0] resonance structure.26,27
In previous reports, ligand radical character in uranium-radical ligand complexes was supported by the observation of an EPR signal at room temperature at a g value around 2.12,15,29,30 Moreover, recently a mixed-valent U(III)/U(IV) triamidoamine complex bridged by the diradical dianion OCP˙2− unit was reported to possess a high magnetic moment (per complex) of 1.35 μB at 2 K and showed EPR signals both from the U(III) center and from the bridging radical (the weak nature of the signals was assigned to the presence of a U-radical antiferromagnetic coupling).31 However, in other reports, compounds were formulated as charge separated [U(IV)L˙−] systems and showed quite high low-temperature magnetic moments because of the contribution of the ligand radical, but no EPR signal could be observed, probably due to coupling between the U center and the radical.4h,32 An EPR-silent ground state arising from the coupling of the U(III) center with the bipy˙− radical was also reported for the charge separated [U(III)(Tp*)bpy˙−] (Tp*: tris(3,5- dimethyl-1-pyrazolyl)borate).25
In order to further investigate the electronic structure of the complexes, X-band EPR spectra were measured (Fig. 5). Complex 1 did not present any measurable EPR signal in the solid state, as expected for a system containing U(IV) ions.33 In turn, as shown in Fig. 5 complex 2 demonstrates a characteristic EPR spectrum with a pronounced narrow signal at low field around g1 = 4.98 (H0 = 1348 Gs) and two overlapping broad features shifted up-field to g2 = 0.65 (H0 = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 327 Gs) and g3 = 0.61 (H0 = 11004 Gs). Similar EPR spectra were reported for mononuclear uranium(III) complexes26,34 or for a radical-bridged dinuclear U(III)/U(IV) complex,31 where an EPR signal associated to the radical was also observed.
327 Gs) and g3 = 0.61 (H0 = 11004 Gs). Similar EPR spectra were reported for mononuclear uranium(III) complexes26,34 or for a radical-bridged dinuclear U(III)/U(IV) complex,31 where an EPR signal associated to the radical was also observed.
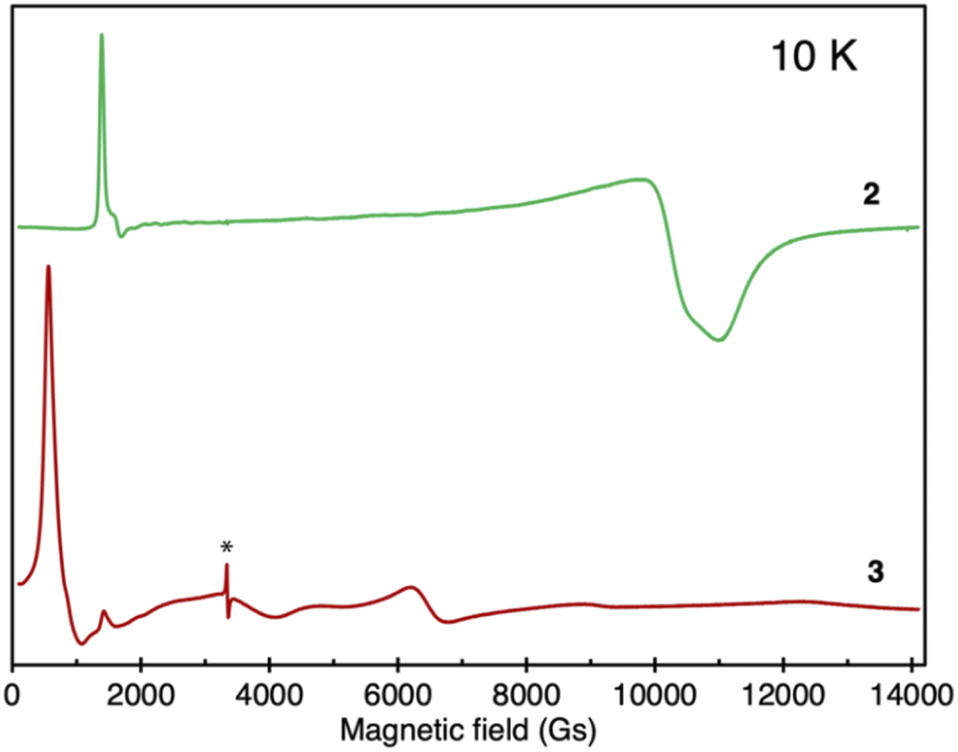 | ||
| Fig. 5 Comparison of solid-state EPR spectra of 2 (green) and 3 (red), recorded at 10 K; the asterisk denotes a bpym˙− radical signal. | ||
Accordingly, the observed EPR spectrum would be consistent with the formulation of 2 as a UIII–bpym2−–UIV species. However, an alternative formulation of 2 as a radical-bridged UIII–bpym˙−–UIII complex, where the bridging radical is not enabling efficient coupling between the two U(III) centers, is also conceivable and is supported by computational studies (vide infra). Notably, the two computed lowest spin states of complex 2 correspond to the presence of two U(III) for complex 2 with either an antiferromagnetically or ferromagnetically coupled spin on the ligand. Since these two states are very close in energy, the spin on the radical flips easily, which may lead to weak magnetic coupling and could explain the lack of an EPR signal. This is also consistent with the observed low magnetic moment.
In contrast, for complex 3 a very different EPR spectrum was observed (Fig. 5) with main features at g1 = 11.77 (H0 = 570 Gs) and g2 = 1.05 (H0 = 6530 Gs). EPR spectra with similar shapes were reported previously for U(III) multimetallic systems and were associated with weak ion-to-ion communication.35 Accordingly, the EPR spectrum recorded for complex 3 is consistent with the presence of two weakly coupled U(III) centers.
Finally, complex 4 is EPR-silent, ruling out the presence of a UIII–bpym0–U(IV) configuration. The absence of EPR signals has already been observed in mononuclear charge-separated U(IV)-radical compounds,4h,32 thus supporting the assignment of 4 as a diuranium(IV) radical-bridged complex.
In sum, complexes 2 and 4 can be assigned as radical-bridged diuranium(III) and diuranium(IV) complexes, respectively, indicating that such systems are accessible for uranium. However, if some radical-promoted magnetic communication between the U centers exists, it could not be unambiguously detected by the variable temperature susceptibility measurements. This suggests that the combination of the bpym ligand with amide supporting ligands, while favoring the formation of radical-bridged species, is not optimal to promote strong magnetic communication between the metal ions. Notably, recent studies on nitride-bridged diuranium(IV) complexes showed that for analogous bridging groups, the nature of the supporting ligand has a decisive influence on the presence or absence of magnetic coupling.23
In order to probe for any slow magnetic relaxation phenomena, compounds 2–4 were investigated by employing alternating current (AC) magnetometry at low temperatures (1.8–2.2 K), under an applied field of 0.2 T. Complex 2 presented a frequency-induced response in the measured range of up to 1 kHz, characterised by a shifting maximum in the out-of-phase (χ′′) magnetic susceptibility (Fig. S50†). The energy barrier, ΔE/kB, was best determined from the linear fit to the Arrhenius law τ = τ0 exp(ΔE/kB T) and was found to be 12.84(3) K (or 8.92(2) cm−1) with τ0 = 8.87(23) · 10−7 s (Fig. S51†). Even though the data only allowed for an approximate estimate, the barrier value is comparable with previously reported U(III) examples, such as (BDI)U(ODipp)2 (Ueff = 14.5(1) cm−1),4l, [U(N(SiMe3)2)4]− (Ueff = 16 cm−1) and [U(OSi(OtBu)3)4]− (Ueff = 18 cm−1)4g [U-(BIPMTMS)(I)2(THF)][BIPMTMS = CH(PPh2NSiMe3)2] (Ueff = 23.4 (0.8) K or 16.3(0.6) cm−1).36 The behavior of complex 2 is compatible either with the presence of a single U(III) ion (formulation of 2 as a UIII–bpym2−–UIV species) or with a UIII–bpym˙−–UIII species with an ineffective coupling between the two UIII centers. In contrast, 3, containing two U(III) centers bridged by dianionic bipyrimidine, did not show any out-of-phase components of the dynamic susceptibility at an applied field of 0.2 T (Fig. S53†). Since no slow relaxation is observed for 3, the behaviour observed for 2 could be assigned to the presence of the bridging radical bpym˙− which enhances coupling and reduces quantum tunneling. Slow magnetic relaxation was previously reported in one case for a mononuclear U(III) complex with a radical ligand and it was shown that U(III)-radical coupling removed quantum tunnelling mechanism under zero static field.11c
Complex 4 manifests a better-defined maximum in out-of-phase (χ′′) susceptibility compared to 2, and the ΔE/kB barrier was estimated to be 2.81(5) K (or 1.95(3) cm−1) with τ0 = 6.38(10) · 10−4 s (Fig. S55 and S56†). A similar SMM behavior was observed for the U(IV) radical-containing SIMs, [{(SiMe2NPh)3-tacn}UIV(η2-N2Ph2˙)].4h
Computational studies
Calculations were carried out at the DFT level (B3PW91) on complexes 1–4. The geometries were fully optimised in all cases for different spin states (see Table S7†). It should be noted that all the electrons of the two metals are considered without including any coupling interaction. For the neutral complex 1, the lowest spin state was found to be a quintet (S = 2), which means four unpaired electrons, consistent with two U(IV) centers and a doubly reduced bpym ligand. A septet (S = 3), which corresponds to six unpaired electrons, in line with two U(III) and an unreduced bpym ligand, was also optimised but lies 7.3 kcal mol−1 above the quintet (at the DFT level, the sextet corresponds to three unpaired electrons at each U(III) and therefore does not necessarily imply any coupling between the two U(III) centers). To confirm the oxidation states of the uranium centers in 1, the unpaired spin density was scrutinised and two unpaired electrons per uranium were found (Table S10 and Fig. S58a†) further corroborating the experimental assignment of the uranium oxidation states. The double reduction of the bpym ligand is highlighted in the molecular orbital diagram of 1 where the HOMO-4 that is doubly occupied corresponds to the bpym π* (Fig. 6a). Moreover, in the optimised geometry of 1 in the quintet spin state, the C–Cinter-ring bond length is 1.35 Å, in excellent agreement with the experimental value, which corresponds to a localised C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1.33 Å in ethylene) and is in line with the bpym π* being doubly occupied.
C (1.33 Å in ethylene) and is in line with the bpym π* being doubly occupied.
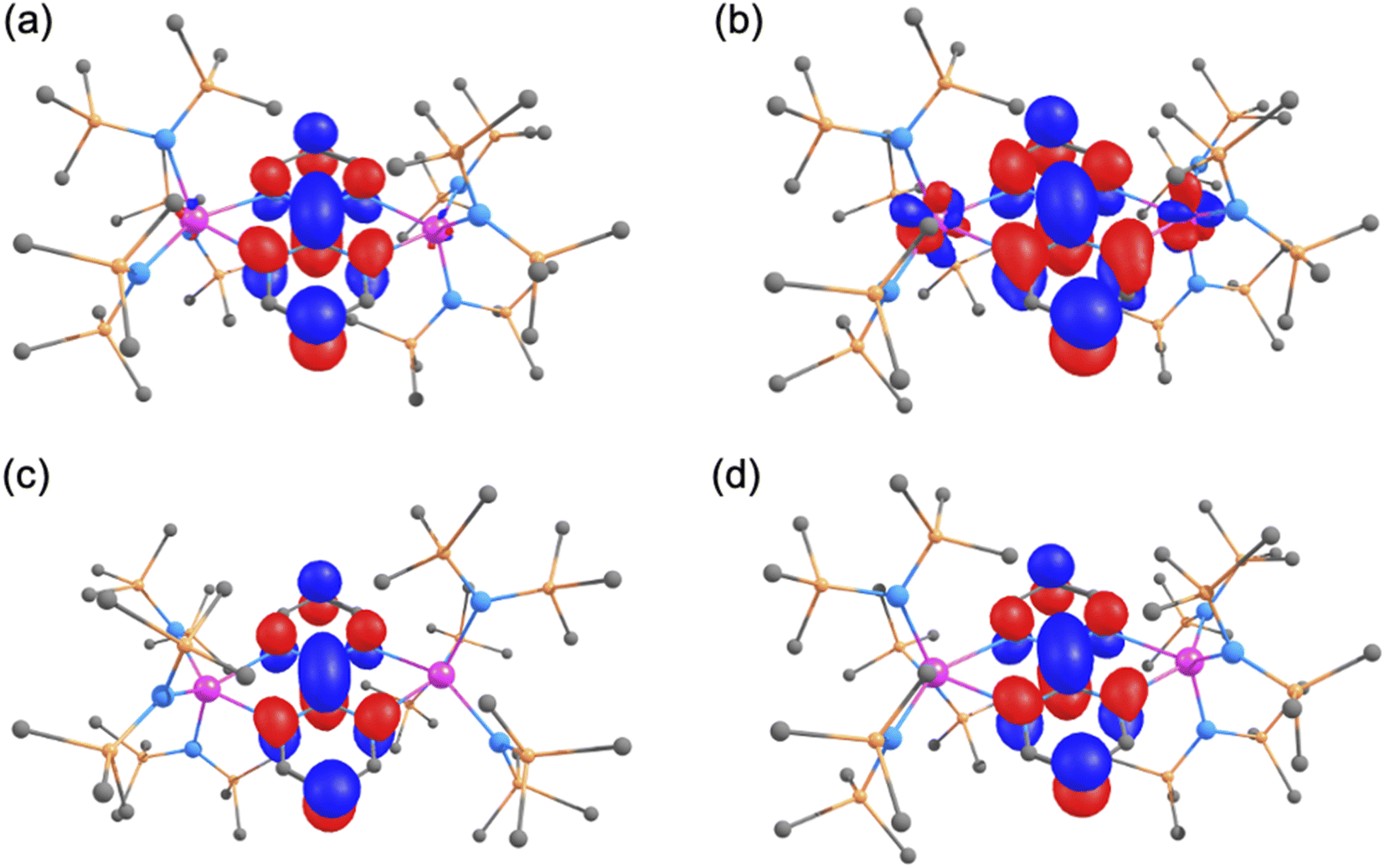 | ||
| Fig. 6 3D depiction of (a) the HOMO-4 of complex 1, (b) the SOMO of complex 2, (c) the HOMO-6 of complex 3 and (d) the SOMO of complex 4. | ||
The singly reduced form of complex 1, complex 2, was investigated using the same methodology. In this case, two spin states (a sextet and an octet) were found to be almost degenerate, the sextet being 1.8 kcal mol−1 lower than the octet. It is interesting to note that both spin states are displaying the presence of two U(III) and a radical anion bpym ligand (an antiferromagnetic coupling between the uranium centers and the ligand radical was found in the sextet and a ferromagnetic coupling in the octet). Due to the relatively small energy difference, multireference (CASSCF) calculations were also carried out in this system by distributing 7 electrons into 7 orbitals. The lowest state is a sextet which corresponds to a mixture of the UIII–bpym˙−–UIII (65%) and UIII–bpym2−–UIV (35%), the octet being 2.3 kcal mol−1 higher in energy. Therefore, the multireference character of the sextet ground state, as well as the very low energy required for the spin flip at the bpym radical do not allow efficient magnetic coupling between the different unpaired electrons. The SOMO of the system (Fig. 6b) is the bpym π* and the unpaired spin densities (Table S10 and Fig. S58b†) highlight the presence of two U(III) and one bpym radical. The optimised C–Cinter-ring bond length is slightly longer than in complex 1 at 1.40 Å (1.37(2) Å experimentally) and similar to that observed in benzene (1.39 Å). This also suggests that the bpym π* is not doubly occupied but rather singly occupied. It is very interesting to note that the reduction of complex 1 appears to occur at the metal center in this case and a concomitant oxidation occurs at the bpym ligand to yield a UIII–bpym˙−–UIII species. This is in line with the reversible electron transfer from f-elements to N-heterocylic derivatives that was demonstrated by Andersen and Nocton in lanthanide chemistry.37
The doubly reduced form, complex 3, displays features similar to complex 1. Indeed, although different spin states were investigated (Table S7†), the ground state is a septet (S = 3) with the quintet (S = 2) far higher in energy (+16.0 kcal mol−1). Both the unpaired spin density (Table S10 and Fig. S58c†) and the nature of the HOMO-6 (Fig. 6c) are indicating the presence of two U(III) centers with a doubly reduced bpym ligand. The optimised C–Cinter-ring of 1.36 Å (experimental value: 1.349(5) Å) is in line with a localised double bond and a doubly occupied bpym π*. Thus, the second reduction takes place at the bpym ligand.
Finally, the oxidised form of complex 1, complex 4, was investigated. In line with our observation for the first reduction, two spin states (quartet and sextet) are lying at the same energy (quartet being 1.5 kcal mol−1 lower than the sextet). Like complex 2, the two spin states only differ by a spin flip at the bpym radical so that two U(IV) centers are present. Therefore, CASSCF calculations were carried out on this system by distributing 5 electrons into 5 orbitals. The ground state is found to be the quartet which is essentially described by a single configuration UIV–bpym˙−–UIV (98%). The sextet is lying 2.1 kcal mol−1 higher in energy. This is further highlighted by the unpaired spin density values of 2.18 on each U (Table S10 and Fig. S58d†) and the nature of the SOMO of the system (Fig. 6d). The optimised C–Cinter-ring of 1.41 Å (experimental value: 1.407(10) Å) is indicative of a partial occupation of the bpym π* as already found in complex 2. This shows that the oxidation occurs at the bpym ligand, as expected. As already mentioned for complex 2, the easy spin flip of the electron at the bpym radical may prevent coupling between the different unpaired electrons and could explain the lack of an organic radical EPR signal in this complex.
Conclusions
The redox-active bpym ligand is a particularly attractive linker that was found to promote magnetic coupling in lanthanides resulting in complexes displaying exchange coupling and single molecule magnet behavior. Here we have identified convenient routes for the synthesis of dinuclear complexes of uranium in low oxidation states where the two uranium centers are bridged by the redox-active bpym ligand. The neutral UIV-bpym2−–UIV complex 1 was prepared by reaction of the UIII complex [U{N(SiMe3)2}3] with neutral bpym. The one and two electron chemical reduction of 1 yielded the mono-anionic and di-anionic complexes 2 and 3, respectively, while one electron oxidation afforded the cationic complex 4. The four complexes show analogous solid-state structures but different metrical parameters. The U–N distances and the value of the inter-ring C–C bond distance of the bpym ligand provide indication of charge localization in these complexes which was further corroborated by magnetic and EPR studies. Magnetic and EPR data are in agreement with the assignment of 3 as a UIII–bpym2−–UIII species and of 4 as a radical-bridged diuranium(IV) complex UIV–bpym˙−–UIV species. The assignment was also corroborated by computational studies. The electron configuration of complex 2 is more ambiguous despite all the structural, magnetic and EPR analysis and both formulations of 2 as a UIII–bpym2−–UIV or a UIII–bpym˙−–UIII species could be possible. However, computational study indicated that complex 2 is best identified as a radical-bridged diuranium(III) complex. Cyclic voltammetry studies show similar quasi-reversible waves for all complexes confirming that they can be reversibly transformed into each other by one electron reduction or oxidation. The large separation in the reduction and oxidation waves is suggestive of different reduction processes (metal and ligand), supporting the assignment of 2 as UIII–bpym˙−–UIII. The measurements of the variable temperature magnetic susceptibility did not show evidence of strong magnetic coupling, but presence of weak magnetic coupling cannot be ruled out. Notably, AC susceptibility measurements revealed a slow magnetic relaxation behavior for the UIII–bpym˙−–UIII complex 2 which is not observed in the UIII–bpym2−–UIII complex 3 and that could be interpreted in terms of a weak radical-based exchange.In summary, these results show that radical-bridged low-valent uranium complexes can be accessed using simple amide supporting ligands and that the radical bridge leads to slow magnetic relaxation in these systems. These findings pave the way for the synthesis of radical-bridged U(III) complexes and suitable tuning of the supporting ligand should lead to improved magnetic communication and exchange-coupled SMM's.
Data availability
All data were deposited in ESI.†Author contributions
D. K. M. and M. S. B. carried out the synthetic experiments and analysed the experimental data. R. S. carried out the X-ray single crystal structure analyses. I. Z. and M. S. B. performed the magnetic measurements and analyzed the magnetic data. A. S. and M. S. B. recorded and interpreted the EPR spectra. T. R. and L. M. carried out and analysed the computational data. M. M. originated the central idea, coordinated the work, and analysed the experimental data. D. K. M., M. S. B. and M. M. wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation 200021_ 178793 and from the École Polytechnique Fédérale de Lausanne (EPFL). We thank Roxane Moinat for carrying out the elemental analyses, and Farzaneh Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses.Notes and references
- (a) S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Nat. Commun., 2017, 8, 1–9 CrossRef CAS PubMed; (b) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed; (c) F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed; (d) C. A. Gould, K. R. McClain, D. Reta, J. G. C. Kragskow, D. A. Marchiori, E. Lachman, E. S. Choi, J. G. Analytis, R. D. Britt, N. F. Chilton, B. G. Harvey and J. R. Long, Science, 2022, 375, 198–202 CrossRef CAS PubMed.
- (a) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed; (b) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed; (c) S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701–4711 RSC.
- (a) J. D. Rinehart, T. D. Harris, S. A. Kozimor, B. M. Bartlett and J. R. Long, Inorg. Chem., 2009, 48, 3382–3395 CrossRef CAS PubMed; (b) R. A. Layfield, Organometallics, 2014, 33, 1084–1099 CrossRef CAS; (c) S. T. Liddle and J. Van Slageren, Lanthanides and Actinides in Molecular Magnetism, Wiley-VCH, 2015 Search PubMed; (d) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC; (e) K. R. Meihaus and J. R. Long, Dalton Trans., 2015, 44, 2517–2528 RSC; (f) S. G. McAdams, A. M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS; (g) S. Dey and G. Rajaraman, J. Chem. Sci., 2019, 131 Search PubMed; (h) S. Dey, G. Velmurugan and G. Rajaraman, Dalton Trans., 2019, 48, 8976–8988 RSC; (i) S. Dey and G. Rajaraman, Inorg. Chem., 2022, 61, 1831–1842 CrossRef CAS PubMed.
- (a) J. D. Rinehart and J. R. Long, J. Am. Chem. Soc., 2009, 131, 12558–12559 CrossRef CAS PubMed; (b) J. D. Rinehart, K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7572–7573 CrossRef CAS PubMed; (c) M. A. Antunes, L. C. J. Pereira, I. C. Santos, M. Mazzanti, J. Marçalo and M. Almeida, Inorg. Chem., 2011, 50, 9915–9917 CrossRef CAS PubMed; (d) K. R. Meihaus, J. D. Rinehart and J. R. Long, Inorg. Chem., 2011, 50, 8484–8489 CrossRef CAS PubMed; (e) J. T. Coutinho, M. A. Antunes, L. C. J. Pereira, H. Bolvin, J. Marçalo, M. Mazzanti and M. Almeida, Dalton Trans., 2012, 41, 13568–13571 RSC; (f) J. D. Rinehart and J. R. Long, Dalton Trans., 2012, 41, 13572–13574 RSC; (g) L. C. J. Pereira, C. Camp, J. T. Coutinho, L. Chatelain, P. Maldivi, M. Almeida and M. Mazzanti, Inorg. Chem., 2014, 53, 11809–11811 CrossRef CAS PubMed; (h) M. A. Antunes, J. T. Coutinho, I. C. Santos, J. Marçalo, M. Almeida, J. J. Baldovi, L. C. J. Pereira, A. Gaita-Ariño and E. Coronado, Chem. - Eur. J., 2015, 21, 17817–17826 CrossRef CAS PubMed; (i) J. J. Le Roy, S. I. Gorelsky, I. Korobkov and M. Murugesu, Organometallics, 2015, 34, 1415–1418 CrossRef CAS; (j) M. A. Boreen, D. J. Lussier, B. A. Skeel, T. D. Lohrey, F. A. Watt, D. K. Shuh, J. R. Long, S. Hohloch and J. Arnold, Inorg. Chem., 2019, 58, 16629–16641 CrossRef CAS PubMed; (k) F. S. Guo, Y. C. Chen, M. L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2019, 58, 10163–10167 CrossRef CAS PubMed; (l) M. A. Boreen, C. A. Gould, C. H. Booth, S. Hohloch and J. Arnold, Dalton Trans., 2020, 49, 7938–7944 RSC.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CrossRef CAS.
- (a) V. Mougel, L. Chatelain, J. Pécaut, R. Caciuffo, E. Colineau, J. C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed; (b) L. Chatelain, J. P. S. Walsh, J. Pécaut, F. Tuna and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 13434–13438 CrossRef CAS PubMed; (c) V. Mougel, L. Chatelain, J. Hermle, R. Caciuffo, E. Colineau, F. Tuna, N. Magnani, A. de Geyer, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2014, 53, 819–823 CrossRef CAS PubMed; (d) L. Chatelain, J. Pécaut, F. Tuna and M. Mazzanti, Chem. - Eur. J., 2015, 21, 18038–18042 CrossRef CAS; (e) L. Chatelain, F. Tuna, J. Pécaut and M. Mazzanti, Dalton Trans., 2017, 46, 5498–5502 RSC.
- (a) R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS; (b) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS; (c) G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS PubMed; (d) V. Mougel, P. Horeglad, G. Nocton, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2009, 48, 8477–8480 CrossRef CAS PubMed; (e) L. P. Spencer, E. J. Schelter, P. Yang, R. L. Gdula, B. L. Scott, J. D. Thompson, J. L. Kiplinger, E. R. Batista and J. M. Boncella, Angew. Chem., Int. Ed., 2009, 48, 3795–3798 CrossRef CAS; (f) B. S. Newell, A. K. Rappé and M. P. Shores, Inorg. Chem., 2010, 49, 1595–1606 CrossRef CAS PubMed; (g) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC; (h) P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed; (i) L. Chatelain, V. Mougel, J. Pécaut and M. Mazzanti, Chem. Sci., 2012, 3, 1075–1079 RSC; (j) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed; (k) B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens, Jr., L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef; (l) A.-C. Schmidt, F. W. Heinemann, W. W. Lukens, Jr. and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed; (m) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC; (n) M. Falcone, L. Barluzzi, J. Andrez, F. Fadaei-Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed.
- (a) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546–18549 CrossRef CAS; (b) S. Demir, I. R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289, 149–176 CrossRef; (c) C. A. Gould, L. E. Darago, M. I. Gonzalez, S. Demir and J. R. Long, Angew. Chem., Int. Ed., 2017, 56, 10103–10107 CrossRef CAS PubMed.
- (a) E. J. Schelter, J. M. Veauthier, J. D. Thompson, B. L. Scott, K. D. John, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2006, 128, 2198–2199 CrossRef CAS; (b) E. J. Schelter, J. M. Veauthier, C. R. Graves, K. D. John, B. L. Scott, J. D. Thompson, J. A. Pool-Davis-Tournear, D. E. Morris and J. L. Kiplinger, Chem. - Eur. J., 2008, 14, 7782–7790 CrossRef CAS PubMed; (c) E. J. Schelter, R. Wu, B. L. Scott, J. D. Thompson, D. E. Morris and J. L. Kiplinger, Angew. Chem., Int. Ed., 2008, 47, 2993–2996 CrossRef CAS PubMed.
- (a) T. Mehdoui, J. C. Berthet, P. Thuéry and M. Ephritikhine, Eur. J. Inorg. Chem., 2004, 1996–2000 CrossRef CAS; (b) J. C. Berthet, P. Thuéry, C. Baudin, B. Boizot and M. Ephritikhine, Dalton Trans., 2009, 7613–7616 RSC.
- (a) S. J. Kraft, P. E. Fanwick and S. C. Bart, Inorg. Chem., 2010, 49, 1103–1110 CrossRef CAS PubMed; (b) T. Mehdoui, J. C. Berthet, P. Thuery and M. Ephritikhine, 2013, Private Communication to the CSD 2013, CCDC 958634; (c) J. T. Coutinho, M. A. Antunes, L. C. J. Pereira, J. Marçalo and M. Almeida, Chem. Commun., 2014, 50, 10262–10264 RSC.
- M. W. Rosenzweig, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2017, 56, 2792–2800 CrossRef CAS PubMed.
- (a) T. Mehdoui, J. C. Berthet, P. Thuéry, L. Salmon, E. Rivière and M. Ephritikhine, Chem. - Eur. J., 2005, 11, 6994–7006 CrossRef CAS PubMed; (b) G. F. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS; (c) M. K. Takase, M. Fang, J. W. Ziller, F. Furche and W. J. Evans, Inorg. Chim. Acta, 2010, 364, 167–171 CrossRef CAS; (d) A. Mohammad, D. P. Cladis, W. P. Forrest, P. E. Fanwick and S. C. Bart, Chem. Commun., 2012, 48, 1671–1673 RSC; (e) P. L. Diaconescu and C. C. Cummins, Dalton Trans., 2015, 44, 2676–2683 RSC; (f) S. Fortier, J. Veleta, A. Pialat, J. Le Roy, K. B. Ghiassi, M. M. Olmstead, A. Metta-Magaña, M. Murugesu and D. Villagrán, Chem. - Eur. J., 2016, 22, 1931–1936 CrossRef CAS PubMed; (g) L. Zhang, C. Zhang, G. Hou, G. Zi and M. D. Walter, Organometallics, 2017, 36, 1179–1187 CrossRef CAS.
- (a) C. Camp, V. Mougel, J. Pécaut, L. Maron and M. Mazzanti, Chem. - Eur. J., 2013, 19, 17528–17540 CrossRef CAS PubMed; (b) S. Hohloch, J. R. Pankhurst, E. E. Jaekel, B. F. Parker, D. J. Lussier, M. E. Garner, C. H. Booth, J. B. Love and J. Arnold, Dalton Trans., 2017, 46, 11615–11625 RSC.
- K. C. Mullane, T. Cheisson, E. Nakamaru-Ogiso, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem. - Eur. J., 2018, 24, 826–837 CrossRef CAS PubMed.
- D. K. Modder, C. T. Palumbo, I. Douair, F. Fadaei-Tirani, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed., 2021, 60, 3737–3744 CrossRef CAS PubMed.
- D. K. Modder, C. T. Palumbo, I. Douair, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2021, 12, 6153–6158 RSC.
- D. J. Berg, J. M. Boncella and R. A. Andersen, Organometallics, 2002, 21, 4622–4631 CrossRef CAS.
- (a) V. Goudy, A. Jaoul, M. Cordier, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2017, 139, 10633–10636 CrossRef CAS PubMed; (b) D. Wang, J. Moutet, M. Tricoire, M. Cordier and G. Nocton, Inorganics, 2019, 7 CAS; (c) C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS PubMed.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed.
- (a) D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865–8882 CrossRef CAS PubMed; (b) S. Mugiraneza and A. M. Hallas, Commun. Phys., 2022, 5, 1–12 CrossRef.
- B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207–6217 RSC.
- C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840–8849 RSC.
- (a) B. S. Newell, T. C. Schwaab and M. P. Shores, Inorg. Chem., 2011, 50, 12108–12115 CrossRef CAS PubMed; (b) S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC; (c) B. M. Gardner, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 7068–7072 CrossRef CAS PubMed; (d) C. T. Palumbo, R. Scopelliti, I. Zivkovic and M. Mazzanti, J. Am. Chem. Soc., 2020, 142, 3149–3157 CrossRef CAS.
- E. M. Matson, J. J. Kiernicki, N. H. Anderson, P. E. Fanwick and S. C. Bart, Dalton Trans., 2014, 43, 17885–17888 RSC.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757–1759 CrossRef CAS PubMed.
- O. P. Lam, C. Anthon, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 6567–6576 CrossRef CAS PubMed.
- N. H. Anderson, S. O. Odoh, Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed.
- N. H. Anderson, S. O. Odoh, U. J. Williams, A. J. Lewis, G. L. Wagner, J. L. Pacheco, S. A. Kozimor, L. Gagliardi, E. J. Schelter and S. C. Bart, J. Am. Chem. Soc., 2015, 137, 4690–4700 CrossRef CAS PubMed.
- J. J. Kiernicki, R. F. Higgins, S. J. Kraft, M. Zeller, M. P. Shores and S. C. Bart, Inorg. Chem., 2016, 55, 11854–11866 CrossRef CAS PubMed.
- R. Magnall, G. Balázs, E. Lu, F. Tuna, A. J. Wooles, M. Scheer and S. T. Liddle, Angew. Chem., Int. Ed., 2019, 58, 10215–10219 CrossRef CAS PubMed.
- O. P. Lam, P. L. Feng, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 2806–2816 CrossRef CAS PubMed.
- E. Soulie, G. Folcher and B. Kanellakopulos, Can. J. Chem., 1980, 58, 2377–2379 CrossRef CAS.
- (a) I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC; (b) N. J. Wolford, X. Yu, S. C. Bart, J. Autschbach and M. L. Neidig, Dalton Trans., 2020, 49, 14401–14410 RSC; (c) A. Formanuik, F. Ortu, I. J. Vitorica-Yrezabal, F. Tuna, E. J. L. McInnes, L. S. Natrajan and D. P. Mills, Inorganics, 2021, 9, 86 CrossRef CAS.
- A. J. Wooles, D. P. Mills, F. Tuna, E. J. L. McInnes, G. T. W. Law, A. J. Fuller, F. Kremer, M. Ridgway, W. Lewis, L. Gagliardi, B. Vlaisavljevich and S. T. Liddle, Nat. Commun., 2018, 9, 2097 CrossRef PubMed.
- F. Moro, D. P. Mills, S. T. Liddle and J. Slangeren, Angew. Chem., Int. Ed., 2013, 52, 3430–3433 CrossRef CAS PubMed.
- (a) M. Schultz, J. M. Boncella, D. J. Berg, T. D. Tilley and R. A. Andersen, Organometallics, 2002, 21, 460–472 CrossRef CAS; (b) C. H. Booth, M. D. Walter, D. Kazhdan, Y. J. Hu, W. W. Lukens, E. D. Bauer, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2009, 131, 6480–6491 CrossRef CAS PubMed; (c) G. Nocton and L. Ricard, Chem. Commun., 2015, 51, 3578–3581 RSC; (d) R. L. Halbach, G. Nocton, J. I. Amaro-Estrada, L. Maron, C. H. Booth and R. A. Andersen, Inorg. Chem., 2019, 58, 12083–12098 CrossRef CAS PubMed; (e) M. Tricoire, N. Mahieu, T. Simler and G. Nocton, Chem. - Eur. J., 2021, 27, 6860–6879 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2181473–2181476, 2181478. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03592h |
| ‡ D. K. M. and M. S. B. contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |