DOI:
10.1039/D2SC03861G
(Edge Article)
Chem. Sci., 2022,
13, 10342-10348
Nucleus-selective codelivery of proteins and drugs for synergistic antitumor therapy†
Received
11th July 2022
, Accepted 2nd August 2022
First published on 22nd August 2022
Abstract
Subcellular organelle targeted transport is of great significance for accurately delivering drugs to active sites for better pharmacological effects, but there are still a lot of challenges due to transport problems. In addition, the killing effect of one kind of drug on cells is limited. Therefore, it is necessary to develop a multifunctional nanoplatform that can co-deliver synergistic therapeutic agents. Here, we prepare a simple amphiphilic nanocarrier (LC) with rapid endosomal escape ability for nucleus-selective delivery of hydrophilic active protein deoxyribonuclease I (DNase I) and hydrophobic anticancer drug doxorubicin (DOX). LC has been applied to effectively encapsulate DNase I just by simply mixing their aqueous solutions together. In addition, DOX modified with adamantane groups via a redox-responsive linker is incorporated into the architecture of DNase I nanoformulations through host–guest interaction. This multi-component nanoplatform can quickly escape from the endolysosomes into the cytoplasm and make DNase I and DOX highly accumulate in the nucleus and consequently induce strong synergistic anticancer efficacy both in vitro and in vivo. This work illustrates a new platform for codelivery of proteins and drugs that target subcellular compartments for functions.
Introduction
In the past few years, organelle targeted therapy has become a hopeful anti-cancer method. Compared with apoptosis-related subcellular compartments such as lysosomes and mitochondria, the nucleus plays an important role in controlling cell proliferation and differentiation. It is highly sensitive to diverse DNA damages and is a promising target for anti-tumor therapy. Therapeutics based on proteins has attracted increasing attention due to its great potential to revolutionize medicine.1–5 However, most of the current clinically applied therapeutic proteins are based on extracellular targets.6–9 Limited by the disadvantages of a fragile tertiary structure, easy degradation, and poor cell membrane permeability, functional proteins with biological activity in cells have not been vastly studied in clinical applications.10,11 Therefore, it is extremely important to develop effective delivery systems to deliver proteins to their intracellular targets for expanding the scope of therapeutic protein applications.12–17 To date, plenty of protein delivery technologies have been explored for assisting cytosolic delivery, such as polymeric nanoparticles,18–20 lipid nanoparticles,21–23 inorganic nanoparticles,24–27 DNA nanomaterials,28,29 and the direct incorporation of cell penetrating peptides (CPPs).30 However, most of these methods are delivering protein just to the cytoplasm. Therefore, it is of great significance to develop carriers which can deliver proteins to the nucleus.
Except for single protein therapy, the combination with small molecule chemotherapeutics offers a novel approach for more effective and safer tumor combination therapy.31–33 However, it is still very challenging to develop an efficient co-delivery strategy to deliver proteins and chemotherapeutics directly into their desired subcellular compartments, such as the nucleus. First, the inherent hydrophilicity of large proteins and the hydrophobic structure of small molecule drugs make it difficult to integrate them together.34,35 Second, the stability of the co-delivery system is essential for effective cancer treatment, since premature release of the cargo can lead to undesirable adverse effects and reduced tumor accumulation. The co-delivery systems based on electrostatic or hydrophobic interactions to incorporate chemotherapeutics into protein nanoformulations often suffer from poor stability and early drug leakage, which restricts their further application.36 Moreover, the most fatal defect of the existing alternative approaches is that they cannot effectively escape from lysosomes to avoid enzymatic degradation,37 which is crucial for maintaining protein activity and promoting their cytoplasmic transport to the required subcellular targets for better biological functions. Therefore, it is urgent to develop efficient co-delivery strategies to deliver native proteins and chemotherapeutics into cells, and even the nucleus.
Here, we present an efficient nucleus-targeted co-delivery nanoplatform with high endosomal escape ability to transport proteins and drugs into the nucleus for synergistically enhanced cancer therapy. Two representative nucleus-targeted agents, DNase I and DOX were used to validate our design strategy. DNase I is a negatively charged protein which can induce tumor cell death by degrading DNA mainly located in the nucleus. And DOX is a hydrophobic small molecule drug that can induce apoptosis by inserting nuclear DNA. The structure of LC and its principal process for intranuclear co-delivery of DNase I and DOX are illustrated in Scheme 1. The multi-arm amphiphilic structure of LC made it easy to effectively encapsulate DNase I through electrostatic and hydrophobic interactions after simply mixing them together. The encapsulation process was very gentle avoiding any organic agents and solvents, and other tedious preparation processes that might inactivate proteins. In addition, DOX bioreversibly linked with adamantane (DOX-A) can successfully modify the architecture of DNase I nanoformulations through host–guest inclusion interactions between cyclodextrin and adamantane (Scheme 1a). The resulting co-delivery system can not only significantly enhance the endocytosis of DNase I and DOX, but also rapidly release them into the cytoplasm through efficient endosomal escape, and make them highly accumulate in the nucleus via Importin (Imp) mediated nuclear transportation,38 thereby greatly enhancing their synergistic cell destructive ability (Scheme 1b). Compared with other nuclear delivery strategies,39,40 this co-delivery nanoplatform avoids the complex modification of nuclear targeting ligands and disruption of nuclear membrane integrity, and can generally be used for intranuclear delivery of other proteins and drugs.
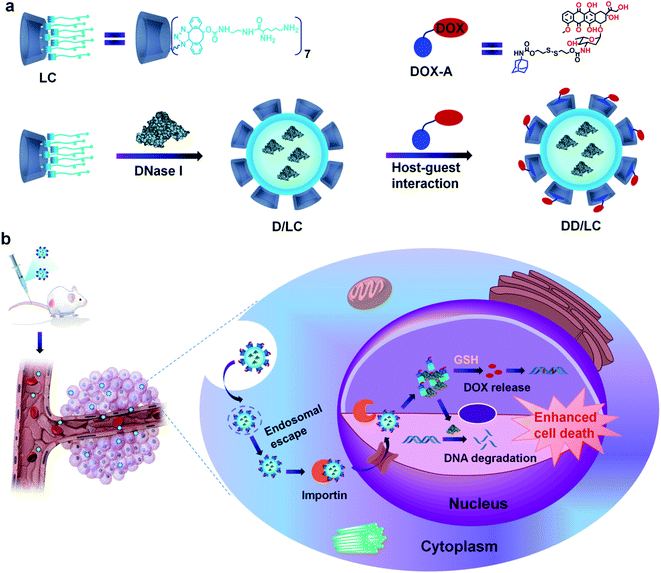 |
| | Scheme 1 Nucleus-targeted protein and chemotherapeutic co-delivery nanoplatform for synergistic antitumor therapy. (a) Fabrication of DNase I and DOX co-delivery nanoformulations. Negatively charged DNase I was effectively encapsulated with amphiphilic LC through electrostatic and hydrophobic interactions. DOX was easily incorporated into the architecture of DNase I nanoformulations through host–guest interactions between cyclodextrin and adamantane. (b) Schematic illustration of DNase I and DOX co-delivery nanoparticles for systemic and nucleus-targeted delivery to enhance the synergistic killing of tumor cells. | |
Results and discussion
Preparation and characterization of D/LC and DD/LC nanoformulations
LC was synthesized according to the literature41 (Fig. S1†). DNase I loaded LC nanocomposites (D/LC) were prepared just by simply mixing them in aqueous solution under mild conditions, avoiding any cumbersome steps that might affect the activity of DNase I. Agarose gel retention experiments showed that when the mass ratio of DNase I to LC was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, DNase I could be completely encapsulated (Fig. 1a), and the loading efficiency was more than 30% and its activity was also well maintained (Fig. S3b†). In addition, native DNase I can be released in PBS with high ionic strength (Fig. S3a†), so as to not affect its intracellular function. In addition, to incorporate DOX into the nanocomposites by cyclodextrin–adamantane inclusion interactions, DOX was modified with adamantane via a redox-responsive linker to obtain DOX-A (Fig. S2†), which can be easily integrated into D/LC with a DOX loading efficiency of about 15% (Fig. S4†). Transmission electron microscopy (TEM) and dynamic light scattering (DLS) showed that D/LC could form nanoparticles in aqueous solution with a size of about 100 nm, and the morphology of DOX and DNase I co-loaded nanoparticles (DD/LC) was more compact and the particle size was slightly increased, which indicated that DOX was successfully encapsulated into the nanocomposites (Fig. 1b and c and S5†). Moreover, compared with D/LC, DD/LC showed a much lower zeta potential (Fig. 1d), further confirming the integration of DOX into the DNase I nanoformulations. Furthermore, due to the higher level of intracellular thiol species,42 adamantane modification in DOX-A can be preferentially removed inside tumor cells, thus avoiding drug preleakage during blood circulation. As shown in Fig. 1e, DD/LC nanoparticles were very stable in PBS in the absence of dithiothreitol (DTT) and almost no DOX release can be observed. However, DOX was rapidly released when the concentration of DTT increased to 5 mM, and the release rate was further accelerated as the concentration increased to 10 mM. These results suggested that it was feasible and reliable to release the drug using a reductive self-immolative linker, which is beneficial in improving the systemic stability of DD/LC nanoparticles.
:2, DNase I could be completely encapsulated (Fig. 1a), and the loading efficiency was more than 30% and its activity was also well maintained (Fig. S3b†). In addition, native DNase I can be released in PBS with high ionic strength (Fig. S3a†), so as to not affect its intracellular function. In addition, to incorporate DOX into the nanocomposites by cyclodextrin–adamantane inclusion interactions, DOX was modified with adamantane via a redox-responsive linker to obtain DOX-A (Fig. S2†), which can be easily integrated into D/LC with a DOX loading efficiency of about 15% (Fig. S4†). Transmission electron microscopy (TEM) and dynamic light scattering (DLS) showed that D/LC could form nanoparticles in aqueous solution with a size of about 100 nm, and the morphology of DOX and DNase I co-loaded nanoparticles (DD/LC) was more compact and the particle size was slightly increased, which indicated that DOX was successfully encapsulated into the nanocomposites (Fig. 1b and c and S5†). Moreover, compared with D/LC, DD/LC showed a much lower zeta potential (Fig. 1d), further confirming the integration of DOX into the DNase I nanoformulations. Furthermore, due to the higher level of intracellular thiol species,42 adamantane modification in DOX-A can be preferentially removed inside tumor cells, thus avoiding drug preleakage during blood circulation. As shown in Fig. 1e, DD/LC nanoparticles were very stable in PBS in the absence of dithiothreitol (DTT) and almost no DOX release can be observed. However, DOX was rapidly released when the concentration of DTT increased to 5 mM, and the release rate was further accelerated as the concentration increased to 10 mM. These results suggested that it was feasible and reliable to release the drug using a reductive self-immolative linker, which is beneficial in improving the systemic stability of DD/LC nanoparticles.
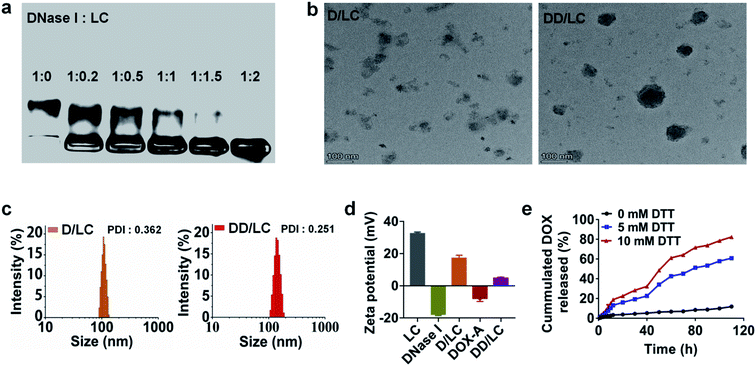 |
| | Fig. 1 Characterization of D/LC and DD/LC nanoformulations. (a) Agarose gel retention assay for analyzing the encapsulation of DNase I with LC nanoparticles. (b) TEM images and (c) size distribution of D/LC and DD/LC nanoparticles. (d) Zeta potentials of various samples. (e) In vitro release of DOX from DD/LC nanoparticles in PBS (pH 7.4) with or without DTT at 37 °C. Data are shown as mean ± SD (n = 3). | |
In vitro cellular uptake and accumulation in the nucleus
To evaluate the efficiency of intracellular co-delivery of DNase I and DOX, 4T1 cells were treated with free DNase I, free DOX, and D/LC or DD/LC nanoparticles. DNase I was labeled with FITC to track its intracellular location by fluorescent microscopy. After 2 h of incubation, strong green fluorescence was observed in the nucleus of cells treated with D/LC or DD/LC nanoparticles (Fig. 2). In contrast, negligible fluorescence was observed from cells that were treated with the free proteins. Quantitative flow cytometry further confirmed that cells treated with D/LC or DD/LC showed a much higher green fluorescence intensity than cells treated with free DNase I (Fig. S6†). These results demonstrated that D/LC or DD/LC nanoparticles could significantly enhance the cellular uptake of DNase I, effectively escape from lysosomes and accumulate in the nucleus. In addition, accumulation of red fluorescence from DOX was also observed in nuclei of cells treated with DD/LC nanoparticles after 2 h of incubation, and green DNase I fluorescence and red DOX fluorescence can well overlap with blue DAPI fluorescence (Fig. S7a†). In addition, the confocal 3D images also showed clear over-lapped fluorescence in the nuclei (Fig. S7b†), and the Pearson correlation coefficient was calculated to be ca. 0.89 for DNase I and 0.88 for DOX (Fig. S8†), suggesting a good nucleus co-localization of DNase I and DOX. All these results collectively confirmed that DD/LC nanoparticles escaped effectively from lysosomes and subsequently imported both DOX and DNase I into the nucleus.
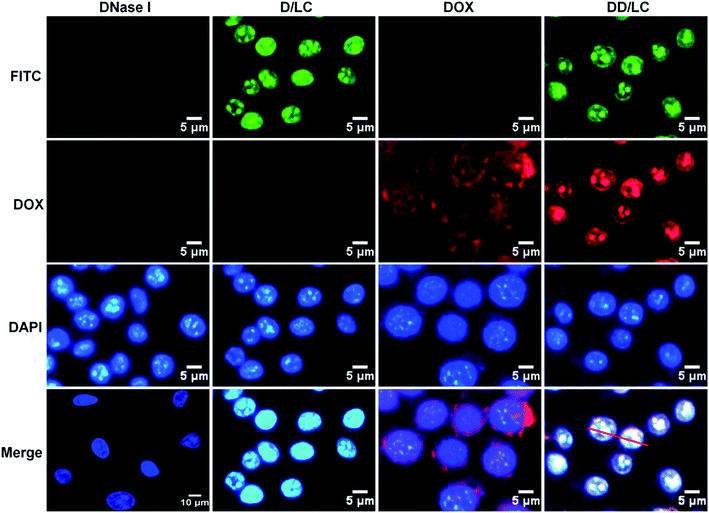 |
| | Fig. 2 Fluorescence images of the intracellular location of different formulations in 4T1 cells. DNase I was labelled with FITC, and nuclei were stained by using DAPI. Scale bars: 5 μm. Both D/LC and DD/LC were mainly concentrated in the nucleus, while free DNase I was almost invisible in the cytoplasm, and free DOX was distributed in the whole cell. | |
Cellular transport mechanism and endosomal escape
Considering that the nuclear accumulation of protein nanoparticles is closely related to endocytosis, we then studied the cellular uptake mechanism of DD/LC nanoparticles. The cellular uptake of DD/LC nanoparticles was significantly reduced at 4 °C or when the cells were pretreated with sodium azide (Fig. 3a). Similarly, reduced endocytosis of DD/LC was also observed in cells pretreated with methyl-β-cyclodextrin (M-β-CD) or genistein, implying that caveolin and lipid raft related pathways were involved in the process of cell internalization. Taken together, these results indicated that DD/LC was actively transported in cells through multiple endocytic pathways, leading to enhanced cellular uptake. In addition, in the presence of Lyso Tracker Red, a marker for secondary endosomes and lysosomes, fluorescence images of cells incubated with D/LC demonstrated that LC nanovehicles could effectively escape from endo-lysosomal compartments (Fig. S9†). Moreover, time-dependent cell imaging (Fig. 3b) showed the emergence of fluorescence in the nucleus after 10 min of incubation, which further indicated that DD/LC nanoparticles were transported rapidly into the nucleus following their uptake. Furthermore, the nuclear transport mechanism of DD/LC nanoparticles was also investigated by pre-treating cells with ivermectin (a specific inhibitor of importin). As shown in Fig. 3c, the nuclear localization of DNase I and DOX was significantly inhibited after blocking importin with ivermectin, revealing that the nuclear transport of DD/LC nanoparticles was mainly mediated by importin.
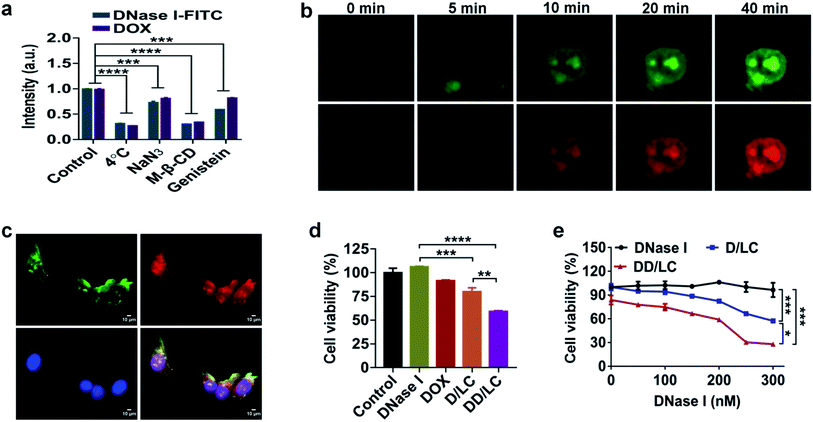 |
| | Fig. 3 (a) Effects of endocytosis inhibitors (4 °C, 10 mM NaN3, 10 mM M-β-CD and 200 μM genistein) on the cellular uptake of the DD/LC nanocomplex. (b) Time-lapsed fluorescence imaging of a 4T1 cell treated with DD/LC. DNase I was labelled with FITC. (c) Fluorescence images of the intracellular location of DD/LC nanoparticles in the presence of ivermectin (15 μM). DNase I was labelled with FITC, and nuclei were stained by using DAPI. Scale bars: 10 μm. (d) Cell viability after different treatments. (e) Concentration dependent cytotoxicity of free DNase I and its different nanoformulations. | |
In vitro antitumor efficacy
To identify the in vitro synergistic anticancer efficacy of DNase I and DOX, the viability of 4T1 cells after different treatments was measured by dimethylthiazolyl-diphenyltetrazolium bromide (MTT) assay. Compared with the untreated control group, native DNase I had no inhibitory effect on tumor cell growth due to its poor cell membrane permeability (Fig. 3d). In contrast, D/LC and DD/LC showed concentration-dependent cytotoxicities (Fig. 3e), and the corresponding concentration of the free LC nanoparticles had almost no cytotoxicity (Fig. S10†), indicating that apoptosis was caused by high accumulation of DNase I in the nucleus. Notably, the cytotoxicity of DD/LC was higher than that of D/LC, demonstrating the importance of incorporation of DOX into DNase I nanoformulations for the synergistic induction of apoptosis (CDI (coefficient of drug interaction) = 0.79) in 4T1 cells.
Biodistribution and in vivo antitumour activity
To investigate the in vivo biodistribution of DNase I and DOX delivered by LC, free DNase I-Cy7, D/LC-Cy7 and DD/LC-Cy7 were intravenously injected into 4T1 tumor-bearing mice and the fluorescence was imaged in vivo and ex vivo. The in vivo fluorescence images showed that D/LC or DD/LC nanoparticles rapidly accumulated in tumor tissues, and generated strong fluorescent signals at 4 h post-injection (Fig. 4a). In contrast, negligible fluorescence was observed in tumors from the free DNase I treated group even after 24 h of injection due to the low tumor uptake and rapid degradation of free proteins, while fluorescence in tumors of the D/LC or DD/LC treated group was still strong, indicating their tumor specificity (Fig. 4b). Furthermore, the fluorescence intensity of D/LC or DD/LC at the tumor site was much higher than that of free DNase I as determined by the quantitative result obtained using the region-of-interest analysis (Fig. 4c). Thus, these results suggested that LC can mediate the high accumulation and retention of DNase I and DOX, thus improving its therapeutic effect in vivo.
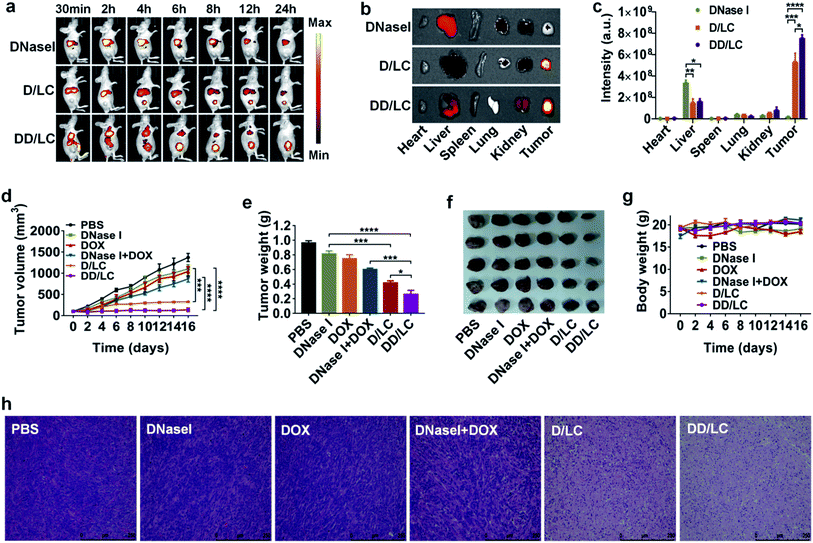 |
| | Fig. 4 (a) In vivo fluorescence imaging of 4T1 tumor-bearing mice at 0.5, 2, 4, 6, 8, 12, and 24 h after the intravenous injection of DNase I-Cy7, D/LC-Cy7 or DD/LC-Cy7. (b) Ex vivo fluorescence imaging and (c) quantification of fluorescence intensities of the excised tumors and major organs of DNase I-Cy7, D/LC-Cy7 or DD/LC-Cy7 treated 4T1 tumor-bearing mice at 24 h post-injection. (d) Tumor volume changes of 4T1-tumor-bearing mice after intravenous injection of PBS, DNase I, DOX, DNase I + DOX, D/LC, and DD/LC on day 0, 3, 6, and 9 (3 mg kg−1 DNase I and 2 mg kg−1 DOX). (e) Photographs of tumors taken from mice at the end of different treatments. (f) Average tumor weights of the mice treated as described in (d) on day 16. (g) Variation in the body weight of mice during different treatments. (h) H&E stained images of tumor slices taken from different treatment groups on day 16. (*p < 0.05, ***p < 0.001, and ****p < 0.0001). | |
Based on the above results, the in vivo synergistic anticancer effect of DNase I and DOX was evaluated in 4T1-tumor-bearing mice by intravenous injection of various formulations (3 mg kg−1 DNase I and 2 mg kg−1 DOX) every three days, for a total of four times. As shown in Fig. 4d, tumor growth was significantly inhibited in the D/LC and DD/LC treated group while minimal tumor inhibition was observed in mice treated with free DNase I or DNase I + DOX, which could be attributed to the enhanced tumor accumulation and retention of D/LC and DD/LC. In addition, compared with D/LC, DD/LC has a higher tumor inhibitory effect, confirming the synergistic anticancer effect of the co-delivered DOX and DNase I (Fig. 4e), which was consistent with the in vitro results that DOX and DNase I delivered by LC can accumulate together in the nucleus of the same cell to enhance cytotoxicity. Besides, tumor weights at the end of the experiment also indicated that DD/LC had a better therapeutic effect than any other groups (Fig. 4f), and DD/LC also significantly prolonged the survival rate (Fig. S11†), which reflected the importance of a suitable co-delivery carrier to improve the synergistic anti-tumor effect of drugs. Moreover, histological images of tumor tissues stained with hematoxylin and eosin (H&E) revealed that D/LC and DD/LC caused significant apoptosis of cancer cells as compared with other groups (Fig. 4h), while no significant reduction in body weight was observed in these nanoparticle treated groups when compared to the PBS control group during the experimental period (Fig. 4g), indicating that D/LC and DD/LC had high biosafety for in vivo cancer treatment. Furthermore, negligible damage to healthy organs (such as the heart, liver, spleen, lung, and kidney) was observed in all the tested formulations (Fig. S12†). This is because a relatively low DOX dose (2 mg kg−1) was used in our experiment to avoid possible significant side effects. Taken together, these results suggested that our nuclear targeted combination therapy provides a promising strategy for synergistic treatment of tumors with high biocompatibility.
Conclusions
In summary, we present a facile and robust nucleus-targeted protein and chemotherapeutic co-delivery nanoplatform to simultaneously deliver DNase I and DOX into the nucleus for enhanced synergistic cancer therapy. An amphiphilic LC was prepared to encapsulate intact DNase I with a high loading efficiency of about 30% just by simply mixing them together, without the introduction of any organic solvent or cumbersome separation steps that might inactivate the proteins. In addition, DOX can be concurrently loaded based on host–guest inclusion interactions. The resultant DD/LC nanoparticles with high endosomal escape ability could efficiently deliver DNase I and DOX into the nucleus of the same cell to significantly enhance their cytotoxicities. Moreover, DD/LC nanoparticles could selectively accumulate at the tumor site with high systemic stability, resulting in greatly improved synergistic anticancer effects of DNase I and DOX in a murine breast cancer model. Furthermore, this nucleus-targeted cooperative nanoplatform can be applied for the delivery of other combinations of intracellular active proteins and drugs. Therefore, our co-delivery strategy may serve as a general and powerful tool for nucleus-targeted delivery of proteins and chemotherapeutics to improve the treatment of cancer and other diseases.
Author contributions
X. F. and N. Z. conceived the project and designed the experiments. L. Y. and H. M. synthesized the chemicals. L. Y. performed the cell experiments and analyzed the data. L. Y., H. M., S. L. and Y. Z. performed the animal experiments. X. F., N. Z. and H. C. supervised the project. All of the authors were involved in the analyses and interpretation of data. X. F., L. Y., N. Z., and H. C. wrote the paper, with the help of the co-authors.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21773268 and 21877010) and the Startup Funding of Chongqing University (0236011104419). All animal experiments were carried out in compliance with the requirements of the National Act on the use of Experimental Animals (People's Republic of China) and were approved by the Experimental Animal Ethical Committee of Chongqing University Cancer Hospital. Female BALB/c-nude mice (6–8 weeks) were supplied by the Animal Center of Chongqing Medical University (Chongqing, China).
Notes and references
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS PubMed.
- Z. Gu, A. Biswas, M. Zhao and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638–3655 RSC.
- M. Yu, J. Wu, J. Shi and O. C. Farokhzad, J. Controlled Release, 2016, 240, 24–37 CrossRef CAS PubMed.
- Y. Lu, W. Sun and Z. Gu, J. Controlled Release, 2014, 194, 1–19 CrossRef CAS PubMed.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- O. Leavy, Nat. Rev. Immunol., 2010, 10, 297 CrossRef CAS PubMed.
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- M. C. P. Sok, N. Baker, C. McClain, H. S. Lim, T. Turner, L. Hymel, M. Ogle, C. Olingy, J. I. Palacios, J. R. Garcia, K. Srithar, A. J. García, P. Qiu and E. A. Botchwey, Biomaterials, 2021, 268, 120475 CrossRef CAS PubMed.
- L. Chen, J. Liu, M. Guan, T. Zhou, X. Duan and Z. Xiang, Int. J. Nanomed., 2020, 15, 6097–6111 CrossRef CAS PubMed.
- Z. Zeng, X. He, C. Li, S. Lin, H. Chen, L. Liu and X. Feng, Biomaterials, 2021, 271, 120753 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- X. Liu, F. Wu, Y. Ji and L. C. Yin, Bioconjugate Chem., 2019, 30, 305–324 CrossRef PubMed.
- F. Scaletti, J. Hardie, Y. W. Lee, D. C. Luther, M. Ray and V. M. Rotello, Chem. Soc. Rev., 2018, 47, 3421–3432 RSC.
- Y. Zhang, J. J. Røise, K. W. Lee, J. Li and N. Murthy, Curr. Opin. Biotechnol., 2018, 52, 25–31 CrossRef CAS PubMed.
- X. Qin, C. Yu, J. Wei, L. Li, C. Zhang, Q. Wu, J. Liu, S. Yao and W. Huang, Adv. Mater., 2019, 31, e1902791 CrossRef PubMed.
- K. A. Mix, J. E. Lomax and R. T. Raines, J. Am. Chem. Soc., 2017, 139, 14396–14398 CrossRef CAS PubMed.
- M. Yu, J. Wu, J. Shi and O. C. Farokhzad, J. Controlled Release, 2016, 240, 24–37 CrossRef CAS PubMed.
- X. He, Q. Long, Z. Zeng, L. Yang, Y. Q. Tang and X. L. Feng, Adv. Funct. Mater., 2019, 29, 1906187 CrossRef CAS.
- P. Yuan, X. Mao, X. Wu, S. S. Liew, L. Li and S. Q. Yao, Angew. Chem., Int. Ed., 2019, 58, 7657–7661 CrossRef CAS PubMed.
- H. Chang, J. Lv, X. Gao, X. Wang, H. Wang, H. Chen, X. He, L. Li and Y. Cheng, Nano Lett., 2017, 17, 1678–1684 CrossRef CAS PubMed.
- M. Wang, K. Alberti, S. Sun, C. L. Arellano and Q. Xu, Angew. Chem., Int. Ed., 2014, 53, 2893–2898 CrossRef CAS PubMed.
- P. Zhang, B. Steinborn, U. Lächelt, S. Zahler and E. Wagner, Biomacromolecules, 2017, 18, 2509–2520 CrossRef CAS PubMed.
- S. Li, J. Zhang, C. Deng, F. H. Meng, L. Yu and Z. Y. Zhong, ACS Appl. Mater. Interfaces, 2016, 8, 21155–21162 CrossRef CAS PubMed.
- R. Mout, M. Ray, T. Tay, K. Sasaki, G. Yesilbag Tonga and V. M. Rotello, ACS Nano, 2017, 11, 6416–6421 CrossRef CAS PubMed.
- B. Liu, W. Ejaz, S. Gong, M. Kurbanov, M. Canakci, F. Anson and S. Thayumanavan, Nano Lett., 2020, 20, 4014–4021 CrossRef CAS PubMed.
- S. Zhang and Y. Cheng, Biomater. Sci., 2020, 8, 3741–3750 RSC.
- H. Omar, J. G. Croissant, K. Alamoudi, S. Alsaiari, I. Alradwan, M. A. Majrashi, D. H. Anjum, P. Martins, R. Laamarti, J. Eppinger, B. Moosa, A. Almalik and N. M. Khashab, J. Controlled Release, 2017, 259, 187–194 CrossRef CAS PubMed.
- W. Sun, W. Ji, Q. Hu, J. Yu, C. Wang, C. Qian, G. Hochu and Z. Gu, Biomaterials, 2016, 96, 1–10 CrossRef CAS PubMed.
- J. Liu, T. Wu, X. Lu, X. Wu, S. Liu, S. Zhao, X. Xu and B. Ding, J. Am. Chem. Soc., 2019, 141, 19032–19037 CrossRef CAS PubMed.
- H. D. Bae, M. Kim, J. Lee and K. Lee, Drug Delivery, 2018, 25, 1579–1584 CrossRef CAS PubMed.
- T. Jiang, R. Mo, A. Bellotti, J. Zhou and Z. Gu, Adv. Funct. Mater., 2014, 24, 2295–2304 CrossRef CAS.
- P. Zhang, Y. Zhang, X. Ding, W. Shen, M. Li, E. Wagner, C. Xiao and X. Chen, Adv. Mater., 2020, 32, e2000013 CrossRef PubMed.
- J. Peng, S. Fumoto, T. Suga, H. Miyamoto, N. Kuroda, S. Kawakami and K. Nishida, J. Controlled Release, 2019, 302, 42–53 CrossRef CAS PubMed.
- C. S. Kim, R. Mout, Y. Zhao, Y.-C. Yeh, R. Tang, Y. Jeong, B. Duncan, J. A. Hardy and V. M. Rotello, Bioconjugate Chem., 2015, 26, 950–954 CrossRef CAS PubMed.
- D. C. González-Toro, J.-H. Ryu, R. T. Chacko, J. Zhuang and S. Thayumanavan, J. Am. Chem. Soc., 2012, 134, 6964–6967 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- J. Lv, Q. Fan, H. Wang and Y. Cheng, Biomaterials, 2019, 218, 119358 CrossRef CAS PubMed.
- Y. Miyamoto, K. Yamada and Y. Yoneda, J. Biochem., 2016, 160, 69–75 CrossRef CAS PubMed.
- Y. Zhu, H. Jia, G. Pan, N. W. Ulrich, Z. Chen and F. Wu, J. Am. Chem. Soc., 2018, 140, 4062–4070 CrossRef CAS PubMed.
- L. Wang, T. Zhang, M. Huo, J. Guo, Y. Chen and H. Xu, Small, 2019, 15, e1903254 CrossRef PubMed.
- J. Tang, Y. Liu, D. Qi, L. Yang, H. Chen, C. Wang and X. Feng, Small, 2021, 17, e2101219 CrossRef PubMed.
- A. Santoro, J. S. Calvo, M. D. Peris-Díaz, A. Krężel, G. Meloni and P. Faller, Angew. Chem., Int. Ed. Engl., 2020, 59, 7830–7835 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a




