
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular tetrazine-acryloyl cycloaddition: chemistry and applications†
Akil
Hamsath
,
Oren L.
Lederberg
,
Qi
Cui
 ,
Meg
Shieh
,
Yannie
Lam
,
Brock J.
Brummett
,
Shi
Xu
,
Jerome R.
Robinson
and
Ming
Xian
*
,
Meg
Shieh
,
Yannie
Lam
,
Brock J.
Brummett
,
Shi
Xu
,
Jerome R.
Robinson
and
Ming
Xian
*
Department of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: ming_xian@brown.edu
First published on 19th August 2022
Abstract
An unprecedented intramolecular [4 + 2] tetrazine-olefin cycloaddition with α,β-unsaturated substrates was discovered. The reaction produces unique coumarin-dihydropyridazine heterocycles that exhibited strong fluorescence with large Stokes shifts and excellent photo- and pH-stability. This property can be used for reaction analysis. The rate of cycloaddition was found to be solvent dependent and was determined using experimental data with a kinetic modeling software (COPASI) as well as DFT calculations (k1 = 0.64 ± 0.019 s−1 and 4.1 s−1, respectively). The effects of steric and electronic properties of both the tetrazine and α,β-unsaturated carbonyl on the reaction were studied and followed the known trends characteristic of the intermolecular reaction. Based on these results, we developed a “release-then-click” strategy for the ROS triggered release of methylselenenic acid (MeSeOH) and a fluorescent tracer. This strategy was demonstrated in HeLa cells via fluorescence imaging.
Introduction
Bioorthogonal reactions enable researchers to study the complexities of biological systems without interfering with natural biochemical processes. 1,2,4,5-Tetrazines have been widely used as powerful bioorthogonal reagents because of their rapid reaction kinetics and high selectivity with olefins like trans-cyclooctene, norbornene and cyclopropene. These tetrazine-olefin ligations greatly impact chemical biology because of their non-metal/non-toxic nature, and impressive efficiency under physiological conditions.1–5 These coupling partners have been employed as in vivo imaging agents, metabolic probes, nanoparticle biomaterials, drug surrogates, etc.6,7 The conventional tetrazine-olefin ligation is illustrated in Scheme 1A, which proceeds via a [4 + 2] inverse electron demand Diels–Alder (IEDDA) mechanism. A subsequent retro-Diels–Alder step releases an equivalent of N2 and the cycloaddition product. Experimental and computational results show that tetrazines containing electron withdrawing groups (EWG) and olefins containing electron donating groups (EDG) are the best coupling partners. However, when the electronics are switched, there is little to no reaction, limiting the utility of the tetrazine platform.2 It is worth noting that in almost all reported cases the tetrazine-olefin ligations are intermolecular reactions that quickly tether a biological target (proteins, antibodies, etc.) to a payload (such as fluorescent probes). On the other hand, intramolecular tetrazine reactions are rarely seen, perhaps because the need for such reactions for biological applications is unjustified. However, an intramolecular reaction can have rate constants orders of magnitude larger than an intermolecular reaction. One notable example was reported by Eising et al. for the coordination-assisted intramolecular tetrazine ligation with vinyl boronic acids (Scheme 1B).8 These reactions were only reported for relatively electron-rich olefins. Thus, the reactivity between tetrazines and electron-poor olefins via an intramolecular mechanism remains unexplored.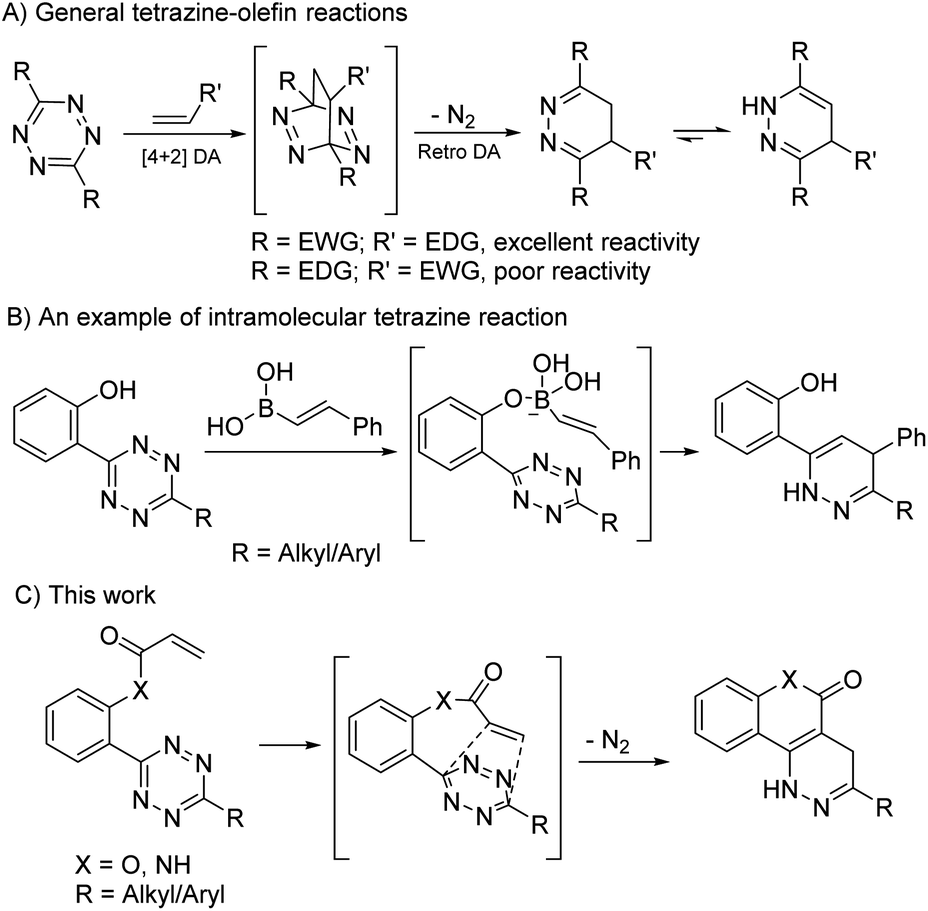 | ||
| Scheme 1 The design of intramolecular tetrazine-acryloyl cycloaddition. | ||
α,β-Unsaturated carbonyls are a class of electron poor olefins that are considered potential cytotoxins. They are potent cellular electrophiles that can cause redox imbalances by depleting cellular cysteine/glutathione and can induce oxidative DNA damage by reacting with DNA bases.9–11 However, some popular prodrug designs release α,β-unsaturated carbonyls as byproducts. For example, phenol-based linkers that undergo 1,4 or 1,6-elimination release ortho- or para-quinone methides and γ-ketothiocarbamates that undergo E1Cb eliminations release acrylates.12,13 The inherent toxicities of these byproducts often complicate the design of prodrugs. We envisioned that if tetrazines could be pushed to react with these byproducts and remove them from the reaction system, it would solve this long-lasting problem and expand the application of tetrazine chemistry. We expected this could be achieved via covalently tethering an acryloyl system to a tetrazine (Scheme 1C). Herein, we report our investigation of such intramolecular tetrazine-acryloyl cycloadditions.
Results and discussion
Model studies of the intramolecular tetrazine-acryloyl cycloaddition
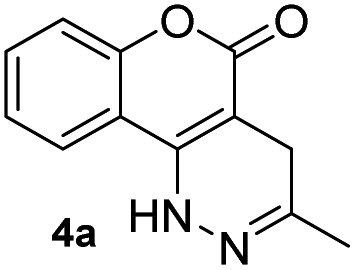
Before testing our hypothesis, we first carried out an intermolecular reaction using tetrazine 1 and methyl/phenyl acrylate (Scheme 2). As expected, no reaction was observed in those cases. To test the intramolecular version, O-hydroxyphenyl methyl tetrazine (2a) was prepared and treated with acryloyl chloride under various conditions as shown in Scheme 2. We expected the intermediate 3a would be produced and cyclize to give the desired product (4a). Indeed, the reaction consistently generated 4a in medium to high yields. This was found to be a clean reaction (monitored by TLC) under mild conditions giving access to a unique fused ring system. Interestingly, compound 4a was found to be fluorescent as it has a coumarin-like structure (λabs: 350 nm, λem: 450, ε = 4320 M−1 cm−1, ϕ = 0.25 in H2O). We took advantage of this property to study reaction kinetics and explored its chemical-/photo-stability and utility in biological systems (vide infra).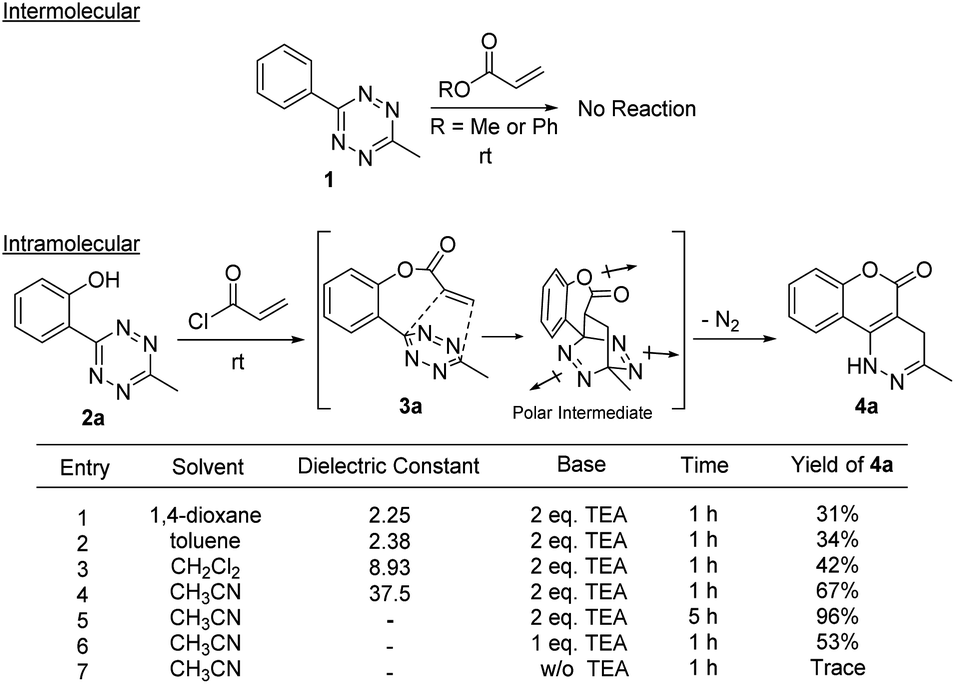 | ||
| Scheme 2 Initial studies of the intramolecular tetrazine-acryloyl cycloaddition. | ||
The reaction yield was found to be solvent dependent with lower yields in relatively non-polar solvents (entries 1–3) and good yields in a relatively polar solvent-CH3CN (entries 4–7). This suggests that a polar transition state was involved in the process. When intermediate 3a undergoes a [4 + 2] cycloaddition a lactone ring is formed along with three dipole moments (illustrated in Scheme 2). This polar transition state could be stabilized by polar solvents with increasing dielectric constants leading to higher reaction efficiency.14 The reaction also showed a clear dependence for triethylamine (TEA) (entries 5–7), indicating that the esterification proceeded before the cycloaddition. Complete conversion of the reaction was achieved in ∼5 h with 2.0 equiv. TEA. It should be noted that the intermediate 3a could be observed on TLC almost quantitatively within minutes. We attempted to isolate 3a but were unsuccessful as it continued to cyclize during workup and purification.
To understand the chemical limitations of this cycloaddition, we next applied the optimized reaction conditions to a series of O-hydroxyphenyl tetrazine substrates. As shown in Fig. 1, high yields of the desired products were obtained from the methyl- and hydrogen-substituted O-hydroxyphenyl tetrazines (products 4a and 4b) while the corresponding bulky t-butyl version was obtained in much decreased yield (product 4c). It is interesting to note that 4b was obtained together with its oxidized pyridazine form (5 shown in Scheme S1†), which suggests that 4b is sensitive to aerobic oxidation. This property could have some applications in biological settings such as to sense reactive oxygen species (ROS). Nevertheless, their mass balance was >95%, indicating a drastic increase in cycloaddition reactivity with less sterically hindered O-hydroxyphenyl tetrazines. This steric trend has also been reported for intermolecular tetrazine cycloadditions.15,16
 | ||
| Fig. 1 Substrate scope containing various O-hydroxyphenyl tetrazines. Tetrazine (2a–h, 0.1 mmol), triethylamine (0.2 mmol), acryloyl chloride (0.2 mmol), CH3CN (1.0 mL). a Reaction was done under argon for 1 h. b Reaction was allowed to run for 24 h. | ||
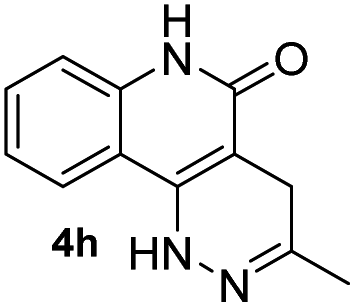
Electron-donating groups (product 4d) drastically decreased the efficiency of the reaction, yielding trace amount of cyclized product even after 24 h. On the other hand, when the tetrazines contained less electron-donating substituents or electron-withdrawing groups the reaction once again became productive albeit at longer reaction times (products 4e and 4f). A single crystal was obtained for 4f, and the core structure confirmed the formation of the fused coumarin-dihydropyridazine moiety shown in Fig. 1 (detailed crystal structure analysis can be found in Fig. S1 and S2† of the X-ray ESI). Next, the phenyl backbone present in these structures was replaced with a two-carbon chain which drastically decreased the reactivity, yielding trace amount of cyclized product even after 24 h (4g). This suggests that structural rigidity is required for this reaction to proceed. Another structural property of these substrates is the ester functional group which could potentially undergo hydrolysis in biological systems, limiting potential applications. Thus, a more stable amide version of these compounds was synthesized, and the expected cycloaddition proceeded in good yield (4h, 85%). We realized that compound 4h could be presented as the lactam (shown in Fig. 1) or its tautomer (2-hydroxyquinoline). Such an equilibrium is known to be solvent dependent, and both forms are found to have little energy difference (∼5 kcal mol−1).17,18 Therefore, we believe the equilibrium will have no effect on applications that produce 4h as a product. For substrates that did not give efficient cycloaddition, the corresponding ester intermediates were isolated and characterized.
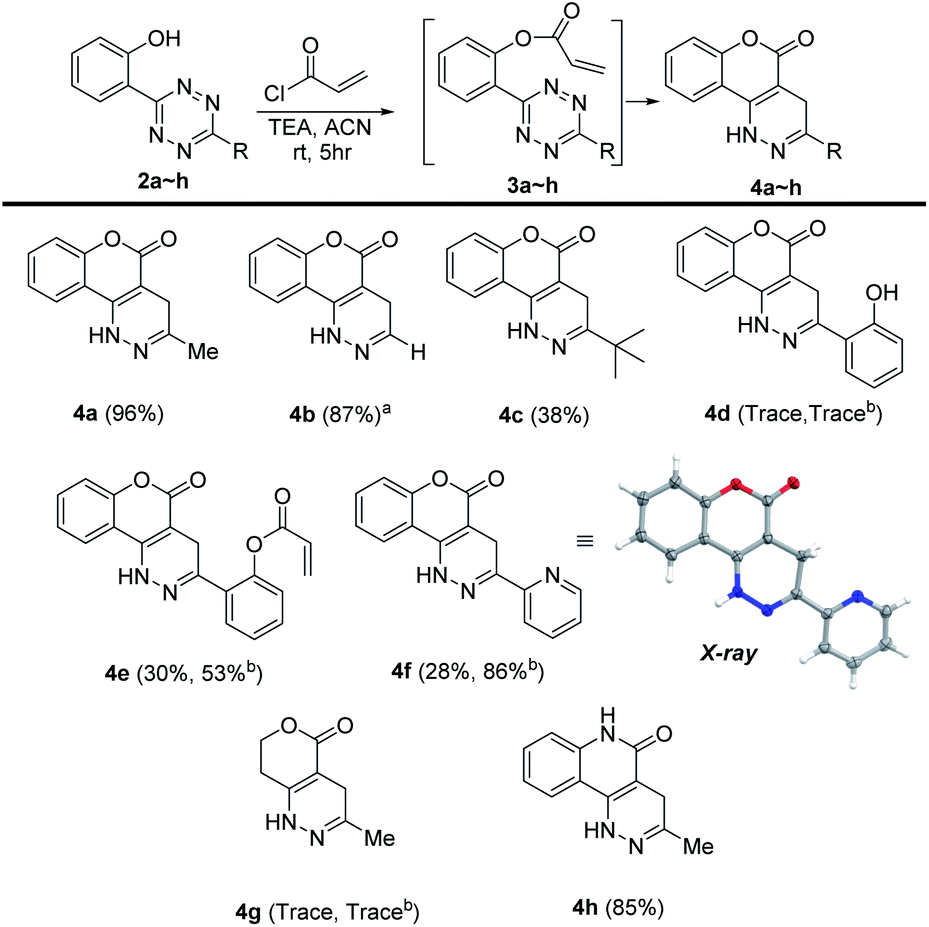
We then set out to understand how the intramolecular cycloaddition would be affected by substitutions at the olefin site. It has been demonstrated that for intermolecular tetrazine reactions terminal olefins react more readily than non-strained internal olefins.19 We wondered if this rule would stand with intramolecular tetrazine reactions. Thus, we tested the reactivity of compounds 8–13 containing various CH3- substituents on the olefin site and H– or CH3– on the tetrazine site (Scheme 3). Among these, only intermediates 12 and 13 provided the cycloaddition products (69% and 51%, respectively) with no over-oxidized pyridazine observed, and all of the rest failed in cycloaddition. The E- and Z-isomers were found to have negligible Gibbs free energy differences (ΔG12 = 20.8, ΔG13 = 20.9 kcal mol−1) calculated at the M06-2X/6-311G(d) level. Thus, the reactivity observed for 12/13 is due to the lack of steric hindrance resulting from a H-substituted tetrazine. Compounds 6–11 were stable enough to be isolated and characterized without noticeable cyclizing even after 24 h. Clearly the introduction of a methyl group on the olefin significantly decreased the reactivity, especially when the tetrazine component was also decorated with a methyl.
 | ||
| Scheme 3 Methyl-decorated acryloyl substrates in cycloaddition. | ||
Photophysical properties of the cycloaddition products
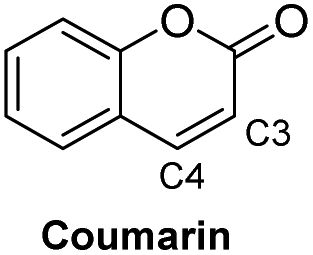
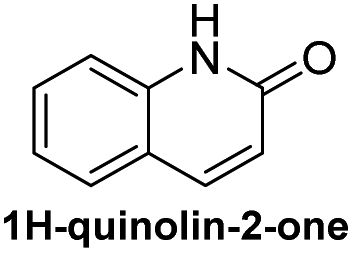
Based on the results acquired thus far, we know that both a sterically unhindered tetrazine and acryloyl group are required for efficient cycloaddition. Any applications using this chemistry will produce either 4a or 4h as products. Therefore, the photophysical properties of these two novel heterocyclic compounds were measured and compiled in Table 1. The experiments were carried out in 1% DMSO and H2O at a concentration of 50 μM. The maximum absorption wavelengths for 4a and 4h were 350 and 320 nm, respectively, which are slightly blue shifted compared to their core coumarin and 1H-quinolin-2-one under the same conditions. The maximum emission wavelengths for 4a (450 mn) and 4h (425 nm) resulted in Stokes shifts of 100 and 105 nm, respectively. Furthermore, 4a showed an increase in quantum yield (ϕ = 0.25) when compared to the non-fluorescent coumarin while 4h showed a 6.8-fold increase in quantum yield when compared to its parent 1H-quinolin-2-one (ϕ = 0.34 vs. 0.05). These results suggest that the dihydropyridazine moiety fused at C3 and C4 has a positive effect on fluorescent intensity attributed to the intramolecular charge transfer between the electron donor and acceptor. The photostability of 4a and 4h was also tested by exciting both compounds with their corresponding absorption wavelengths at 5 min intervals for 16 h (shown in Fig. S2†). Both 4a and 4h were found to be highly photostable with negligible decrease in fluorescence intensity. Moreover, we measured their fluorescence under different pHs (from 6 to 9) and again found negligible changes, suggesting high pH stability as well (Fig. S3†).| Compound | λ abs (nm) | λ em (nm) | ε (M−1 cm−1) | Stokes shift (nm) | ϕ |
|---|---|---|---|---|---|
|
367 | NA | 21.5 (at 367 nm) | NA | NA |
|
350 | 450 | 4320 (at 350 nm) | 100 | 0.25 |
|
325 | 375 | 7920 (at 325 nm) | 50 | 0.05 |
|
320 | 425 | 8333 (at 320 nm) | 105 | 0.34 |
Investigation of the reaction kinetics
Given the strong fluorescence of 4a and 4h, we envisioned that the progress of the cycloaddition could be monitored by fluorescence. To this end, we synthesized a model compound 15 (Fig. 2). 15 can be considered the precursor of the active cycloaddition substrate 16. In this study, 15 (100 μM) was saturated with varying concentrations of NaOH (with 10% DMSO) to force the elimination to generate 16. The formation of 4 h was monitored by its fluorescence. It was anticipated that the total tetrazine concentration would be present as 16 due to the saturation conditions, resulting in a pseudo-first order reaction. As shown in Figure 2, 4h was efficiently produced within 5 minutes with 0.1 M NaOH and within seconds with 1.0 M NaOH, suggesting a rapid cycloaddition rate in aqueous conditions. The overall second-order rate constant for this reaction was determined to be 0.18 M−1 s−1. This experimental rate constant was then used to model the overall reaction using the complex pathway simulator (COPASI) software (version 4.20).20 From this model, the first-order rate constant for the cycloaddition step (16 to 4 h) was calculated to be 0.64 ± 0.019 s−1 (Fig. S4†).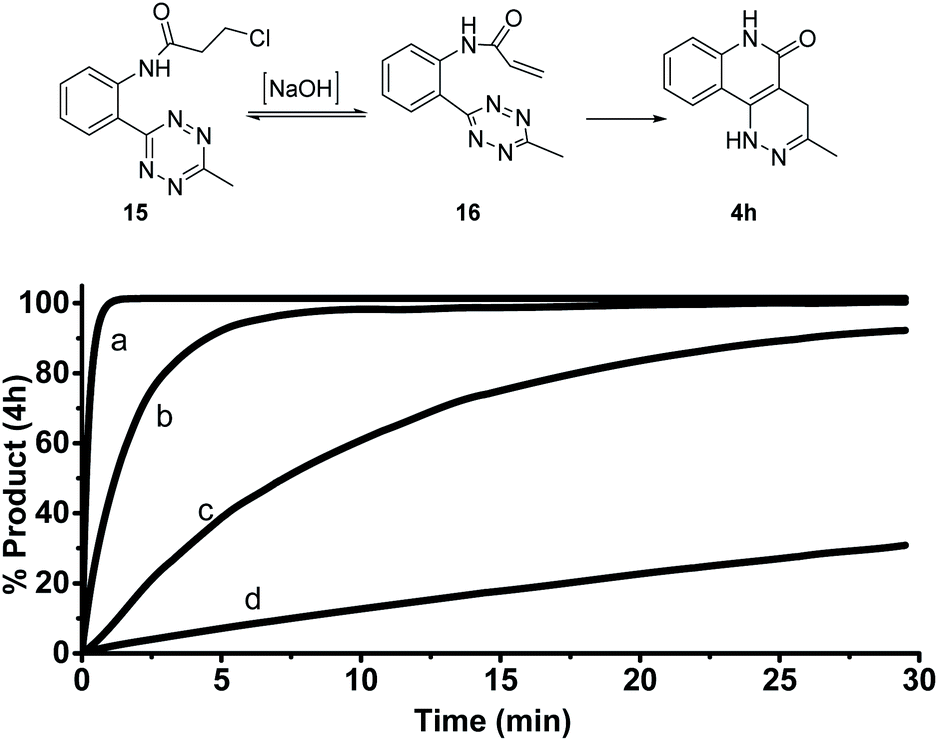 | ||
| Fig. 2 Formation of 4h from 15 monitored by fluorescence. Compound 15 (100 μM) was incubated in 10% DMSO and [NaOH] at 25 °C. λem: 425 nm, λex: 320 nm. (a) 1.0 M NaOH, (b) 0.1 M NaOH, (c) 0.01 M NaOH, (d) 0.001 M NaOH. | ||
To further understand the kinetics of the cycloaddition step alone, we used density functional theory (DFT) calculations to predict the Gibbs free energies of the intermediates and transition states in both the [4 + 2] Diels–Alder step and its subsequent retro-DA steps (Fig. 3). Starting from 16, the Gibbs free energies (ΔG) for the intermediates (17 and 18), the transition structures (TS1 and TS2), and the final product (4h) were calculated at the M06-2X/6-311G(d) level using H2O as the solvent. The ΔG of all structures after TS1 were found to be substantially negative (ΔG = −14.9 to −85.0 kcal mol−1) suggesting a very exergonic reaction. The activation free energy (ΔG‡ = 16.6 kcal mol−1) for TS1 corresponded to a first-order rate constant (k1) of 4.1 s−1 computed from the Eyring transition state theory at room temperature.21 This equated to a half-life (t1/2) for compound 16 of 0.17 seconds. This indicates a 6.4-fold increase in the first-order rate constant when compared to the experimentally calculated 0.64 ± 0.019 s−1 (in water with 10% DMSO). It is worth noting that the rate information shown in Fig. 2 and 3 suggests that the cycloaddition should complete rapidly while the reaction conditions in Fig. 1 show cycloaddition completion >1 hour. These results suggest a solvent dependence on reaction rates. It can be explained by the enhanced hydrophobic effect known to drastically increase reaction rates of Diels–Alder reactions in aqueous media over organic media by 200–5800 times.22 Thus, we believe this reaction is suitable for bioorthogonal applications.
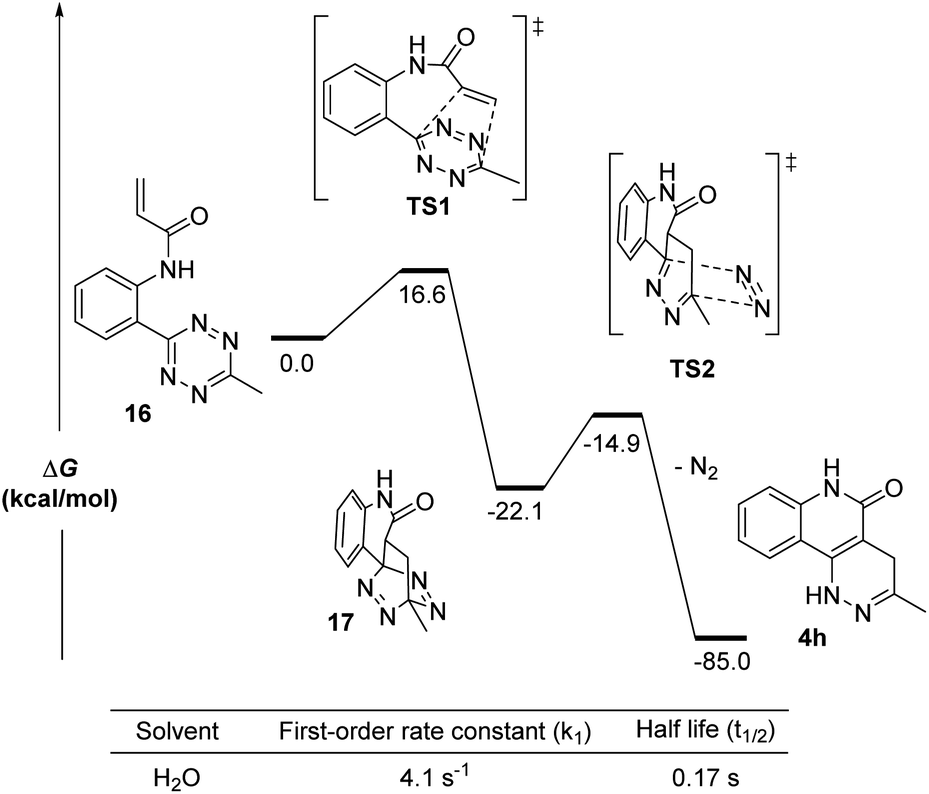 | ||
| Fig. 3 DFT calculations for the intramolecular cycloaddition of 16. Calculations were done on SMD(H2O)/M06-2X/6-311G(d). | ||
A ROS-triggered “release-then-click” system for MeSeOH release
With these results, we envisioned that the formation of 4h-like fluorescent products through a bioorthogonal process could have some applications. To this end, we explored appropriate precursors of tethered tetrazine-acrylamides and triggering strategies to form this pair under biologically relevant conditions. We expected that oxidative selenoxide formation and elimination could be used for this purpose. Endogenously, methylselenocysteine (MSC) is hypothesized to undergo selenoxide formation and elimination to produce dehydroalanine (for pyruvate synthesis) in oxidative environments.23 In this process, methylselenenic acid (MeSeOH) is produced as a critical selenium metabolite because it is a precursor for methylseleninic acid (MeSeO2H) and methylselenol (MeSeH); two compounds known for their chemo-preventative properties.24–27 Though MeSeOH is hypothesized to play an important role in biology, its true function is still unclear because it is too unstable to study. Thus, the development of small molecule donors that generate MeSeOH is needed to understand its biological functions. Using the intra-molecular tetrazine cycloaddition discovered herein, we propose a ROS-triggered “release-then-click” strategy that utilizes oxidative stress to release MeSeOH and a fluorescent tracer. This strategy can potentially slow down tumor progression as well as monitor the release of MeSeOH through fluorescence imaging. As a proof-of-concept, we synthesized a model tetrazine substrate (19) and tested its reactivity under different oxidative environments. As shown in Fig. 4, the oxidation of 19 should produce the selenoxide intermediate 20 followed by elimination and subsequent tetrazine cycloaddition to form 4h.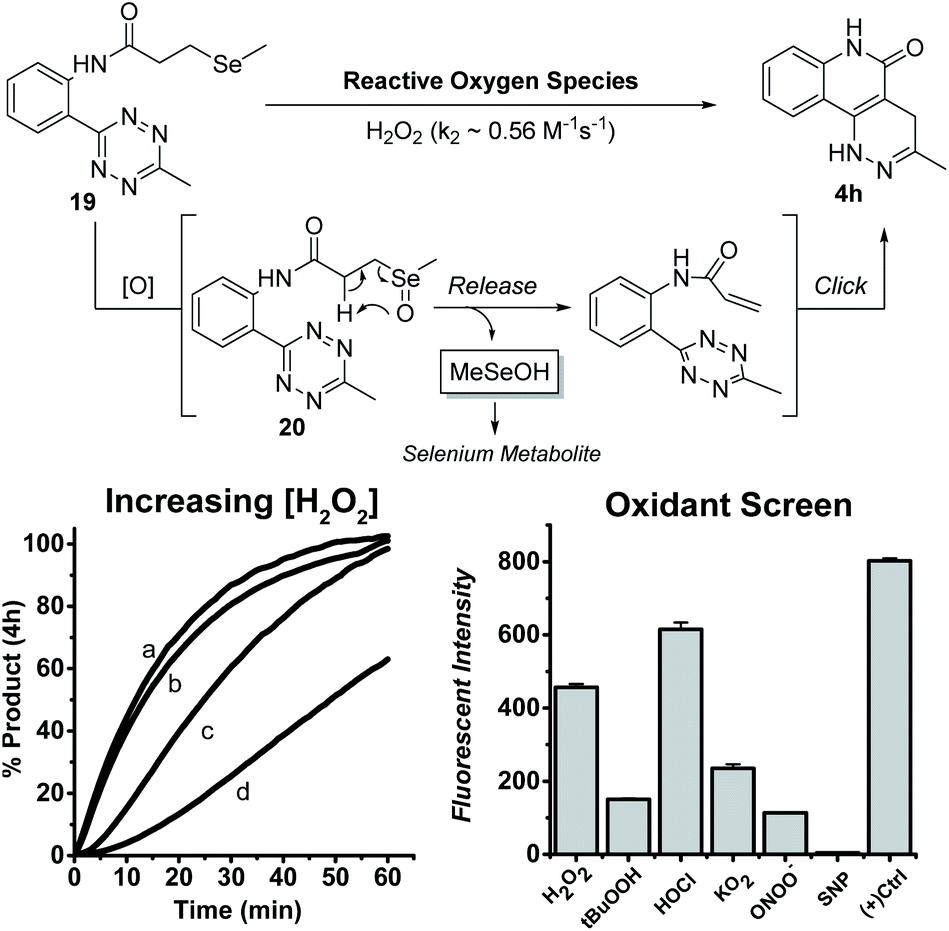 | ||
| Fig. 4 Increasing [H2O2]: 19 (100 μM), (a) 100.0 mM H2O2, (b) 50.0 mM H2O2 (c) 10.0 mM H2O2, (d) 1.0 mM H2O2 in 10% DMSO and 10 mM PBS buffer (pH = 7.4) at 25 °C. Oxidant Screen: 19 (100 μM) and oxidant (200 μM) in 10% DMSO and 10 mM PBS buffer (pH = 7.4) incubated at 37 °C for 1 h (+) Control: the positive control is reported as the average maximum intensities with pure compound 4h in 10% DMSO and 10.0 mM PBS buffer (pH = 7.4) incubated with each oxidant (200 μM) individually at 37 °C for 1 h. | ||
We first tested the reaction viability and 4h progression over time (monitored by fluorescence) with increasing H2O2 concentrations at rt in 10%DMSO/PBS buffer. Compound 19 (100 μM) was indeed activated by H2O2 to yield the fluorescent product 4h in quantitative yields within 1 h at excess H2O2 concentrations (50 and 100 mM). The second order rate constant (k2) for this reaction was calculated to be 0.56 M−1 s−1. Compound 19 (100 μM) was also screened against various oxidants at a more relevant concentration (200 μM) in the same solvent conditions at 37 °C for 1 h. Under these conditions, 19 could be activated by H2O2, hypochlorite (HOCl), tert-butyl hydroperoxide (t-BuOOH), superoxide (O2−), and peroxynitrite (ONOO−), with H2O2 and HOCl being the most reactive triggers. Nitric oxide (produced from sodium nitroprusside, SNP) did not trigger the reaction and this is expectable because NO is known as a weak oxidant compared to other ROS tested.28 The formation of MeSeOH in this process was proved by the observation of its decomposition product dimethyl diselenide (MeSeSeMe).
Having confirmed that compound 19 could release 4hin vitro in the presence of ROS, we wondered if this reaction could take place in response to intracellular ROS. It is known that there are elevated levels of ROS in cancer cells.29 Thus, HeLa cells were used in our studies. As shown in Fig. 5, HeLa cells pretreated with N-acetylcysteine (NAC) to quench intracellular ROS30,31 showed almost no fluorescence when treated with 19 (a and b), while cells not pretreated with NAC showed some fluorescent enhancement (c and d) attributed to endogenous ROS levels present in HeLa cells. This suggests that compounds like 19 can potentially detect endogenous ROS. When 200 μM H2O2 (e and f) or HOCl (g and h) were added exogenously, dramatic fluorescence enhancement was observed after a 30 min incubation. These results suggest that the intramolecular tetrazine-acryloyl cycloaddition does proceed in cellular media. As a result, compounds like 19 have potential as ROS-triggered fluorescent probes/MeSeOH donors. It is worth noting that 19 and 4h did not exhibit obvious cytotoxicity under concentrations up to 300 μM after 24 h (Fig. S5†).
 | ||
| Fig. 5 Fluorescent images of 4h release from 19 in HeLa cells: (a and b) cells were pre-treated with NAC (100 μM) and then treated with 19 (200 μM) for 1 h at 37 °C, (c and d) cells were treated with 19 for 1 h at 37 °C, (e and f) cells were treated with 19 for 1 h then washed and treated with H2O2 (200 μM) for 30 min at 37 °C, (g and h) cells were treated with 19 for 1 h then washed and treated with HOCl (200 μM) for 30 min at 37 °C. | ||
Conclusions
In summary, an intramolecular tetrazine-olefin cycloaddition between an acryloyl ester/amide and 1,2,4,5-tetrazine was discovered. This reaction produced unique coumarin-like heterocyclic products that showed strong fluorescence and excellent photo- and pH-stability. This property can be used to monitor the progress of the reaction. Structure reactivity relationship studies revealed that less sterically hindered tetrazines and non-substituted acrylate/acrylamides worked the best. The rate of cycloaddition was found to be solvent dependent and was determined using experimental data with a kinetic modelling software (COPASI) as well as DFT calculations (k1 = 0.64 ± 0.019 and 4.1 s−1, respectively). With these results, a “release-then-click” strategy was developed using compound 19 for the ROS-triggered release of MeSeOH and 4h in cellular media, suggesting that the rate of cycloaddition is suitable for bioorthogonal applications. Further bioorthogonal applications based on this intramolecular tetrazine-cycloaddition are under investigation in our lab.Data availability
All supporting data are provided in the ESI.†Author contributions
A. H. and M. X. conceived the project. A. H. carried out the experiments of synthesis, reactions, and spectroscopic measurements. O. L. L., Y. L., B. J. B., S. X. helped with the experiments. Q. C. performed the DFT calculation. M. S. performed cell imaging. J. R. R. performed X-ray crystallography. A. H. and M. X. wrote the paper. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by NSF (CHE2100870) and NIH (R01GM125968 to M. X.; 1F31HL158202 to A. H.). DFT calculation was conducted using resources and services at the Center for Computation and Visualization (CCV) at Brown University. X-Ray crystallography was supported in part by NSF (CHE2117549).References
- K. Porte, M. Riberaud, R. Châtre, D. Audisio, S. Papot and F. Taran, ChemBioChem, 2021, 22, 100–113 CrossRef CAS PubMed.
- V. Rigolot, C. Biot and C. Lion, Angew. Chem., Int. Ed., 2021, 60, 23084–23105 CrossRef CAS PubMed.
- M. Smeenk, J. Agramunt and K. M. Bonger, Curr. Opin. Chem. Biol., 2021, 60, 79–88 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed.
- S. Eising, A. H. J. Engwerda, X. Riedijk, F. M. Bickelhaupt and K. M. Bonger, Bioconjugate Chem., 2018, 29, 3054–3059 CrossRef CAS PubMed.
- C. Janzowski, V. Glaab, C. Mueller, U. Straesser, H. G. Kamp and G. Eisenbrand, Mutagenesis, 2003, 18, 465–470 CrossRef CAS PubMed.
- Y. K. Koleva, J. C. Madden and M. T. D. Cronin, Chem. Res. Toxicol., 2008, 21, 2300–2312 Search PubMed.
- J. Ou, J. Zheng, J. Huang, C. T. Ho and S. Ou, J. Agric. Food Chem., 2020, 68, 5039–5048 CrossRef CAS PubMed.
- L. J. Bolton, Curr. Org. Chem., 2014, 18, 61–69 CrossRef PubMed.
- Y. Zhao, A. K. Steiger and M. D. Pluth, Angew. Chem., Int. Ed., 2018, 57, 13101–13105 CrossRef CAS PubMed.
- M. E. Jung and J. Gervay, J. Am. Chem. Soc., 1989, 111, 5469–5470 CrossRef CAS.
- M. R. Karver, R. Weissleder and S. A. Hilderbrand, Bioconjugate Chem., 2011, 22, 2263–2270 CrossRef CAS PubMed.
- J. A. Wagner, D. Mercadante, I. Nikić, E. A. Lemke and F. Gräter, Chem.–Eur. J., 2015, 21, 12431–12435 CrossRef CAS PubMed.
- M. R. Nimlos, D. F. Kelley and E. R. Bernstein, J. Phys. Chem., 1987, 91, 6610–6614 CrossRef CAS.
- A. R. Todorov, T. Wirtanen and J. Helaja, J. Org. Chem., 2017, 82, 13756–13767 CrossRef CAS PubMed.
- A. C. Knall, M. Hollauf and C. Slugovc, Tetrahedron Lett., 2014, 55, 4763–4766 CrossRef CAS PubMed.
- A. Das, S. C. Mandal and B. Pathak, Phys. Chem. Chem. Phys., 2022, 24, 8387–8397 RSC.
- F. Liu, Y. Liang and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 11483–11493 CrossRef CAS PubMed.
- W. Blokzijl, M. J. Blandamer and J. B. F. N. Engberts, J. Am. Chem. Soc., 1991, 113, 4241–4246 CrossRef CAS.
- M. Rooseboom, J. N. M. Commandeur, G. C. Floor, A. E. Rettie and N. P. E. Vermeulen, Chem. Res. Toxicol., 2001, 14, 127–134 Search PubMed.
- C. Ip, Y. Dong and H. E. Ganther, Cancer Metastasis Rev., 2002, 21, 281–289 CrossRef CAS PubMed.
- J. T. Pinto, J. I. Lee, R. Sinha, M. E. MacEwan and A. J. L. Cooper, Amino Acids, 2011, 41, 29–41 CrossRef CAS PubMed.
- R. R. Ramoutar and J. L. Brumaghim, Cell Biochem. Biophys., 2010, 58, 1–23 CrossRef CAS PubMed.
- E. G. Varlamova and E. A. Turovsky, Int. J. Mol. Sci., 2021, 22, 6614 CrossRef CAS PubMed.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5839–5848 CrossRef CAS PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- Y. H. Han, Y. M. Yang, S. Z. Kim and W. H. Park, Anticancer Res., 2010, 30, 2107–2112 CAS.
- G. Spagnuolo, V. D'Antò, C. Cosentino, G. Schmalz, H. Schweikl and S. Rengo, Biomaterials, 2006, 27, 1803–1809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, compound characterizations, NMR spectra. CCDC 2193153. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04331a |
| This journal is © The Royal Society of Chemistry 2022 |