
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Symmetry-breaking synthesis of Janus Au/CeO2 nanostructures for visible-light nitrogen photofixation†
Henglei
Jia‡
 ,
Mengxuan
Zhao‡
,
Aoxuan
Du‡
,
Yanrong
Dou
and
Chun-yang
Zhang
*
,
Mengxuan
Zhao‡
,
Aoxuan
Du‡
,
Yanrong
Dou
and
Chun-yang
Zhang
*
College of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, China. E-mail: cyzhang@sdnu.edu.cn
First published on 24th October 2022
Abstract
Precise manipulation of the reactive site spatial distribution in plasmonic metal/semiconductor photocatalysts is crucial to their photocatalytic performance, but the construction of Janus nanostructures through symmetry-breaking synthesis remains a significant challenge. Here we demonstrate a synthetic strategy for the selective growth of a CeO2 semi-shell on Au nanospheres (NSs) to fabricate Janus Au NS/CeO2 nanostructures with the assistance of a SiO2 hard template and autoredox reaction between Ag+ ions and a ceria precursor. The obtained Janus nanostructures possess a spatially separated architecture and exhibit excellent photocatalytic performance toward N2 photofixation under visible-light illumination. In this scenario, N2 molecules are reduced by hot electrons on the CeO2 semi-shell, while hole scavengers are consumed by hot holes on the exposed Au NS surface, greatly promoting the charge carrier separation. Moreover, the exposed Au NS surface in the Janus structures offers an additional opportunity for the fabrication of ternary Janus noble metal/Au NS/CeO2 nanostructures. This work highlights the genuine superiority of the spatially separated nanoarchitectures in the photocatalytic reaction, offering instructive guidance for the design and construction of novel plasmonic photocatalysts.
Introduction
Artificial N2 fixation stands out as a potential strategy to convert naturally abundant N2 to NH3, since NH3 is an indispensable compound for all forms of life and modern societies.1–3 NH3 has recently intrigued interest as an alternative hydrogen carrier owing to its high hydrogen capacity (17.6 wt%) and ease of storage and transportation.4,5 Industrially, N2 fixation to NH3 is achieved via the classical Haber–Bosch process with N2 and H2 as feeding gases, but high temperature (>300 °C) and high pressure (>200 atm) conditions are required to overcome the rate-determining step that is the cleavage of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond, suffering from its intrinsic energy-intensive nature. Artificial photosynthesis of NH3 holds great promise as a flexible alternative to the Haber–Bosch process, because earth-abundant H2O other than H2 is employed as the reductant and the sustainable solar energy is the only energy source in the N2 photofixation process.6–10 Among various photocatalysts, semiconductor nanomaterials, especially those contain oxygen vacancies (OVs), have been widely explored for N2 photofixation, because the OVs in these catalysts can weaken the NN triple bond and activate N2 molecules via electron donation from the adjacent transition metal complexes, and ultimately promote the reduction of N2.11–19 An obstacle for the individual semiconductor photocatalyst is the recombination of the generated carriers. To address this issue, the formation of a Schottky barrier at the interface through integrating a semiconductor with a plasmonic metal has proved promising and very useful.20–25 Plasmonic metals (e.g., Au nanocrystals) possess unique properties of localized surface plasmon resonance (LSPR), which can not only improve the charge carrier separation but also promote the visible and near-infrared light activity of wide-bandgap semiconductors.26–28
N triple bond, suffering from its intrinsic energy-intensive nature. Artificial photosynthesis of NH3 holds great promise as a flexible alternative to the Haber–Bosch process, because earth-abundant H2O other than H2 is employed as the reductant and the sustainable solar energy is the only energy source in the N2 photofixation process.6–10 Among various photocatalysts, semiconductor nanomaterials, especially those contain oxygen vacancies (OVs), have been widely explored for N2 photofixation, because the OVs in these catalysts can weaken the NN triple bond and activate N2 molecules via electron donation from the adjacent transition metal complexes, and ultimately promote the reduction of N2.11–19 An obstacle for the individual semiconductor photocatalyst is the recombination of the generated carriers. To address this issue, the formation of a Schottky barrier at the interface through integrating a semiconductor with a plasmonic metal has proved promising and very useful.20–25 Plasmonic metals (e.g., Au nanocrystals) possess unique properties of localized surface plasmon resonance (LSPR), which can not only improve the charge carrier separation but also promote the visible and near-infrared light activity of wide-bandgap semiconductors.26–28
However, another challenge arises from the spatial architecture of plasmonic metal/semiconductor photocatalysts.29–33 For example, considerable efforts have been devoted to the synthesis of (plasmonic metal)@semiconductor core@shell nanostructures during the past few decades. A disastrous consequence for these symmetric nanostructures lies in that the hot holes generated in plasmonic metals can hardly be accessible by the reactant molecules, because the plasmonic metals are totally buried in the semiconductor shell,32 which leads to the recombination of charge carriers. A feasible way to achieve effective charge separation is breaking the symmetry in the growth process to construct asymmetric metal/semiconductor nanostructures, such as dumbbell-shaped or Janus nanostructures.34–41 We have previously reported dumbbell-shaped Au/end-CeO2 nanostructures through the site-selective growth of CeO2 on the ends of Au nanorods.37 In contrast to the selective growth on the ends of Au nanorods by virtue of the curvature difference, the selective growth of a semiconductor on the partial surface of Au NSs is also a grant challenge due to the spherically symmetric structures.
In this work, we developed a strategy for the construction of Janus Au NS/CeO2 nanostructures with the assistance of a SiO2 hard template in combination with an autoredox reaction between Ag+ and a ceria precursor. A CeO2 semi-shell with sufficient OVs is successfully grown on the partial surface of the Au NS core. In addition, the Janus Au NS/CeO2 nanostructures offer an additional opportunity for the selective growth of another noble metal on the exposed surface of the Au NSs to obtain ternary Janus noble metal/Au NS/CeO2 nanostructures. Moreover, the Janus Au NS/CeO2 nanostructures exhibit excellent photocatalytic performance toward N2 photofixation under visible-light illumination, benefiting from the spatially separated reaction active sites. The N2 photofixation rate of the Janus Au NS/CeO2 nanostructures is 5.3 times higher than that of the core@shell counterparts.
Results and discussion
Fig. 1 illustrates the synthesis process for the construction of Janus Au NS/CeO2 nanostructures. The whole process can be divided into four steps. (1) Growth of the Au NSs: cetyltrimethylammonium bromide (CTAB)-stabilized Au NSs were prepared using a seed-mediated growth method.42 (2) Growth of the Janus Au NS/SiO2 nanostructures: a SiO2 semi-shell is selectively grown on the partial surface of the Au NSs to obtain the Janus Au NS/SiO2 nanostructures through competitive ligand coordination between 4-mercaptophenylacetic acid (4-MPAA) and poly(acrylic acid) (PAA).43 Specifically, 4-MPAA and PAA ligands self-assemble over Au NSs, resulting in efficient phase separation on the Au NS surface. Since the surface functionalized with the 4-MPAA ligand possesses higher interface energy than the other surface coated with polymer PAA, a SiO2 semi-shell preferentially grows on the half surface functionalized with 4-MPAA upon the hydrolysis of the SiO2 precursor, leaving the PAA-coated half surface exposed. The SiO2 semi-shell is selected as the hard template because the growth of SiO2 on Au nanocrystals has been fully exploited.44–46 (3) Growth of the ternary Janus Au NS/SiO2/CeO2 nanostructures: a CeO2 semi-shell is therefore selectively grown on the exposed Au NS surface that is functionalized with the PAA ligand via an autoredox reaction between Ag+ ions and the ceria precursor.37,47 Ce(AC)3 is employed as the precursor of CeO2. It hydrolyzes into Ce(OH)3 at a temperature higher than 60 °C. The autoredox reaction is thereafter triggered following the reaction: Ag+ + Ce(OH)3 = Ag + CeO2.37 The Ag+ ion is selected as the oxidant because the product Ag has a small lattice mismatch with Au (0.2%).48 Due to the involvement of a small number of Ag+ ions, small Ag crystal nuclei are generated upon the rapid reduction by L-ascorbic acid (AA). These small nuclei can infiltrate the PAA ligand layer and grow on the Au NS surface. In addition, the PAA molecules can function as the stabilizing agent, facilitating the generation of Ag nanocrystals and a CeO2 semi-shell. The produced Ag can direct the preferential nucleation of CeO2 on the exposed Au NS surface. Once the CeO2 nuclei are generated, the ternary Janus Au NS/SiO2/CeO2 nanostructures are obtained through the overgrowth of the CeO2 semi-shell. (4) Etching of the SiO2 semi-shell to get the Janus Au NS/CeO2 nanostructures: due to the weak alkaline environment and the reaction system temperature (90 °C), the SiO2 semi-shell will be gradually etched after the growth of the CeO2 semi-shell in about 40 minutes without the addition of an etching agent.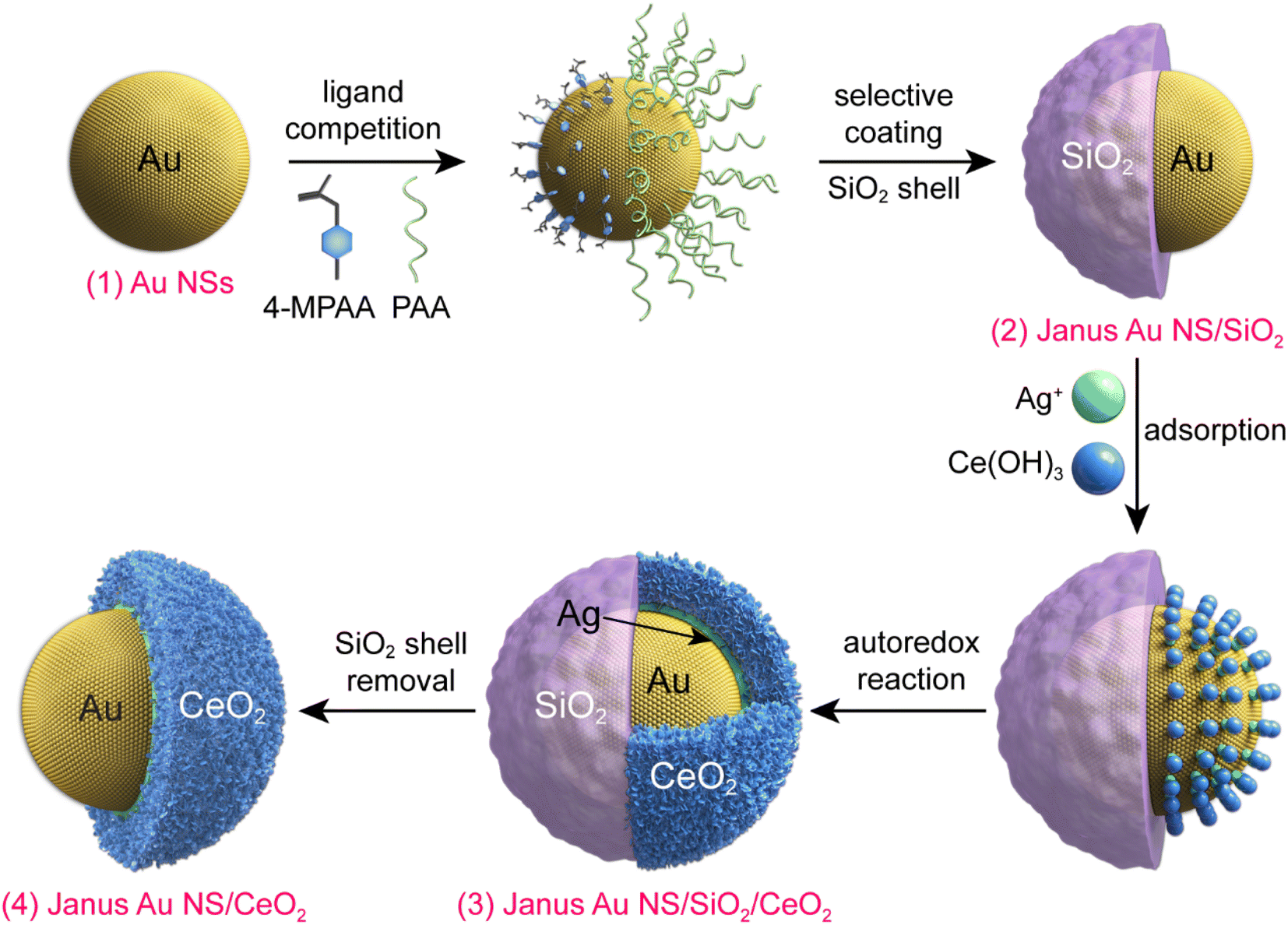 | ||
| Fig. 1 Schematic illustrating the synthesis process of the Janus Au NS/CeO2 nanostructures. | ||
The representative transmission electron microscopy (TEM) images of the Au NSs, Janus Au NS/SiO2 nanostructures, ternary Janus Au NS/SiO2/CeO2 nanostructures, and Janus Au NS/CeO2 nanostructures are displayed in Fig. 2a–d. The pre-grown Au NSs possess high shape and size monodispersity, with a diameter of 50.5 ± 2.5 nm (Fig. 2a). After the surface functionalization with two competing ligands, 4-MPAA and PAA, a SiO2 semi-shell with a thickness of 39.7 ± 3.6 nm is selectively grown on the partial surface of Au NSs to generate the Janus Au NS/SiO2 nanostructures (Fig. 2b). Next, the selective growth of the CeO2 semi-shell takes place on the exposed Au NS surface with the assistance of an autoredox reaction and the ternary Janus Au NS/SiO2/CeO2 nanostructures are obtained in high yield (Fig. 2c). In contrast to the SiO2 semi-shell, the CeO2 semi-shell looks darker owing to the relatively large atomic number. The successful overgrowth of a CeO2 semi-shell on the Janus Au NS/SiO2 nanostructures is further confirmed by high-resolution TEM (HRTEM) imaging (Fig. S1†), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging, and energy-dispersive X-ray (EDX) elemental mapping (Fig. S2†), respectively. Intriguingly, the SiO2 semi-shell in the ternary Janus Au NS/SiO2/CeO2 nanostructures is not stable in the reaction system due to the weak alkaline environment and the reaction temperature (90 °C). Eventually, the SiO2 semi-shell is etched in about 40 minutes (Fig. S3†). As depicted in Fig. 3d, a CeO2 semi-shell with a thickness of 12.3 ± 1.8 nm is selectively grown on the partial surface of the Au NSs to produce the final Janus Au NS/CeO2 nanostructures. The thickness of the CeO2 semi-shell may be readily tuned in the range of 6.9–21.8 nm by adjusting the amount of ceria precursor (Fig. S4†). The growth process can also be vividly observed from the extinction spectra (Fig. S5†), because the change of the surrounding medium can induce a shift of the resonance peak of Au nanocrystals.49 Owing to the significantly larger refractive index of CeO2 (2.2) than those of H2O (1.33) and SiO2 (1.46),50 the plasmon resonance peak of Au NSs exhibits a clear redshift (from 525.8 nm to 552.8 nm) after the growth of the CeO2 semi-shell, suggesting the formation of the Janus Au NS/CeO2 nanostructures.
 | ||
| Fig. 2 (a–d) TEM images of the Au NSs (a), Janus Au NS/SiO2 (b), ternary Janus Au NS/SiO2/CeO2 (c), and Janus Au NS/CeO2 nanostructures (d), respectively. (e) HAADF-STEM image and corresponding elemental maps of a representative Janus Au NS/CeO2 nanostructure. (f) HRTEM images of a single Janus Au NS/CeO2 nanostructure. | ||
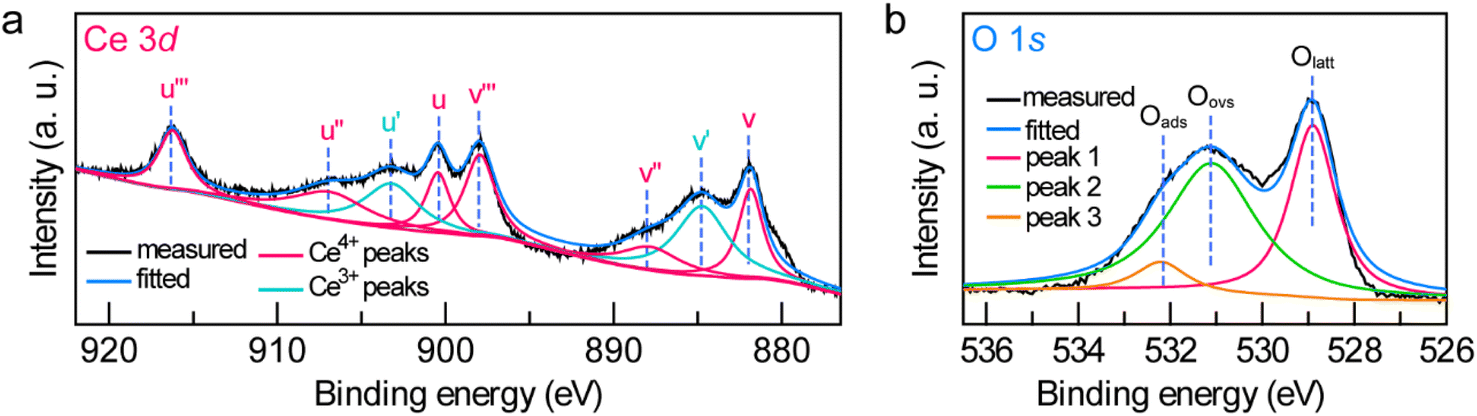 | ||
| Fig. 3 (a and b) High-resolution Ce 3d (a) and O 1s (b) XPS spectra of the Janus Au NS/CeO2 nanostructures. | ||
To gain a better understanding on the structural feature of the Janus Au NS/CeO2 nanostructures, HAADF-STEM imaging, EDX elemental mapping, and HRTEM imaging were conducted (Fig. 2e and f and S6†). HAADF-STEM and EDX elemental mapping results clearly demonstrate the Janus feature of the obtained sample (Fig. 2e), consistent with the elemental profile results (Fig. S6†). As displayed in the HRTEM images, the CeO2 semi-shell is loosely bound together with small CeO2 nanocrystals (Fig. 2f). The formation of crystalline CeO2 nanocrystals at a moderate temperature (90 °C) benefits from the autoredox reaction, since a temperature as high as about 400 °C is required to produce crystalline CeO2 in traditional synthesis strategies.
To further examine the crystalline nature and chemical states of the Janus Au NS/CeO2 nanostructures, X-ray diffraction (XRD) (Fig. S7†) and X-ray photoelectron spectroscopy (XPS) (Fig. 3a and b and S8†) were conducted. The XRD patterns of the Janus Au NS/CeO2 nanostructures present two distinct sets of diffraction peaks, one is assigned to the cubic Au phase, and the other belongs to the cubic CeO2 phase (Fig. S7†). Ag peaks are absent in the XRD results due to the small amount of metallic Ag in the Janus nanostructures. The presence of elements Au, Ag, Ce, and O in the Janus nanostructures is further ascertained by XPS results (Fig. S8†). The high-resolution Au 4f (Au 4f7/2 at 83.3 eV and Au 4f5/2 at 87.0 eV) and Ag 3d (Ag 3d5/2 at 367.2 eV and Ag 3d3/2 at 373.2 eV) XPS spectra suggest metallic Au0 and Ag0 states in the Janus nanostructures (Fig. S8†).37,51 The high-resolution Ce 3d XPS spectrum can be deconvoluted into eight peaks (Fig. 3a). Six peaks belong to the Ce(Iv) states, while the other two originate from the Ce(III) states.37 Specifically, there are two series of spin–orbit lines in the Ce 3d XPS spectrum, including Ce 3d5/2 (labelled v in Fig. 3a) and Ce 3d3/2 (labelled u in Fig. 3a) components.52,53 For Ce 3d5/2 levels, the v (Ce 3d94f2Ln−2, 881.85 eV), v′′ (Ce 3d94f1Ln−1, 887.8 eV), and v′′′ (Ce 3d94f0Ln, 897.95 eV) peaks belong to the main and satellite characteristic peaks of the Ce(IV) state, while the v′ (Ce 3d94f1Ln, 884.74 eV) peak is assigned to the Ce(III) state.54 The Ce 3d3/2 levels with the u structures have an identical assignment. The u (900.45 eV), u′′ (906.9 eV), and u′′′ (916.24 eV) peaks are assigned to the Ce(IV) state, while the u′ (903.2 eV) peak is assigned to the Ce(III) state. The other two peaks (v0 and u0) corresponding to the Ce 3d94f2Ln−1 state are absent in our sample due to tiny distinction with the 3d94f2Ln−2 state. The Ce(III)/Ce(IV) ratio is 0.35/0.65 calculated by integrating the peak areas. The existence of a high concentration of Ce(III) states (35%) in the CeO2 semi-shell corroborates the formation of abundant OVs in CeO2 nanocrystals. In addition, the Au/Ce elemental ratio calculated by integrating the XPS peak area is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.52. The O 1s spectrum can be fitted with three peaks (Fig. 3b). Peak 1 (528.9 eV) and peak 3 (532.2 eV) represent the crystal lattice oxygen and adsorbed oxygen or hydroxide ions, while peak 2 (531.1 eV) is assigned to the OVs in the CeO2 semi-shell,55 further confirming the presence of OVs in the as-prepared sample.
:2.52. The O 1s spectrum can be fitted with three peaks (Fig. 3b). Peak 1 (528.9 eV) and peak 3 (532.2 eV) represent the crystal lattice oxygen and adsorbed oxygen or hydroxide ions, while peak 2 (531.1 eV) is assigned to the OVs in the CeO2 semi-shell,55 further confirming the presence of OVs in the as-prepared sample.
The asymmetric Janus Au NS/CeO2 nanostructures offer an additional opportunity for the overgrowth of other noble metal nanocrystals (e.g., Ag, Pd, and Pt) on the exposed Au NS surface to produce ternary Janus noble metal/Au NS/CeO2 nanostructures. Because the lattice mismatch between the noble metal and Au is much smaller than that between the noble metal and CeO2, the noble metal tends to preferentially nucleate on the exposed Au NS surface. As a proof-of-concept, Ag nanocrystals were selected for the overgrowth to fabricate the ternary Janus Ag/Au NS/CeO2 nanostructures through the reduction of AgNO3 with AA in the presence of polyvinylpyrrolidone (PVP). As displayed in Fig. 4a, Ag nanocrystals with a diameter of about 46.7 ± 4.2 nm are selectively grown on the exposed Au NS surface to obtain the ternary Janus Ag/Au NS/CeO2 nanostructures. After the overgrowth of Ag nanocrystals, two new peaks emerge in the extinction spectrum, one is assigned to the plasmon resonance of Ag nanocrystals (426 nm) and the other is originated from the plasmon coupling between Au NSs and Ag nanocrystals (734 nm), suggesting the formation of Ag nanocrystals (Fig. S9†). The successful construction of the ternary Janus Ag/Au NS/CeO2 nanostructures is further revealed by the EDX elemental mapping and HRTEM results (Fig. 4b and c). Clearly, element Ag is located on one side of Au NSs, while elements Ce and O are mainly on the other side (Fig. 4b). This is in good agreement with the observation from the elemental profile results (Fig. S10†). In addition, XPS results confirm that the chemical states in the Janus nanostructures remain unchanged after the growth of Ag nanocrystals (Fig. S11†). Similar to the Ag overgrowth, Pd overgrowth on the Janus Au NS/CeO2 nanocrystals can be achieved by using cetyltrimethylammonium chloride (CTAC) as the stabilizing agent (Fig. S12†), because CTAC can stabilize the metal ions through the formation of complexes.45 The controllable growth of two different types of catalysts (noble metal and CeO2) on the plasmonic Au NSs opens a new avenue for exploring diverse photocatalytic applications as bifunctional photocatalysts.
 | ||
| Fig. 4 Ternary Janus Ag/Au NS/CeO2 nanostructures. (a) TEM image. (b) HAADF-STEM image and corresponding elemental maps. (c) HRTEM images. | ||
The spatially separated properties of the Janus Au NS/CeO2 nanostructures are favourable for charge separation, because the hot electrons and holes can participate in the reduction and oxidation reactions on the CeO2 semi-shell and exposed Au NS surface independently. The photocatalytic performance of the Janus Au NS/CeO2 nanostructures was evaluated using N2 photofixation under visible-light illumination. Methanol was employed as the hole scavenger. For comparison, four other catalysts, Au NSs (Fig. 2a), CeO2 nanocrystals (Fig. S13a†), a mixture of Au NSs with CeO2 nanocrystals, and Au NS@CeO2 core@shell nanostructures (Fig. S13b†), were also prepared. The produced NH3 amount was determined using the indophenol-blue method.24 A linear calibration relationship was obtained before the photocatalytic experiments (Fig. S14†).
Under visible-light illumination, Au NSs and CeO2 nanocrystals exhibit very weak N2 photofixation activity due to the lack of catalytic active sites or the weak light-harvesting capability (Fig. 5a). The catalytic activity is weak for simply mixing the Au NSs with CeO2 nanocrystals, suggesting that the direct contact of Au with CeO2 is of great importance to their synergistic effect. In the Au NS/CeO2 nanostructures, a Schottky barrier is formed at the interface that facilitates charge separation. The Au NS@CeO2 core@shell nanostructures display a slight improvement in the catalytic activity. In contrast, a significant improvement in the photocatalytic activity is obtained for the Janus Au NS/CeO2 nanostructures. The generation rate of NH3 on the Janus Au NS/CeO2 nanostructures is 52.7 ± 9.0 μmol h−1 g−1, which is 5.3 times higher than that of the core@shell counterparts (Fig. 5a). We compared the photocatalytic activity of the binary (Janus Au NS/CeO2) with that of the ternary (Janus Ag/Au NS/CeO2) Janus nanostructures as well. The binary Janus nanostructures exhibit higher photocatalytic activity toward the N2 photofixation (Fig. S15†). The excellent N2 photofixation activity benefits from the spatially separated active sites and the plasmonic properties of the Janus nanostructures. To investigate whether the N2 photofixation is driven by the LSPR of Au NSs, we calculated the apparent quantum efficiencies (AQE) by conducting photocatalytic experiments on the Janus Au NS/CeO2 nanostructures under the illumination of different monochromatic lights. As displayed in the action spectrum (Fig. S16†), the AQE values faithfully follow the LSPR of the Au NSs, with a maximum at about the LSPR wavelength. The above results confirm that the excellent N2 photofixation activity of the Janus nanostructures is driven by the LSPR of Au NSs. To trace the source of NH3, control experiments were conducted (Fig. S17†). No NH3 is detected under these conditions: (a) without catalysts, (b) in the dark, and (c) under an Ar atmosphere (Fig. S17†), implying that the generation of NH3 is originated from the N2 photofixation on the Janus nanostructures. The NH3 production rate is 9.0 ± 1.3 μmol h−1 g−1 in the absence of a sacrificial agent, which is much smaller than that obtained in the presence of a sacrificial agent, suggesting that the presence of a sacrificial agent facilitates charge separation and promotes the N2 photofixation activity. To further verify the nitrogen source, we performed 15N2 isotope labelling experiments. As displayed in Fig. 5b, N2 photofixation experiments are conducted under 14N2 and 15N2 atmospheres, respectively. When 14N2 is used as the feeding gas, three peaks can be detected in the 1H NMR spectrum of the reaction solution, which coincides with the standard 14NH4+ sample. In contrast, when 15N2 is employed as the feeding gas, two peaks associated with the standard 15NH4+ appear in the 1H NMR spectrum. The isotope labelling experiments reveal that the produced NH3 in the photocatalytic process is derived from the N2 reduction rather than contamination or other sources.56 To examine the stability of the Janus nanostructures, TEM imaging and XPS were performed after the typical photocatalytic process (Fig. S18 and S19†). TEM results suggest no morphological changes or aggregation after the photocatalytic process (Fig. S18†). In addition, the chemical states remain unchanged in the Janus nanostructures, as revealed by the XPS results. Moreover, the ratio of Ce(III) to Ce(IV) is about 0.34/0.66 and is close to that of the as-prepared sample, implying the stability of the OVs on the CeO2 semi-shell. To examine the recyclability, we compared the photocatalytic activities of the Janus nanostructures in three successive cycles (Fig. S20†). The Janus nanostructures exhibit excellent recyclability in the N2 photofixation. The above results confirm the excellent stability and recyclability of the Janus nanostructures during the photocatalytic process.
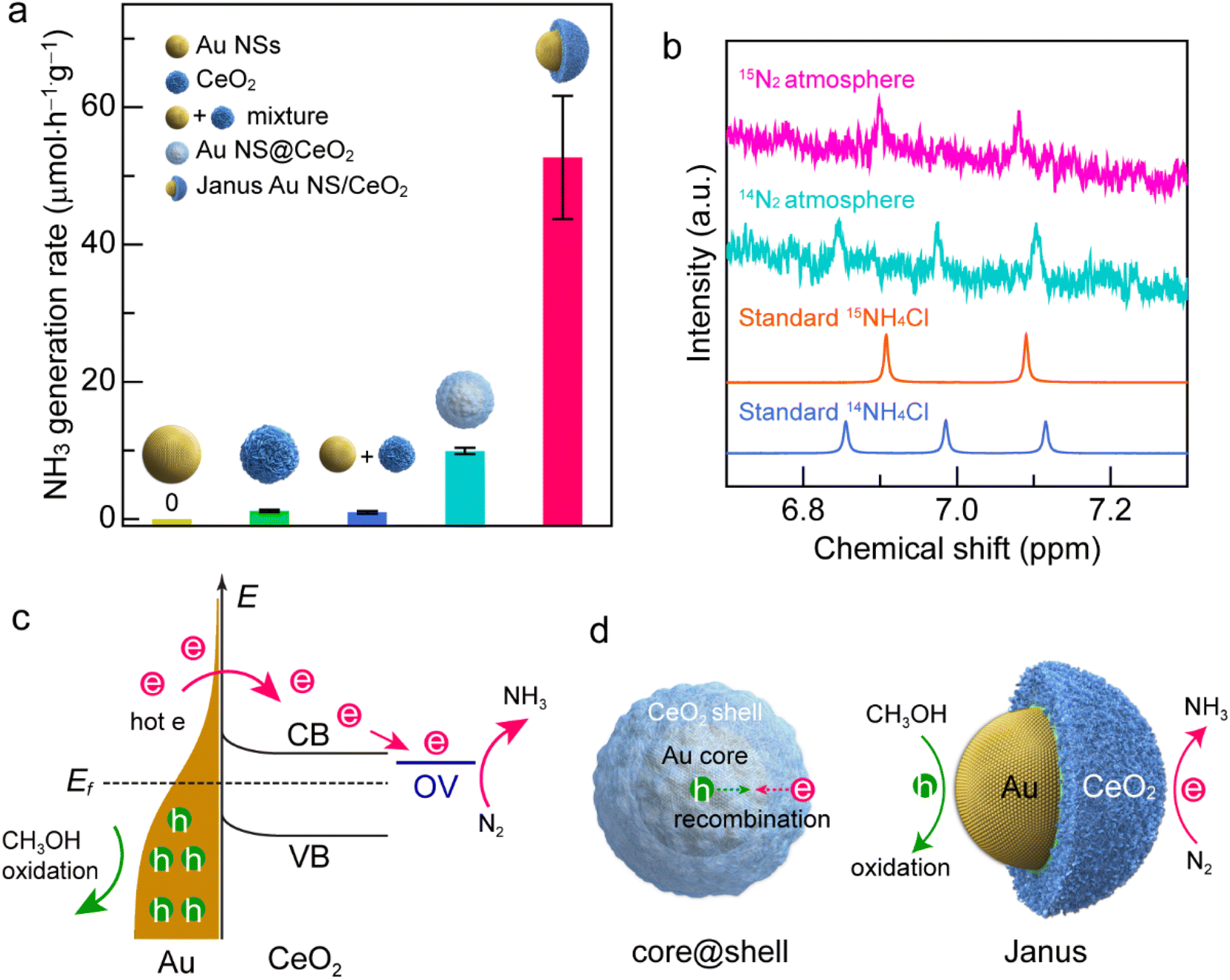 | ||
| Fig. 5 N2 photofixation under visible-light illumination. (a) Comparison of the NH3 generation rates on the different catalysts. (b) 1H NMR spectra of the reaction solution after a 2 h photocatalytic N2 reduction reaction using the Janus Au NS/CeO2 nanostructures as the catalyst in 14N2 and 15N2 atmospheres, respectively. (c) Photocatalytic N2 fixation mechanism on the Au NS/CeO2 nanostructures. CB, conduction band; VB, valence band; OV, oxygen vacancy states; Ef, Fermi level; e, hot electrons; h, hot holes. (d) Schematics illustrating the hot carrier separation behaviors of the core@shell nanostructure (left) and the Janus Au NS/CeO2 nanostructure (right). | ||
The excellent N2 photofixation activity of the Janus Au NS/CeO2 nanostructures is attributed to their unique architecture and spatially separated active sites. To investigate the activation sites for N2 fixation, we performed N2 temperature-programmed desorption (TPD) analysis (Fig. S21†). The Janus Au NS/CeO2 sample exhibits two peaks. The peak at a relatively low temperature is attributed to N2 physisorption, and the strong peak at a higher temperature is originated from N2 chemisorption. The presence of the chemisorption peak reveals that the activation sites for N2 fixation are the OV sites on the CeO2 nanocrystals. In regard to the plasmonic metal/semiconductor photocatalyst, it is commonly known that the Schottky barrier can efficiently promote the charge carrier separation.20 Under plasmon excitation, Au nanocrystals harvest light and produce hot electrons and hot holes (Fig. 5c). The hot electrons with sufficient energy can cross the Schottky barrier and inject into the CB of CeO2, leaving hot holes in the Au nanocrystals.37 Hot electrons and hot holes take part in the reduction and oxidation reactions on the active sites of CeO2 and Au nanocrystals, respectively. Since OVs are extensively present on the CeO2 surface, a localized electronic state (OV state) is formed and lies below the CB of CeO2. Hot electrons are prone to be trapped by the OV-induced defect states. In addition, N2 molecules tend to be adsorbed and activated at OV states, followed by reduction with hot electrons.24 Meanwhile, hot holes are consumed on Au nanocrystals to close the photocatalytic cycle. Nevertheless, electron–hole recombination occurs if the hot carriers are not consumed quickly.37,40 Because the surface of the Au NSs in core@shell nanostructures is totally covered by the CeO2 shell, the hot holes left in Au NSs can hardly be accessible and consumed by the hole scavengers, resulting in the recombination of the hot carriers (Fig. 5d, left). In contrast, the active sites for both oxidation and reduction reactions on the Janus Au NS/CeO2 nanostructures are fully exposed. N2 molecules are reduced by hot electrons on the OVs of the CeO2 semi-shell, while hole scavengers (CH3OH) are oxidized by hot holes on Au NSs, achieving efficient electron–hole separation (Fig. 5d, right).
Conclusions
In summary, we have demonstrated a synthetic strategy to construct an asymmetric metal/semiconductor plasmonic photocatalyst for visible-light N2 photofixation. With the assistant of a SiO2 hard template and an autoredox reaction between Ag+ ions and a CeO2 precursor, Janus Au NS/CeO2 nanostructures featuring spatially separated active sites are successfully obtained. The unique asymmetric nanostructures allow the hot electrons and hot holes to participate in the photocatalytic reactions at the CeO2 semi-shell and Au NS surface independently. In addition, the exposed Au NS surface in the Janus nanostructures offers more room for the overgrowth of another noble metal to fabricate ternary Janus noble metal/Au NS/CeO2 nanostructures as bifunctional photocatalysts. Moreover, the Janus Au NS/CeO2 nanostructures exhibit excellent N2 photofixation performance under visible-light illumination. The superior N2 photofixation activity benefits from the unique design and spatially separated active sites of the catalyst. This work offers a new strategy for rational arrangement of the active site spatial distribution in plasmonic photocatalysts, shedding new light on the design and construction of spatially separated nanostructures for photocatalytic applications.Author contributions
H. L. Jia, M. X. Zhao and A. X. Du contributed equally to this work. H. L. Jia and C.-y. Zhang designed and supervised the project. M. X. Zhao and A. X. Du prepared the materials and performed the photocatalytic tests. Y. R. Dou carried out the product detection. H. L. Jia and C.-y. Zhang wrote the paper. All authors revised the manuscript and discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21735003), the Natural Science Foundation of Shandong Province (No. ZR2020MB040), and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China.Notes and references
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed
.
- M. Nazemi and M. A. El-Sayed, Acc. Chem. Res., 2021, 54, 4294–4304 CrossRef PubMed
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef PubMed
- J. H. Yang, Y. Z. Guo, W. Z. Lu, R. B. Jiang and J. F. Wang, Adv. Mater., 2018, 30, 1802227 CrossRef PubMed
- E. Contreras, R. Nixon, C. Litts, W. X. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS
- S.-L. Meng, X.-B. Li, C.-H. Tung and L.-Z. Wu, Chem, 2021, 7, 1431–1450 CAS
- M. Cheng, C. Xiao and Y. Xie, Adv. Mater., 2021, 33, 2007891 CrossRef CAS
- C. Y. Hu, X. Chen, J. B. Jin, Y. Han, S. M. Chen, H. X. Ju, J. Cai, Y. R. Qiu, C. Gao, C. M. Wang, Z. M. Qi, R. Long, L. Song, Z. Liu and Y. J. Xiong, J. Am. Chem. Soc., 2019, 141, 7807–7814 CrossRef CAS PubMed
- B. B. Chang, Y. Z. Guo, D. H. Wu, L. Li, B. C. Yang and J. F. Wang, Chem. Sci., 2021, 12, 11213–11224 RSC
- S. Zhang, Y. X. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, Z. Wang, Y. X. Weng and T. R. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2554–2560 CrossRef CAS PubMed
- J. Li, H. Li, G. M. Zhan and L. Z. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS
- S. Y. Wang, X. Hai, X. Ding, K. Chang, Y. G. Xiang, X. G. Meng, Z. X. Yang, H. Chen and J. H. Ye, Adv. Mater., 2017, 29, 1701774 CrossRef
- Y. X. Zhao, Y. F. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. R. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef
- H. Y. Bai, S. H. Lam, J. H. Yang, X. Z. Cheng, S. S. Li, R. B. Jiang, L. Shao and J. F. Wang, Adv. Mater., 2022, 34, 2104226 CrossRef PubMed
- J. S. Lee, X. H. Liu, A. Kumar, Y. Hwang, E. Lee, J. M. Yu, Y. D. Kim and H. Lee, Chem. Sci., 2021, 12, 9619–9629 RSC
- P. S. Li, Z. A. Zhou, Q. Wang, M. Guo, S. W. Chen, J. X. Low, R. Long, W. Liu, P. R. Ding, Y. Y. Wu and Y. J. Xiong, J. Am. Chem. Soc., 2020, 142, 12430–12439 CrossRef PubMed
- Y. N. Sun, W. Pei, M. C. Xie, S. Xu, S. Zhou, J. J. Zhao, K. Xiao and Y. Zhu, Chem. Sci., 2020, 11, 2440–2447 RSC
- G. Q. Liu, Z. Q. Cui, M. M. Han, S. B. Zhang, C. J. Zhao, C. Chen, G. Z. Wang and H. M. Zhang, Chem. - Eur. J., 2019, 25, 5904–5911 CrossRef CAS
- R. B. Jiang, B. X. Li, C. H. Fang and J. F. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CrossRef CAS PubMed
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed
- E. Cortés, L. V. Besteiro, A. Alabastri, A. Baldi, G. Tagliabue, A. Demetriadou and P. Narang, ACS Nano, 2020, 14, 16202–16219 CrossRef
- J. H. Yang, Y. Z. Guo, R. B. Jiang, F. Qin, H. Zhang, W. Z. Lu, J. F. Wang and J. C. Yu, J. Am. Chem. Soc., 2018, 140, 8497–8508 CrossRef CAS
- S. Q. Luo, X. H. Ren, H. W. Lin, H. Song and J. H. Ye, Chem. Sci., 2021, 12, 5701–5719 RSC
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS
- Y. C. Zhang, S. He, W. X. Guo, Y. Hu, J. W. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927–2954 CrossRef CAS
- S. S. Li, H. Huang, L. Shao and J. F. Wang, ACS Nano, 2021, 15, 10759–10768 CrossRef CAS
- M. Ha, J.-H. Kim, M. You, Q. Li, C. H. Fan and J.-M. Nam, Chem. Rev., 2019, 119, 12208–12278 CrossRef
- D. D. Zheng, X.-N. Cao and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef PubMed
- J. W. Hong, D. H. Wi, S.-U. Lee and S. W. Han, J. Am. Chem. Soc., 2016, 138, 15766–15773 CrossRef PubMed
- S.-i. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef
- J. Piella, A. Gónzalez-Febles, J. Patarroyo, J. Arbiol, N. G. Bastús and V. Puntes, Chem. Mater., 2019, 31, 7922–7932 CrossRef CAS
- Z. Q. Huang, J. L. Gong and Z. H. Nie, Acc. Chem. Res., 2019, 52, 1125–1133 CrossRef CAS PubMed
- X. Q. Yang, Y. Lu, Y. Liu, J. Wang, L. Shao and J. F. Wang, Small Struct., 2021, 2100101 CrossRef CAS
- B. H. Wu, D. Y. Liu, S. Mubeen, T. T. Chuong, M. Moskovits and G. D. Stucky, J. Am. Chem. Soc., 2016, 138, 1114–1117 CrossRef CAS
- H. L. Jia, A. X. Du, H. Zhang, J. H. Yang, R. B. Jiang, J. F. Wang and C.-y. Zhang, J. Am. Chem. Soc., 2019, 141, 5083–5086 CrossRef CAS
- H. L. Jia, F. Li, T. H. Chow, X. Y. Liu, H. Zhang, Y. Lu, J. F. Wang and C.-y. Zhang, Nano Lett., 2022, 22, 7268–7274 CrossRef
- L. Weng, H. Zhang, A. O. Govorov and M. Ouyang, Nat. Commun., 2014, 5, 4792 CrossRef
- Z. W. Seh, S. H. Liu, M. Low, S.-Y. Zhang, Z. L. Liu, A. Mlayah and M.-Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef
- W. J. Xu, J. Jia, T. Wang, C. Li, B. W. He, J. P. Zong, Y. W. Wang, H. J. Fan, H. X. Xu, Y. H. Feng and H. Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 22246–22251 CrossRef PubMed
- Q. F. Ruan, L. Shao, Y. W. Shu, J. F. Wang and H. K. Wu, Adv. Opt. Mater., 2014, 2, 65–73 CrossRef
- T. Chen, G. Chen, S. X. Xing, T. Wu and H. Y. Chen, Chem. Mater., 2010, 22, 3826–3828 CrossRef CAS
- C. Hanske, M. N. Sanz-Ortiz and L. M. Liz-Marzán, Adv. Mater., 2018, 30, 1707003 CrossRef
- F. Wang, S. Cheng, Z. H. Bao and J. F. Wang, Angew. Chem., Int. Ed., 2013, 52, 10344–10348 CrossRef CAS PubMed
- X. Z. Zhu, H. L. Jia, X.-M. Zhu, S. Cheng, X. L. Zhuo, F. Qin, Z. Yang and J. F. Wang, Adv. Funct. Mater., 2017, 27, 1700016 CrossRef
- X. Wang, S. Y. Song and H. J. Zhang, Chem. Soc. Rev., 2020, 49, 736–764 RSC
- J. Liu and J. T. Zhang, Chem. Rev., 2020, 120, 2123–2170 CrossRef CAS
- J. P. Zheng, X. Z. Cheng, H. Zhang, X. P. Bai, R. Q. Ai, L. Shao and J. F. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed
- A. ¡ncel, T. Güner, O. Parlak and M. M. Demir, ACS Appl. Mater. Interfaces, 2015, 7, 27539–27546 CrossRef
- Y. H. Zheng, C. Q. Chen, Y. Y. Zhan, X. Y. Lin, Q. Zheng, K. M. Wei and J. F. Zhu, J. Phys. Chem. C, 2008, 112, 10773–10777 CrossRef CAS
- B. X. Li, T. Gu, T. Ming, J. X. Wang, P. Wang, J. F. Wang and J. C. Yu, ACS Nano, 2014, 8, 8152–8162 CrossRef PubMed
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28 CrossRef
- C. Ho, J. C. Yu, T. Kwong, A. C. Mak and S. Lai, Chem. Mater., 2005, 17, 4514–4522 CrossRef
- Q. Y. Wang, Y. X. Li, A. Serrano-Lotina, W. Han, R. Portela, R. X. Wang, M. A. Bañares and K. L. Yeung, J. Am. Chem. Soc., 2021, 143, 196–205 CrossRef PubMed
- J. H. Yang, H. Y. Bai, Y. Z. Guo, H. Zhang, R. B. Jiang, B. C. Yang, J. F. Wang and J. C. Yu, Angew. Chem., Int. Ed., 2021, 60, 927–936 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03863c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |