
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxy-directed iridium-catalyzed enantioselective formal β-C(sp2)–H allylic alkylation of α,β-unsaturated carbonyls†
Sankash
Mitra
 ,
Rahul
Sarkar‡
,
Aditya
Chakrabarty‡
and
Santanu
Mukherjee
*
,
Rahul
Sarkar‡
,
Aditya
Chakrabarty‡
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@iisc.ac.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 13th October 2022
Abstract
Hydroxy-directed iridium-catalyzed enantioselective formal β-C(sp2)–H allylic alkylation of kojic acid and structurally related α,β-unsaturated carbonyl compounds is developed. This reaction, catalyzed by an Ir(I)/(P,olefin) complex, utilizes the nucleophilic character of α-hydroxy α,β-unsaturated carbonyls, to introduce an allyl group at its β-position in a branched-selective manner in good to excellent yield with uniformly high enantioselectivity (up to >99.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 er). To the best of our knowledge, this report represents the first example of the use of kojic acid in a transition metal catalyzed highly enantioselective transformation.
:0.1 er). To the best of our knowledge, this report represents the first example of the use of kojic acid in a transition metal catalyzed highly enantioselective transformation.
Introduction
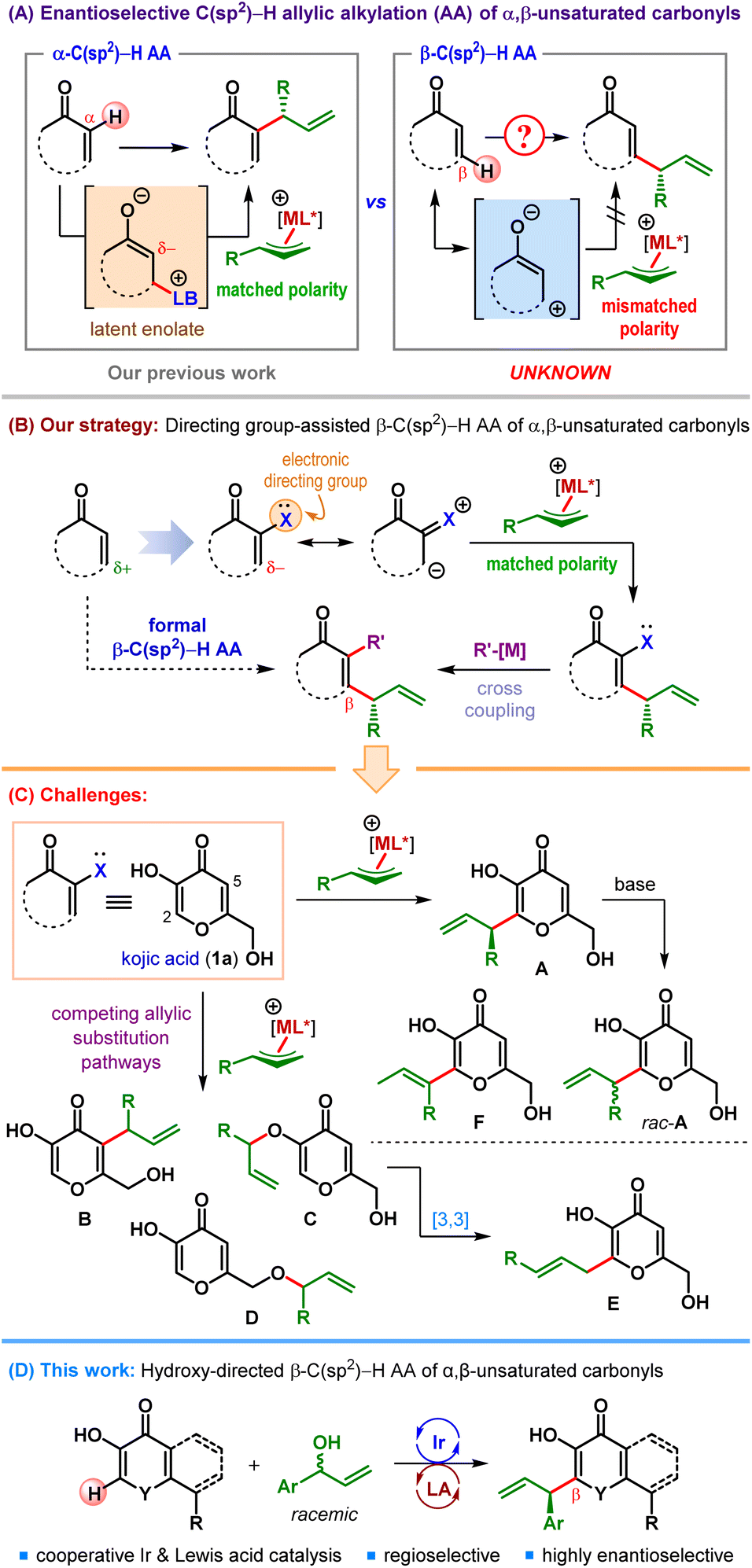
Iridium-catalyzed asymmetric allylic substitution (AAS) reaction has been established as a reliable method for enantioselective construction of carbon–carbon and carbon–heteroatom bonds using various classes of nucleophiles.1 In the realm of carbon-centered nucleophiles, both stabilized as well as unstabilized nucleophiles were successfully employed and led to several formal C(sp3)–H allylic alkylation (AA) reactions.2 Ir-catalyzed enantioselective C(sp2)–H AA reactions, in contrast, are far less developed and confined primarily to electron-rich (hetero)aromatic C(sp2)–H bonds.3The ability to selectively introduce an allyl group either at the α- or at the β-position of α,β-unsaturated carbonyl compounds while retaining the unsaturation marks a synthetic strategy having the potential to access a relatively less explored chemical space. Such C(sp2)–H allylic alkylation reactions are exceedingly rare.4 Although sporadically known since 2003,4d we have recently developed the first enantioselective formal α-C(sp2)–H allylic alkylation under cooperative Lewis base (LB) and Ir-catalysis (Scheme 1A).4a These α-selective allylic alkylation reactions of α,β-unsaturated carbonyl compounds were made possible due to their latent enolate character, which renders their α-position nucleophilic.5
 | ||
| Scheme 1 Catalytic enantioselective C(sp2)–H allylic alkylation of α,β-unsaturated carbonyls. | ||
The β-position of α,β-unsaturated carbonyls, on the other hand, is intrinsically electrophilic (Scheme 1A). Consequently, β-C(sp2)–H allylic alkylation of α,β-unsaturated carbonyls using an electrophilic allyl fragment is challenging due to the inherent polarity mismatch and therefore remains elusive.6
With the aim of expanding the synthetic toolbox involving β-C(sp2)–H functionalization of α,β-unsaturated carbonyl compounds, we became interested in conceptual development of this intriguing transformation. We surmised that a suitably positioned and easily modifiable electronic directing group (X in Scheme 1B) within the core structure of α,β-unsaturated carbonyl might impart sufficient nucleophilicity at its β-position to allow for the introduction of an electrophilic fragment.
To this end, our initial thoughts revolved around α-hydroxy α,β-unsaturated carbonyls (X = OH, Scheme 1B) since the hydroxyl functionality, after serving its role as the electronic directing group, can be transformed into a leaving group, and engage in various cross-coupling reactions. The overall process would retain the unsaturation and represent a formal β-C(sp2)–H functionalization of α,β-unsaturated carbonyl compounds.
Our search for suitable α-hydroxy α,β-unsaturated carbonyl compounds led us to kojic acid (1a) – a naturally occurring chelating agent produced primarily by fungi (Scheme 1C).7 Due to its high bioavailability, encouraging toxicity profile as well as high metal binding affinity,8 this class of 3-hydroxy-4-pyrones found a wide range of medicinal applications.9
Despite its widespread prevalence and densely functionalized scaffold, catalytic enantioselective reactions involving kojic acid remain surprisingly underdeveloped.10 In 2010 Bode et al. reported an elegant enantioselective Coates–Claisen rearrangement of kojic acid derivatives catalyzed by N-heterocyclic carbenes.11 Later on, Zlotin and Reddy groups independently disclosed kojic acid as a nucleophile in organocatalytic asymmetric Michael reactions with electrophilic partners such as nitroolefins and β,γ-unsaturated α-ketoesters.12 Nonetheless, compared to these organocatalytic reactions, metal catalyzed transformations of kojic acid are rare,13 let alone their enantioselective variants.14
A possible reason for this paucity is the presence of several Lewis basic functionalities in kojic acid, which can open up multiple reaction pathways apart from chelating the metal ions. For example, a reaction of unprotected kojic acid (1a) with an allylic electrophile can potentially result in two different allylic etherification products15 (C and D) and a C5-allylic alkylation product (B) apart from the desired C2-allylic alkylation product A (Scheme 1C). In fact, multiple allylic alkylation of kojic acid is also possible. The allylic ether C can subsequently undergo Claisen rearrangement13,14,16 to generate a linear C2-allylic alkylation product E. The competing branched vs. linear selectivity in the allylic substitution step can further complicate the scenario. In addition, the adjacent α,β-unsaturated carbonyl functionality renders the proton present at the stereocenter in A reasonably acidic, which can cause racemization under basic conditions and even stereoablation through olefin isomerization to thermodynamically favored F. Therefore, a successful β-C(sp2)–H allylic alkylation of kojic acid necessitates the circumvention of all these competing pathways.
Herein, we present the results of a study which culminated in the first catalytic enantioselective β-C(sp2)–H allylic alkylation of kojic acid, its derivatives and structurally related α-hydroxy α,β-unsaturated carbonyl compounds (Scheme 1D).
Results and discussion
To address the challenges discussed above, we initially focused on finding reaction conditions which would allow for the access of C2-allylic kojic acid selectively. Our preliminary experiments were aimed on probing the effects of different combinations of substrates with ligands, promoters and solvents on the efficiency and selectivity of the reaction.17 Gratifyingly, in the presence of 3 mol% [Ir(COD)Cl]2 and 12 mol% Carreira's (P,olefin) ligand (S)-L1c along with 20 mol% of Zn(OTf)2 as the Lewis acidic promoter in THF at 25 °C, the desired C2-allylic alkylation of kojic acid 1a took place using branched allylic alcohol rac-2a as the electrophilic partner (Table 1). Under these conditions, β-C(sp2)–H allylic kojic acid 3aa was isolated in 50% yield after 48 h with 99.8:0.2 er (entry 1). We were pleased to find that neither any of the allylic etherification products (C and D, Scheme 1C) nor the C5-allylic alkylation product B could be detected and 3aa was isolated exclusively as a single regioisomer (>20:1 b/l). Despite rate enhancement, no significant improvement in yield was observed when the reaction was carried out at an elevated temperature (entry 2). Switching the promoter to Fe(OTf)2 led to an increase in reaction rate and yield while retaining the enantioselectivity (entry 3). The use of other Lewis acidic promoters and solvents proved deleterious to the yield of this reaction (entries 4–7). As expected, no reaction took place in the absence of a Lewis acid promoter.17 Increasing the amount of allylic electrophile 2a turned out to be beneficial, and a 1:1.5 ratio of 1a and 2a proved to be the best, furnishing 3aa essentially as a single enantiomer in 91% yield (entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Promoter | Solvent | t [h] | Yielda [%] | erb |
|
a Yields were determined by 1H-NMR spectroscopy with mesitylene as an internal standard.
b Enantiomeric ratio (er) of the desired product was determined by HPLC analysis on a chiral stationary phase.
c Yield corresponds to the isolated product after chromatographic purification.
d Reaction at 50 °C.
e Reaction on a 0.2 mmol scale with 1:1.5 ratio of 1a and 2a.
|
|||||
| 1 | Zn(OTf)2 | THF | 48 | 50c | 99.8:0.2 |
| 2d | Zn(OTf)2 | THF | 24 | 54 | 99.9:0.1 |
| 3 | Fe(OTf)2 | THF | 24 | 81c | 99.9:0.1 |
| 4 | Sc(OTf)3 | THF | 36 | 62 | 99.9:0.1 |
| 5 | Fe(OTf)2 | 2-Me THF | 24 | 72c | 99.9:0.1 |
| 6 | Fe(OTf)2 | Toluene | 24 | 50c | >99.5:0.5 |
| 7 | Fe(OTf)2 | Acetone | 24 | 75 | 99.9:0.1 |
| 8e | Fe(OTf)2 | THF | 22 | 91c | 99.9:0.1 |

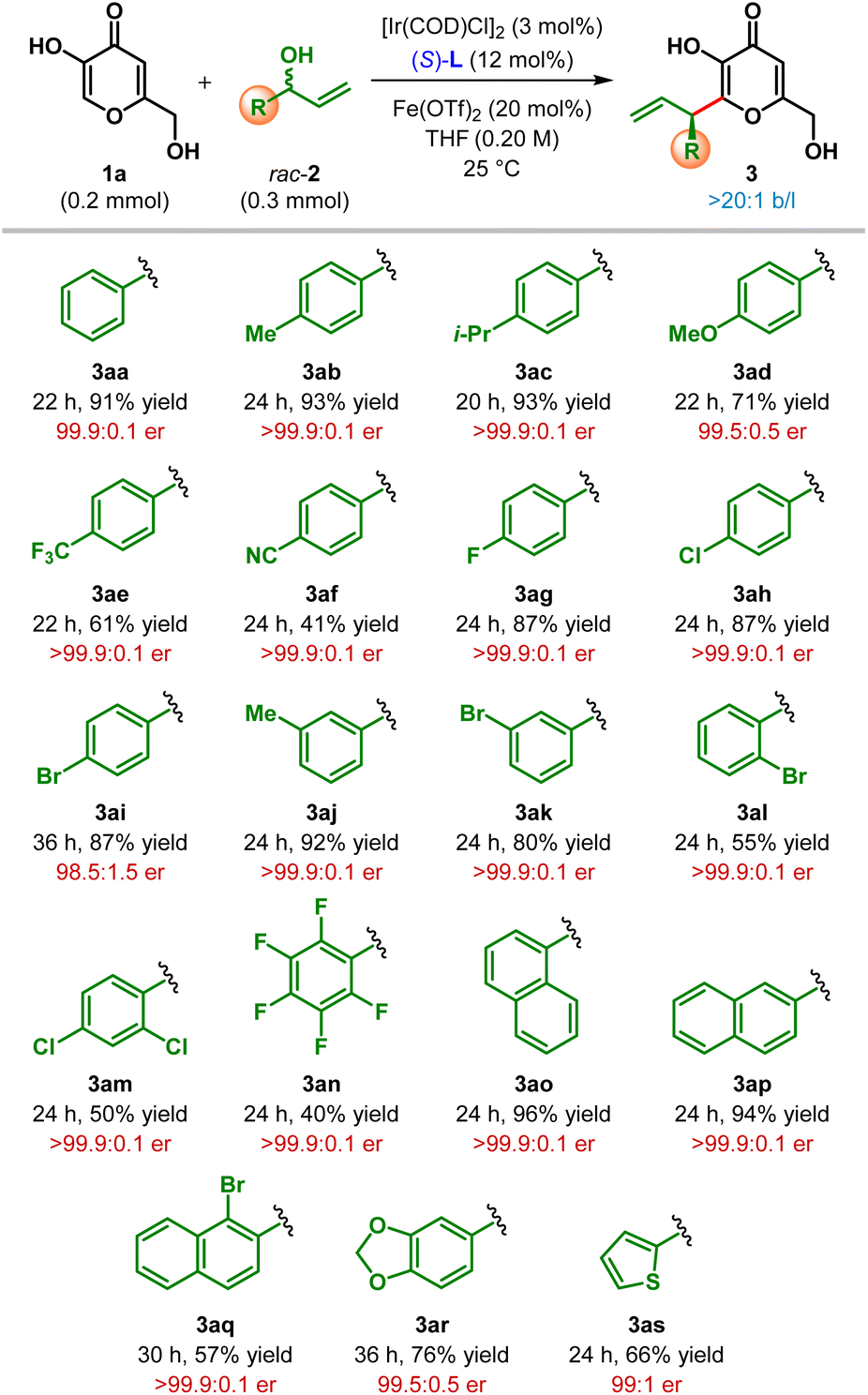
Having optimized the catalyst, promoter and the other reaction parameters (Table 1, entry 8), we assessed the generality of this hydroxy-directed β-C(sp2)–H allylic alkylation of α,β-unsaturated carbonyl compounds. These conditions were found to be suitable for a variety of branched allylic alcohols 2. As shown in Table 2, allylic alcohols (2b–2s) bearing both electron-rich as well as electron-deficient aryl substituents were tolerated and delivered the products (3ab–as) in moderate to excellent yield with uniformly high enantioselectivity. Allylic alcohols with electron-rich aryl substituents generally resulted in the products with higher yield compared to those having electron-deficient aryl groups. Highly electron deficient p-cyanophenyl and pentafluorophenyl substituted allylic alcohols (2f and 2n, respectively) adversely affected the yield of the reaction. Similarly, ortho-substituents on the aryl ring of allylic alcohols resulted the products (3al, 3am, 3an and 3aq) with only diminished yield, even though outstanding enantioselectivity was retained in each of these cases.
| a Yields correspond to the isolated product after chromatographic purification. Enantiomeric ratios (er) were determined by HPLC analysis on a chiral stationary phase. |
|---|
|
Heterocyclic substituents such as dioxolane and thiophene could also be introduced into the product under our standard reaction conditions with moderate to good yield and excellent enantioselectivity (3ar–as). In all cases, the products were obtained exclusively as a branched isomer. No competing side reaction (cf.Scheme 1C) was detected in any of these cases. Unfortunately, alkyl substituted allylic alcohols failed to participate in this allylic alkylation reaction and remained as a limitation of our protocol.17
After demonstrating the generality and limitations of allylic electrophile, we set out to explore the scope of nucleophile in this enantioselective AA reaction. Accordingly, a number of kojic acid derivatives and structurally related α-hydroxy α,β-unsaturated carbonyl compounds were tested. We were pleased to find that kojic acid derivatives bearing a variety of substituents at the C6 position underwent facile allylic alkylation under our optimized reaction conditions (Table 3). For example, maltol (1b), devoid of a hydroxyl group, furnished the desired product 3ba in excellent yield with 99.9:0.1 er. To showcase the functional group tolerance of this protocol, the hydroxyl unit of the hydroxymethyl group at the C6 position of kojic acid was replaced with other functionalities. These examples include chloro (1c), azide (1d), thiophenol (1e) and benzoyl ester (1h), all of which were found to be competent substrates in this reaction and underwent highly enantioselective allylic alkylation. These products (3da, 3ea and 3ha) contain synthetically relevant functional groups, which can serve as handles for further manipulation (see below). Allylic alkylation of structurally related α-hydroxy α,β-unsaturated carbonyl compounds such as 3-hydroxy chromone (1f) and 2-hydroxy naphthoquinone (1g) also proceeded without any difficulty, affording the products (3fa–ga) with excellent enantioselectivities. The success of the benzoyl-protected kojic acid (1h) in this reaction opened the possibility of tagging biologically relevant compounds to our products, for testing further functional group tolerance and stereochemical outcome. Consequently, the C6-hydroxymethyl group of kojic acid was acylated with pharmaceuticals such as indomethacin, naproxen, dehydrocholic acid and cis-pinonic acid. All these kojic acid derivatives (1i–l) were found to be suitable substrates in this AA reaction and furnished the corresponding products (3ia–la) in good to excellent yields with outstanding stereoselectivities, while retaining the existing functionalities and the molecular complexity. The existing stereocenters in these substrates (1j–l) did not exert any influence on the stereochemical outcome as these reactions were found to be completely catalyst controlled.
| a Yields correspond to the isolated product after chromatographic purification. Enantiomeric ratios (er) were determined by HPLC analysis on a chiral stationary phase. Diastereomeric ratios (dr) were determined by 1H NMR analysis of the crude reaction mixtures. |
|---|

|
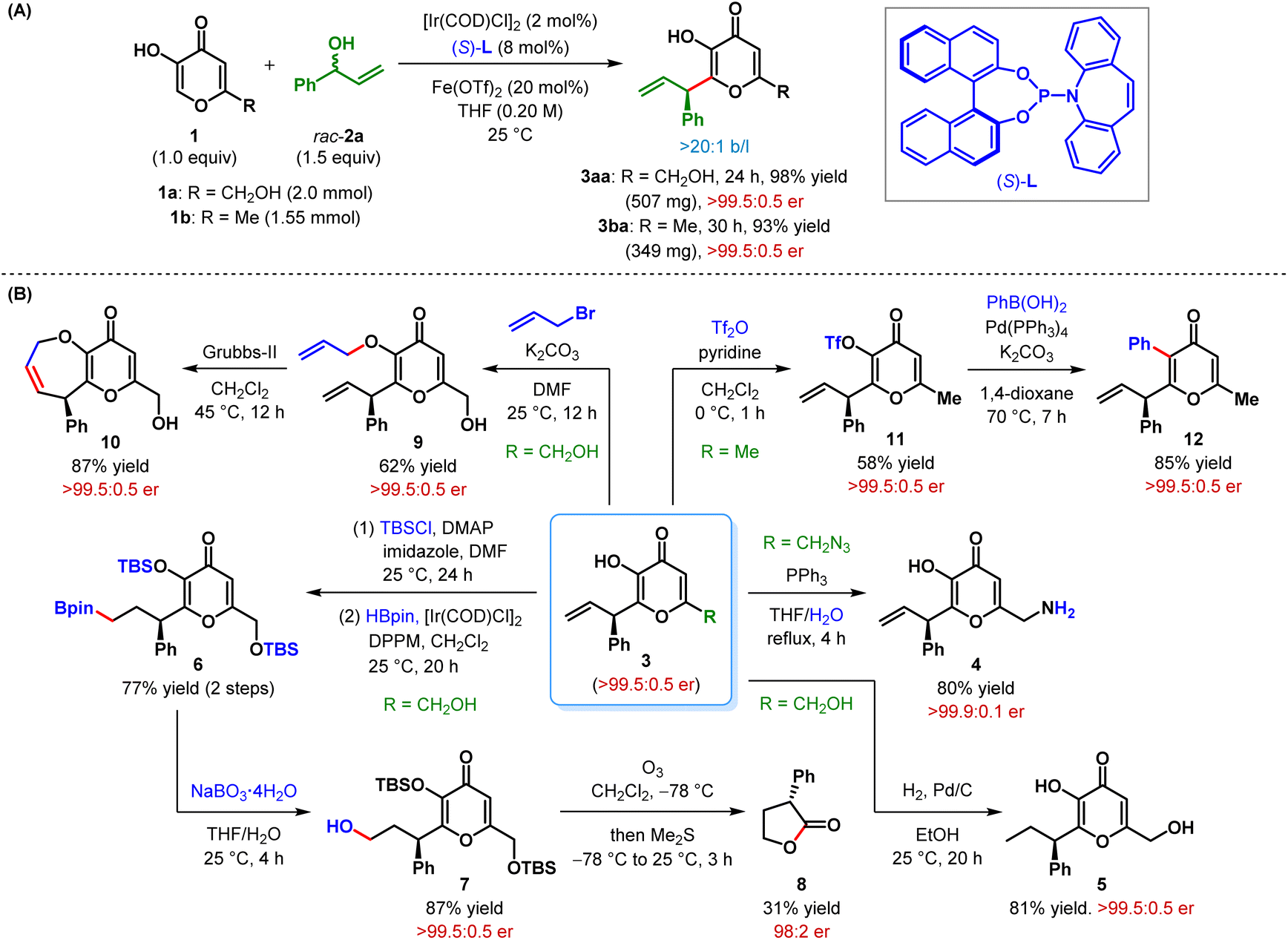
The scalability of our protocol was displayed by larger scale experiments on kojic acid (1a) and maltol (1b) (Scheme 2A). Using a slightly lower catalyst loading, these reactions, with 2a as the allylic electrophile, furnished the products (3aa–ba) in somewhat improved yield without any erosion in enantioselectivity.
 | ||
| Scheme 2 (A) Scale-up experiments and (B) synthetic elaborations of β-allyl 3-hydroxypyranone derivatives. | ||
The prospective potential of this β-C(sp2)–H AA reaction was further illustrated by successful elaboration of some of these intricately functionalized β-allyl 3-hydroxypyranone derivatives (Scheme 2B). For example, Staudinger reduction of 3da delivered the corresponding kojic amine derivative 4 in 80% yield. Notably, kojic amine is known as a novel γ-aminobutyric acid analogue and exhibits skeletal muscle relaxant as well as antinociceptive activities.18 Selective hydrogenation of the terminal olefin in 3aa with palladium on carbon afforded 5, featuring an aliphatic side chain, in 81% yield. Although the Ir-catalyzed hydroboration19 of the terminal olefin could not be performed directly on 3aa, the reaction proceeded smoothly on the double-TBS-protected 3aa to furnish the alkyl boronate 6 in 77% yield over two steps. Oxidation of 6 under sodium perborate resulted in an overall anti-Markovnikov hydration product 7 in 87% yield. The alcohol 7, when exposed to ozonolytic conditions, furnished 3-phenyldihydrofuranone 8via oxidative cleavage of the pyranone ring and cyclization. Notwithstanding the poor yield of butanolide 8 and slight erosion of its enantiopurity during the ozonolysis step, its formation helped us to determine the absolute configuration of the allylic alkylation product 3aa in retrospect, as the enantioselective synthesis of 8 was previously reported by MacMillan et al.20 The absolute stereochemistry of the other products (3), shown in Tables 2 and 3, were inferred as the same by analogy.
The enolic hydroxy group of 3aa could be selectively alkylated with allyl bromide to produce the O-allyl kojic acid derivative 9 in 62% yield. Ring-closing metathesis of this diallyl derivative under Grubbs-II furnished chiral dihydropyrano-oxepinone derivative 10 in high yield without any loss of its stereochemical integrity. Upon treatment with triflic anhydride, 3ba was converted to enol triflate 11 in moderate yield. This triflate derivative 11 was then successfully subjected to Pd-catalyzed Suzuki cross-coupling with phenyl boronic acid to obtain α-phenyl β-allyl pyranone derivative 12 in high yield while maintaining the er. The overall process represents a formal β-C(sp2)–H allylic alkylation of a pyranone derivative, which is inherently electrophilic at its β-position. Introduction of other (herero)aryl, alkenyl, alkynyl and even alkyl substituents at the α-position by cross-coupling of triflate such as 11 is in principle possible.21
We have conducted a control experiment to ascertain the role of the α-hydroxy group in this regioselective allylic alkylation reaction. Thus, α-methoxy kojic acid 13, when subjected to our standard reaction conditions, failed to deliver the desired C2-allylic alkylation product, even at 50 °C (Scheme 3). No allylic etherification product, arising out of the reaction of C6-hydroxymethyl group with electrophilic π-allyl–Ir complex, could be detected either under these conditions.
 | ||
| Scheme 3 Control experiment with α-methoxy kojic acid. | ||
The outcome of this experiment clearly highlights the importance of α-hydroxyl in α,β-unsaturated carbonyl compounds as the electronic directing group in driving this enantioselective allylic alkylation reaction.
Conclusions
In conclusion, we have developed the first asymmetric allylic alkylation of kojic acid and structurally related α-hydroxy α,β-unsaturated carbonyl compounds. This hydroxy-directed AA reaction is catalyzed by an in situ generated Ir(I)/(P,olefin) complex and employs Fe(OTf)2 as the Lewis acid co-catalyst to activate easily accessible racemic branched allylic alcohols, which are used as the allylic electrophile. The resulting β-allyl 3-hydroxypyranone derivatives, even adorned with pharmaceutically important structural motifs, are generally obtained in good to excellent yield with outstanding enantioselectivity. These enantioenriched kojic acid derivatives, as demonstrated with various transformations, can serve as useful building blocks in organic synthesis. The α-hydroxy group in the AA product is transformed into a phenyl group and the overall process represents the first formal β-C(sp2)–H allylic alkylation of α,β-unsaturated carbonyl compounds. To the best of our knowledge, this report represents the first example of the use of kojic acid in a transition metal catalyzed highly enantioselective transformation. Considering the ubiquity of kojic acid in pharmaceuticals, we believe our protocol will find useful applications.Data availability
The experimental details, characterization data, NMR spectra, and HPLC chromatograms associated with this article are provided in the ESI.†Author contributions
S. M. conceived the project and R. S. initiated the optimization studies. S. M. conducted the experiments. A. C. helped with the optimization studies and a part of the substrate scope. S. M. and S. M. wrote the manuscript together with the inputs from A. C. and R. S. All authors have given the approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the Science and Engineering Research Board (SERB) in the form of a Core Research Grant [Grant No. CRG/2020/000176] and the Science and Technology Award for Research (SERB-STAR) [Grant No. STR/2021/000009] are gratefully acknowledged. We also thank the Council of Scientific and Industrial Research (CSIR) [Grant no. 02(0385)/19/EMR-II] for funding our research. S. M. and R. S. thank Indian Institute of Science, Bangalore and CSIR, New Delhi for their respective doctoral fellowships. A. C. thanks the Ministry of Education, Government of India for his doctoral fellowship through the Prime Minister's Research Fellowship (PMRF) scheme. High resolution mass spectra (HR-MS) were recorded on an equipment procured under the Department of Science and Technology (DST)-FIST grant [Grant No. SR/FST/CS II-040/2015].Notes and references
- For selected reviews, see: (a) T. Sawano and R. Takeuchi, Catal. Sci. Technol., 2022, 12, 4100–4112, 10.1039/d2cy00316c; (b) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed; (c) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed; (d) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef CAS PubMed; (e) J. C. Hethcox, S. E. Shockley and B. M. Stoltz, ACS Catal., 2016, 6, 6207–6213 CrossRef CAS PubMed; (f) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC; (g) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed.
- For selected recent examples of Ir-catalyzed formal C(sp3)–H allylic alkylation, see: (a) F. A. Moghadam, E. F. Hicks, Z. P. Sercel, A. Q. Cusumano, M. D. Bartberger and B. M. Stoltz, J. Am. Chem. Soc., 2022, 144, 7983–7987 CrossRef CAS PubMed; (b) X. Cheng, C. Shen, X.-Q. Dong and C.-J. Wang, Chem. Commun., 2022, 58, 3142–3145 RSC; (c) S. Mitra and S. Mukherjee, Org. Lett., 2021, 23, 3021–3026 CrossRef CAS PubMed; (d) A. Chakrabarty and S. Mukherjee, Org. Lett., 2020, 22, 7752–7756 CrossRef CAS PubMed; (e) M. Han, M. Yang, R. Wu, Y. Li, T. Jia, Y. Gao, H.-L. Ni, P. Hu, B.-Q. Wang and P. Cao, J. Am. Chem. Soc., 2020, 142, 13398–13405 CrossRef CAS PubMed; (f) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 2039–2043 CrossRef CAS PubMed; (g) Y. Sempere, J. L. Alfke, S. L. Rössler and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 9537–9541 CrossRef CAS PubMed; (h) R. Sarkar and S. Mukherjee, Org. Lett., 2019, 21, 5315–5320 CrossRef CAS PubMed; (i) B.-B. Yue, Y. Deng, Y. Zheng, K. Wei and Y.-R. Yang, Org. Lett., 2019, 21, 2449–2452 CrossRef CAS PubMed; (j) C.-Y. Shi, J.-Z. Xiao and L. Yin, Chem. Commun., 2018, 54, 11957–11960 RSC; (k) R. Sarkar, S. Mitra and S. Mukherjee, Chem. Sci., 2018, 9, 5767–5772 RSC; (l) S. E. Shockley, J. C. Hethcox and B. M. Stoltz, Angew. Chem., Int. Ed., 2017, 56, 11545–11548 CrossRef CAS PubMed; (m) X. Jiang and J. F. Hartwig, Angew. Chem., Int. Ed., 2017, 56, 8887–8891 CrossRef CAS PubMed; (n) M. Bos and E. Riguet, Chem. Commun., 2017, 53, 4997–5000 RSC; (o) X.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4002–4005 CrossRef CAS PubMed; (p) M. Zhan, R.-Z. Li, Z.-D. Mou, C.-G. Cao, J. Liu, Y.-W. Chen and D. Niu, ACS Catal., 2016, 6, 3381–3386 CrossRef CAS; (q) J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 7644–7647 CrossRef CAS PubMed; (r) W. Chen, M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 15825–15828 CrossRef CAS PubMed; (s) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2068–2071 CrossRef CAS PubMed.
- For selected recent examples of Ir-catalyzed formal C(sp2)–H allylic alkylation, see: (a) R. J. Qing-Ru Zhao and S.-L. You, Acta Chim. Sin., 2021, 79, 1107–1112 CrossRef; (b) C.-X. Liu, P. Yang, W.-W. Zhang and S.-L. You, Acta Chim. Sin., 2021, 79, 742–746 CrossRef; (c) J. Lu, R. Xu, H. Zeng, G. Zhong, M. Wang, Z. Ni and X. Zeng, Org. Lett., 2021, 23, 3426–3431 CrossRef CAS PubMed; (d) J. A. Rossi-Ashton, A. K. Clarke, J. R. Donald, C. Zheng, R. J. K. Taylor, W. P. Unsworth and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 7598–7604 CrossRef CAS PubMed; (e) H. Tian, P. Zhang, F. Peng, H. Yang and H. Fu, Org. Lett., 2017, 19, 3775–3778 CrossRef CAS PubMed; (f) M. A. Schafroth, S. M. Rummelt, D. Sarlah and E. M. Carreira, Org. Lett., 2017, 19, 3235–3238 CrossRef CAS PubMed; (g) Z.-L. Zhao, Q. Gu, X.-Y. Wu and S.-L. You, Chem.–Asian J., 2017, 12, 2680–2683 CrossRef CAS PubMed; (h) Q.-L. Xu, C.-X. Zhuo, L.-X. Dai and S.-L. You, Org. Lett., 2013, 15, 5909–5911 CrossRef CAS PubMed; (i) Q.-L. Xu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 2579–2581 CrossRef CAS PubMed; (j) W.-B. Liu, H. He, L.-X. Dai and S.-L. You, Org. Lett., 2008, 10, 1815–1818 CrossRef CAS PubMed.
- (a) R. Sarkar and S. Mukherjee, Chem. Sci., 2021, 12, 3070–3075 RSC; (b) Y. Hu, W. Shi, B. Zheng, J. Liao, W. Wang, Y. Wu and H. Guo, Angew. Chem., Int. Ed., 2020, 59, 19820–19824 CrossRef CAS PubMed; (c) Y.-Q. Li, H.-J. Wang and Z.-Z. Huang, J. Org. Chem., 2016, 81, 4429–4433 CrossRef CAS PubMed; (d) B. G. Jellerichs, J.-R. Kong and M. J. Krische, J. Am. Chem. Soc., 2003, 125, 7758–7759 CrossRef CAS PubMed. For an indirect approach using dienolsilane, see: (e) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15249–15252 CrossRef CAS PubMed.
- (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447–5674 CrossRef CAS PubMed; (c) R. R. Huddleston and M. J. Krische, Synlett, 2003, 12–21 CAS.
- For related β C(sp2)–H halogenation see: T. Huber, D. Kaiser, J. Rickmeier and T. Magauer, J. Org. Chem., 2015, 80, 2281–2294 CrossRef CAS PubMed.
- (a) R. Yamada, T. Yoshie, S. Wakai, N. Asai-Nakashima, F. Okazaki, C. Ogino, H. Hisada, H. Tsutsumi, Y. Hata and A. Kondo, Microb. Cell Fact., 2014, 13, 71 CrossRef PubMed; (b) R. Bentley, Nat. Prod. Rep., 2006, 23, 1046–1062 RSC; (c) H. R. V. Arnstein and R. Bentley, Nature, 1950, 166, 948–949 CrossRef CAS PubMed; (d) O. E. May, A. J. Moyer, P. A. Wells and H. T. Herrick, J. Am. Chem. Soc., 1931, 53, 774–782 CrossRef CAS.
- (a) K. H. Thompson, C. A. Barta and C. Orvig, Chem. Soc. Rev., 2006, 35, 545–556 RSC; (b) M. Amélia Santos, Coord. Chem. Rev., 2002, 228, 187–203 CrossRef; (c) B. E. Bryant and C. Fernelius, J. Am. Chem. Soc., 1954, 76, 5351–5352 CrossRef CAS.
- For selected examples, see: (a) H. S. Rho, S. M. Ahn, D. S. Yoo, M. K. Kim, D. H. Cho and J. Y. Cho, Bioorg. Med. Chem. Lett., 2010, 20, 6569–6571 CrossRef CAS PubMed; (b) D. T. Puerta, M. Botta, C. J. Jocher, E. J. Werner, S. Avedano, K. N. Raymond and S. M. Cohen, J. Am. Chem. Soc., 2006, 128, 2222–2223 CrossRef CAS PubMed; (c) D. T. Puerta, J. Mongan, B. L. Tran, J. A. McCammon and S. M. Cohen, J. Am. Chem. Soc., 2005, 127, 14148–14149 CrossRef CAS PubMed; (d) K. Saatchi, K. H. Thompson, B. O. Patrick, M. Pink, V. G. Yuen, J. H. McNeill and C. Orvig, Inorg. Chem., 2005, 44, 2689–2697 CrossRef CAS PubMed; (e) J. Bransová, J. Brtko, M. Uher and L. Novotný, Int. J. Biochem. Cell Biol., 1995, 27, 701–706 CrossRef; (f) M. M. Finnegan, S. J. Rettig and C. Orvig, J. Am. Chem. Soc., 1986, 108, 5033–5035 CrossRef CAS.
- M. Zirak and B. Eftekhari-Sis, Turk. J. Chem., 2015, 39, 439–496 CrossRef CAS.
- (a) J. Mahatthananchai, J. Kaeobamrung and J. W. Bode, ACS Catal., 2012, 2, 494–503 CrossRef CAS PubMed; (b) J. Kaeobamrung, J. Mahatthananchai, P. Zheng and J. W. Bode, J. Am. Chem. Soc., 2010, 132, 8810–8812 CrossRef CAS PubMed.
- (a) R. A. Kovalevsky, M. V. Smirnov, A. S. Kucherenko, K. A. Bykova, E. V. Shikina and S. G. Zlotin, Eur. J. Org. Chem., 2022, e202101435 CAS; (b) A. S. Kucherenko, A. A. Kostenko, A. N. Komogortsev, B. V. Lichitsky, M. Y. Fedotov and S. G. Zlotin, J. Org. Chem., 2019, 84, 4304–4311 CrossRef CAS PubMed; (c) A. A. Kostenko, A. S. Kucherenko, A. N. Komogortsev, B. V. Lichitsky and S. G. Zlotin, Org. Biomol. Chem., 2018, 16, 9314–9318 RSC; (d) B. V. S. Reddy, S. M. Reddy, M. Swain, S. Dudem, S. V. Kalivendi and C. S. Reddy, RSC Adv., 2014, 4, 9107–9111 RSC; (e) J. Wang, Q. Zhang, H. Zhang, Y. Feng, W. Yuan and X. Zhang, Org. Biomol. Chem., 2012, 10, 2950–2954 RSC.
- M. C. Pirrung and J. N. Nalbandian, Tetrahedron Lett., 2013, 54, 3752–3754 CrossRef CAS PubMed.
- An attempted Pybox-Zn(OTf)2-catalyzed asymmetric Claisen rearrangement by Wender et al. met with poor enantioselectivity. See: P. A. Wender, N. D'Angelo, V. I. Elitzin, M. Ernst, E. E. Jackson-Ugueto, J. A. Kowalski, S. McKendry, M. Rehfeuter, R. Sun and D. Voigtlaender, Org. Lett., 2007, 9, 1829–1832 CrossRef CAS PubMed.
- (a) Y. Wang, W.-Y. Zhang and S.-L. You, J. Am. Chem. Soc., 2019, 141, 2228–2232 CrossRef CAS PubMed; (b) J. Y. Hamilton, S. L. Rössler and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 8082–8085 CrossRef CAS PubMed; (c) M. A. Schafroth, S. M. Rummelt, D. Sarlah and E. M. Carreira, Org. Lett., 2017, 19, 3235–3238 CrossRef CAS PubMed; (d) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568–5571 CrossRef CAS PubMed.
- For selected examples of this type of Claisen rearrangement, see: (a) P. A. Wender, N. Buschmann, N. B. Cardin, L. R. Jones, C. Kan, J.-M. Kee, J. A. Kowalski and K. E. Longcore, Nat. Chem., 2011, 3, 615–619 CrossRef CAS PubMed; (b) X. Xiong and M. C. Pirrung, Org. Lett., 2008, 10, 1151–1154 CrossRef CAS PubMed; (c) P. A. Wender and J. L. Mascarenas, J. Org. Chem., 1991, 56, 6267–6269 CrossRef CAS; (d) P. A. Wender and F. E. McDonald, J. Am. Chem. Soc., 1990, 112, 4956–4958 CrossRef CAS.
- See the ESI† for details.
- (a) K. A. Pelley and J. L. Vaught, Neuropharmacology, 1987, 26, 301–307 CrossRef CAS PubMed; (b) J. W. Ferkany, T. H. Andree, D. E. Ciarke and S. J. Enna, Neuropharmacology, 1981, 20, 1177–1182 CrossRef CAS PubMed; (c) J. G. Atkinson, Y. Girard, J. Rokach, C. S. Rooney, C. S. McFarlane, A. Rackham and N. N. Share, J. Med. Chem., 1979, 22, 99–106 CrossRef CAS PubMed.
- Y. Yamamoto, R. Fujikawa, T. Umemoto and N. Miyaura, Tetrahedron, 2004, 60, 10695–10700 CrossRef CAS.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370–4371 CrossRef CAS PubMed.
- For selected examples of such cross-coupling reactions, see: (a) Y. Qu, A. V. Ananin and G. A. Kraus, Tetrahedron Lett., 2020, 61, 151591 CrossRef CAS; (b) J. Duan, Y.-F. Du, X. Pang and X.-Z. Shu, Chem. Sci., 2019, 10, 8706–8712 RSC; (c) T. Kamino, K. Kuramochi and S. Kobayashi, Tetrahedron Lett., 2003, 44, 7349–7351 CrossRef CAS; (d) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. See DOI: https://doi.org/10.1039/d2sc03966d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |