
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh spin Fe3+-related bonding strength and electron transfer for sensitive and stable SERS detection†
Xinlu
Zheng
,
Xiao
Wu
,
Letian
Zhang
,
Jianjian
Kang
,
Man
Zhou
,
Yang
Zhong
,
Jinlong
Zhang
 and
Lingzhi
Wang
*
and
Lingzhi
Wang
*
Shanghai Engineering Research Center for Multi-Media Environmental Catalysis and Resource Utilization, Key Lab for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science & Technology, 130 Meilong Road, Shanghai, 200237, China. E-mail: wlz@ecust.edu.cn
First published on 12th October 2022
Abstract
The intrinsic electronic states of transition metal-containing SERS substrates, especially the effect of spin state on the detection sensitivity, still remain unknown. Herein, we propose a simple co-precipitation approach to form trimetallic MIL-101(FeNiTi) with high-spin (HS) Fe3+ as a result of geometric distortion of the octahedral symmetry. Using methylene blue as a demonstration, the trimetallic MIL-101(FeNiTi) shows a high enhancement factor (EF) of 6.1 × 106, a low detection limit of 10−9 M and excellent detection stability after long-term preservation. X-ray absorption fine structure and photoelectron spectra demonstrate that coupling between high-spin Fe3+ and aliovalent transition metals Ni2+ and Ti4+ with different filling degree of 3d eg-orbitals results in electron delocalization. The DFT calculation suggests that MIL-101(FeNiTi) with high-spin Fe3+ favors molecular adsorption and the charge transfer from the molecule to MIL-101(FeNiTi) is promoted, benefitting from the enhanced electron delocalization, which both contribute to the distinguished SERS performance of MIL-101(FeNiTi). This finding provides in-depth mechanistic understanding of the effect of the spin state of transition metals on mediating SERS activity, which is expected to efficiently promote the development of SERS platforms based on non-noble metals.
Introduction
Surface enhanced Raman spectroscopy (SERS), as a non-destructive analytical technique with a fingerprint spectrum, has attracted tremendous interest in many research fields.1–3 Noble metal substrates (Au, Ag) based on the surface plasmon resonance (SPR) effect have been widely adopted due to the high enhancement efficiency.4–8 However, due to the unavoidable limitations of noble metals, including high cost, poor stability and low biocompatibility,9–11 increasing attention has been focused on semiconductors or other materials such as metal–organic frameworks (MOFs) and graphene.12–15 For these materials, the charge transfer (CT) process between the substrate and adsorbate is well accepted as the dominant SERS mechanism.16–19 To date, great progress has been achieved in developing sensitive SERS substrates based on non-noble metals. For example, photonic crystals with unique inverse opal structure and yolk–shell nanomaterials have been exploited as superior SRES substrates via enhancing the light–matter interaction,20,21 however they are restricted by the tedious preparation process. Meanwhile, reducible metal oxides with abundant oxygen vacancies (MoO3−x, TiO2−x, WO3−x) have proven efficient in promoting CT between the molecule and substrate due to the formation of extra energy levels and improved resonance absorption.22–25 However, surface defects on these materials are unstable under laser irradiation and easy to oxidize even under ambient conditions, which significantly deteriorates the practical SERS performance. Therefore, great effort still needs to be devoted to developing flexible strategies for robust SERS substrates.Compared with defect engineering, the effect of other intrinsic electronic states of non-noble metal substrates including the orbital filling degree (spin state) and electron localization/delocalization on the SERS performance has been scarcely explored to date. It is known that the bonding strength and electron transfer between the crystal and adsorbates can be tuned through regulating the spin state of transition metal ions in the fields of electrocatalysis and photocatalysis. For example, Zou and coworkers proposed the crystal-size-restricting strategy for finely modulating the high-to-low spin transformation of Fe3+, which boosted the bonding strength with adsorbates and facilitated the electron transfer process.26 Xie et al. realized delocalized spin states in Ni-based compounds through introducing structural distortion to the confined 2D atomic layers, leading to modification of the electronic structure and improved electron transport.27 Compared with the commonly observed instability of vacancy defects, the inherent spin characteristics of transition metals are supposed to be more attractive since they simultaneously possess chemical stability under ambient conditions and field-induced reversibility.28–31 To date, the correlation between the SERS performance and electronic spin state of transition metal-based materials still remains unknown. There is a lack of efficient strategies to regulate the SERS activity through tuning the spin state.
Herein, we demonstrated that the low-spin to high-spin transition of Fe3+ in a series of MIL-101 composites has a significant effect on SERS performance, which was achieved via introducing the aliovalent metal dopants Ni2+ and Ti4+ with different electronic configuration. Using methylene blue (MB) as the model molecule, distinguished sensitivity with an enhancement factor (EF) of up to 6.1 × 106 and excellent stability is revealed for trimetallic MIL-101(FeNiTi) with high-spin (HS) Fe3+. By combining electronic structure analyses and density functional theory (DFT) calculations, the SERS performance and the spin state of Fe3+ were positively correlated. The SERS mechanism was explored regarding the bonding strength and CT between molecules and MIL-101 with different spin states of Fe3+. It is expected that the strategy based on spin-regulation would provide a brand-new idea for the development of sensitive non-noble metal SERS technology.
Results and discussion
Sample fabrication and characterization
Tuning of the spin state of MIL-101(Fe) was attempted through adding aliovalent transition metal ions with different electronic configuration and valence state. A series of MIL-101 materials with different ratios of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni, Fe:Ti and Fe:Ni:Ti were fabricated through a co-precipitation method (Fig. S1, Table S1†). The X-ray diffraction (XRD) pattern indicates that trimetallic MIL-101(FeNiTi) and bimetallic MIL-101(FeNi) maintain the characteristic peaks of MIL-101(Fe) (Fig. 1a),32,33 suggesting that the introduction of Ni2+ and Ti4+ ions does not alter the crystalline phase although MIL-101(FeNiTi) has a lower crystallinity than the other composites. The scanning electron microscope (SEM) image of trimetallic MIL-101(FeNiTi) presents a uniform shuttle shape (Fig. 1b), which is slightly different from MIL-101(Fe) and bimetallic MIL-101(FeNi) with an octahedral geometry (Fig. S2†).34 Considering the similar particle size of ca. 1.2 μm of all three composites, the morphological change should be attributed to the decreased crystallinity of MIL-101(FeNiTi). Moreover, the transmission electron microscope (TEM) mapping of MIL-101(FeNiTi) reveals the uniform distribution of elements Fe, Ni, Ti, C and O (Fig. 1c).
:Ni, Fe:Ti and Fe:Ni:Ti were fabricated through a co-precipitation method (Fig. S1, Table S1†). The X-ray diffraction (XRD) pattern indicates that trimetallic MIL-101(FeNiTi) and bimetallic MIL-101(FeNi) maintain the characteristic peaks of MIL-101(Fe) (Fig. 1a),32,33 suggesting that the introduction of Ni2+ and Ti4+ ions does not alter the crystalline phase although MIL-101(FeNiTi) has a lower crystallinity than the other composites. The scanning electron microscope (SEM) image of trimetallic MIL-101(FeNiTi) presents a uniform shuttle shape (Fig. 1b), which is slightly different from MIL-101(Fe) and bimetallic MIL-101(FeNi) with an octahedral geometry (Fig. S2†).34 Considering the similar particle size of ca. 1.2 μm of all three composites, the morphological change should be attributed to the decreased crystallinity of MIL-101(FeNiTi). Moreover, the transmission electron microscope (TEM) mapping of MIL-101(FeNiTi) reveals the uniform distribution of elements Fe, Ni, Ti, C and O (Fig. 1c).
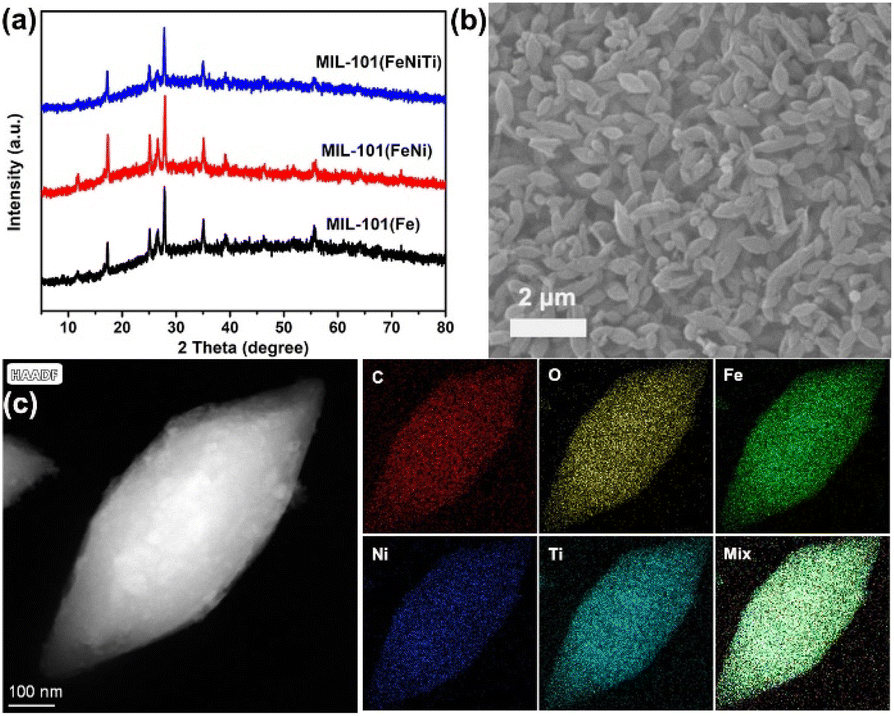 | ||
| Fig. 1 Crystalline structure and morphology characterization of MIL-101. (a) XRD of three kinds of MIL-101; (b) SEM of trimetallic MIL-101(FeNiTi); (c) TEM-mapping of trimetallic MIL-101(FeNiTi). | ||
Transmission 57Fe Mössbauer spectroscopy was used to characterize the spin states of Fe in three kinds of MIL-101 considering its sensitivity towards the spin states of Fe species.35–37Fig. 2a–c show the 57Fe Mössbauer spectra of the three samples, and Table S2† lists the Mössbauer parameters as well as the corresponding areas of different spin states of Fe. For all MIL-101 samples, the spectra can be well fitted into two doublets. Based on the values of isomer shift (IS) and quadrupole splitting (QS), doublets D1 and D2 are assigned to the LS state and the HS state of Fe3+, respectively. It can be seen in Fig. 2a that the pristine MIL-101(Fe) mainly exhibits the LS state (the proportion is ca. 63.0%) with hyperfine magnetic field of 515.4 kOe, IS of 0.404 mm s−1, and QS of 0.486 mm s−1 (Table S1†). The proportion of the HS state in pristine MIL-101(Fe) is much smaller, and is only 37.0% with IS of 0.414 mm s−1, and QS of 0.901 mm s−1. The above results indicate that most Fe3+ in MIL-101(Fe) is in the LS state. But for bimetallic MIL-101(FeNi) (Fig. 2b), it can be clearly observed that the content of the HS state (D2) is increased from 37.0% to 67.9%, indicating that Fe3+ is transformed from the LS state to the HS state due to the introduction of Ni2+. As presented in Fig. 2c, trimetallic MIL-101(FeNiTi) formed through the further introduction of Ti4+ exhibits the complete conversion of Fe3+ to the HS state. Moreover, the HS state of MIL-101(FeTi) (D2, 65.8%) occupies a higher proportion than the LS state (D1, 34.2%), but a lower proportion than the HS state in trimetallic MIL-101(FeNiTi), which suggests that both Ni and Ti elements play an indispensable role in the low-to-high spin transition of Fe3+ (Fig. S4†). Overall, 100% ferric iron can be modulated from the LS state to the HS state through the simultaneous introduction of Ni2+ and Ti4+ into MIL-101(Fe) (Fig. S5†).
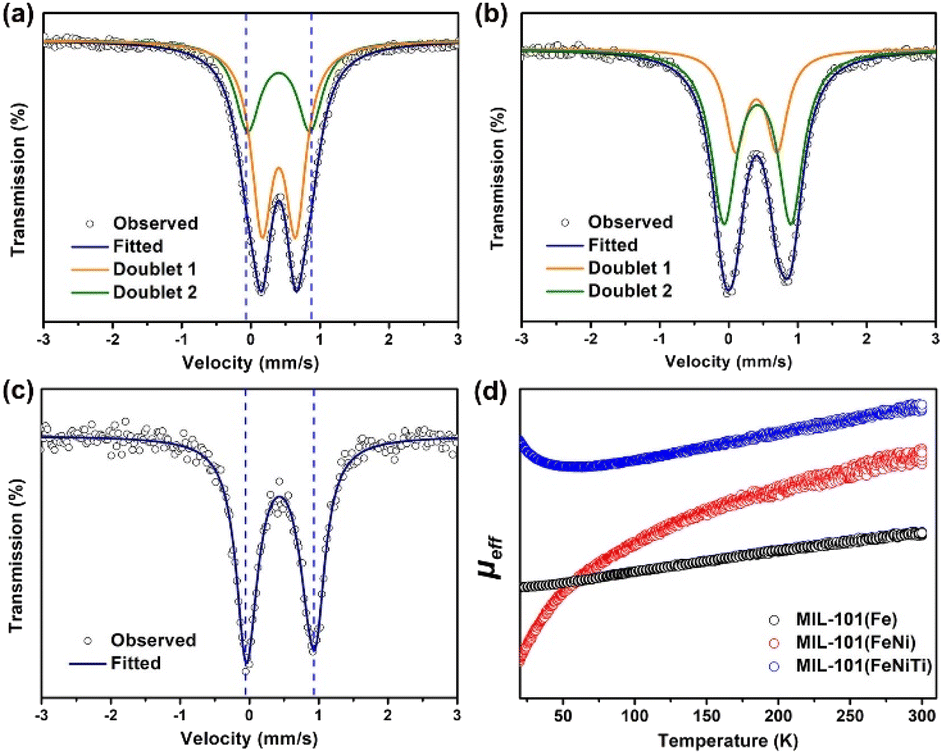 | ||
| Fig. 2 Spin states of MIL-101(Fe), MIL-101(FeNi) and MIL-101(FeNiTi). 57Fe-Mössbauer spectroscopy of (a) MIL-101(Fe); (b) MIL-101(FeNi) and (c) MIL-101(FeNiTi) at room temperature. (d) The comparison of μeff for the three kinds of MOFs. | ||
To more explicitly demonstrate the spin state transformation of the prepared MIL-101, the 3d orbital electron configurations were described by performing the temperature-dependent magnetic susceptibility (M–T) measurement due to the high sensitivity of magnetism to unpaired electrons.38,39 Temperature-dependent paramagnetism can be seen for all MIL-101 materials (Fig. S6†). And the total effective magnetic moment (μeff), which intimately relates to the number of unpaired 3d electrons (n) of Fe3+, can be further derived from linear fittings of the χ−1–T plots according to the following equation:40,41
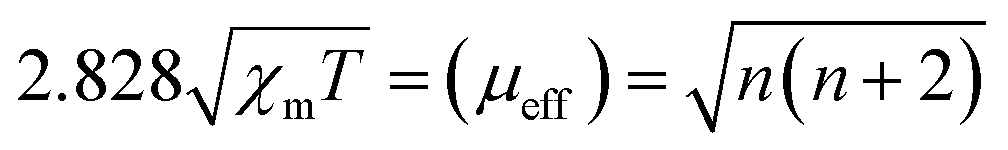
It is worth noting that the value of μeff increases with the introduction of Ni and Ti elements (Fig. 2d), which confirms that the number of singly paired 3d electrons of Fe3+ in trimetallic MIL-101(FeNiTi) is indeed increased compared with that of the pure MIL-101(Fe) and bimetallic MIL-101(FeNi) counterparts, corresponding to an effectively improved local spin state of Fe3+.
SERS performance of trimetallic MIL-101(FeNiTi)
Fig. 3a shows the SERS spectra of MB molecules adsorbed on MIL-101(Fe), bimetallic MIL-101(FeNi) and trimetallic MIL-101(FeNiTi) substrates under 532 nm irradiation. The strongest peak at 1625 cm−1 is attributed to the C–C stretching vibration mode of the thionine ring.42 It can be found that the characteristic peaks of MB can be obviously observed on all three substrates, and the SERS activity is positively correlated with the spin state of MIL-101. The EF was quantitatively estimated based on the peak at 1625 cm−1 using eqn (S1)–(S3).† The calculated EF of trimetallic MIL-101(FeNiTi) can reach as high as 6.1 × 106, which is comparable to that of traditional noble metal-based SERS platforms. In addition, a 785 nm laser was further utilized to investigate the SERS performance of the prepared MIL-101 (Fig. S8†). It can be seen that the intensities under 785 nm irradiation are weaker than those under a 532 nm laser but the characteristic peaks assigned to MB are clearly observed. The limit of detection (LOD) was further investigated, as shown in Fig. 3b. It can be seen that the SERS signal of MB can still be detected even when the concentration of MB is decreased to 10−9 M, indicating that trimetallic MIL-101(FeNiTi) has excellent SERS detection ability. In addition, the universality of trimetallic MIL-101(FeNiTi) as a SERS substrate was verified for various dye molecules, including rose bengal (RB, 10−5 M), acid blue (AB, 10−5 M) and Congo red (CR, 10−5 M) (Fig. S9†). It is widely considered that the detection uniformity is a significant criterion for evaluating the practical application performance of SERS-active platforms. Therefore, the SERS signals of MB molecules from 10 randomly selected positions were collected (Fig. S10†). The relative standard deviation (RSD) for the characteristic band (1625 cm−1) was further calculated to quantitatively assess the uniformity of MIL-101(FeNiTi), which was determined to be ca. 5.6% (Fig. 3c), demonstrating the ideal detection uniformity of trimetallic MOFs. In addition, the SERS intensities obtained for MIL-(FeNiTi) kept for 2 weeks, 1 month and 3 months were nearly the same as those of the as-grown trimetallic MIL-101(FeNiTi). Even when the duration was extended to 5 months, the Raman peaks of the MB molecule still did not show any obvious decrease, exhibiting the excellent long-term chemical stability of trimetallic MIL-101(FeNiTi) (Fig. 3d). Considering the easy aggregation of noble metal nanoparticles and the high oxidative susceptibility of defective oxides under ambient conditions, this outstanding stability indicates the robust application potential of MIL-101(FeNiTi) as a SERS substrate.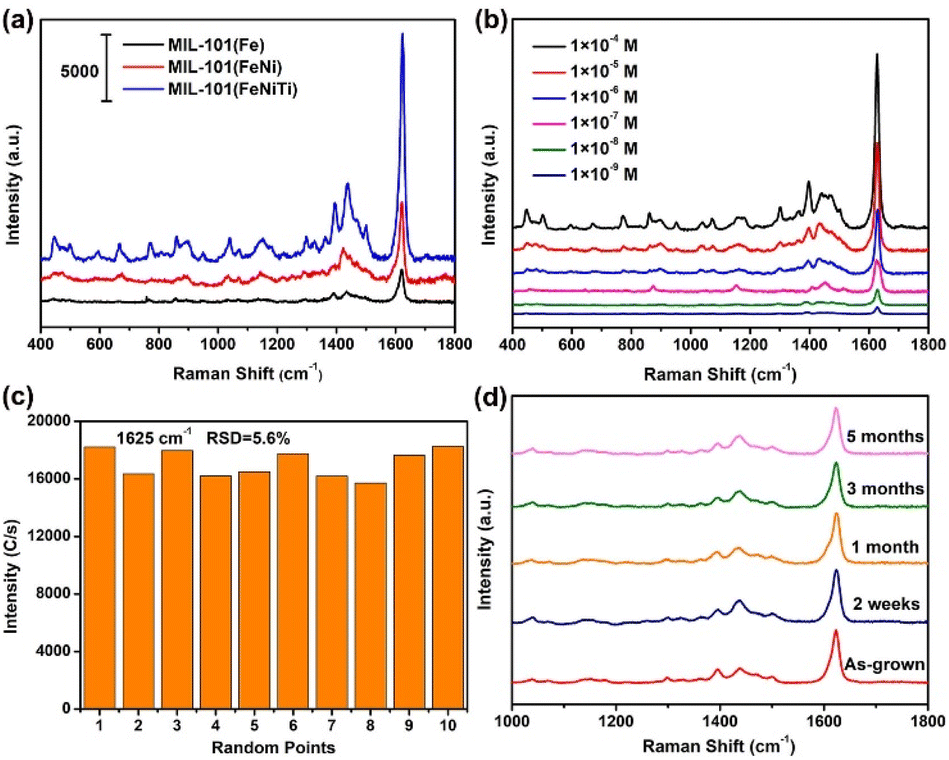 | ||
| Fig. 3 SERS performance of MIL-101(Fe), MIL-101(FeNi) and MIL-101(FeNiTi). (a) The SERS spectra of MB (10−5 M) adsorbed on the three kinds of MOF under 532 nm irradiation; (b) the SERS spectra of MB with different concentrations on trimetallic MIL-101(FeNiTi); (c) the corresponding relative standard deviation (RSD) for the characteristic band (1625 cm−1) based on 10 random positions; (d) the SERS spectra of MB on trimetallic MIL-101(FeNiTi) preserved for different periods. All Raman intensities of detected molecules were obtained by deducting the peaks of the MOF substrates. | ||
The origin of high-spin Fe in MIL-101(FeNiTi)
To understand the origin of the high-spin Fe in trimetallic MIL-101(FeNiTi) and its relationship with the SERS performance, X-ray absorption near edge structure (XANES) spectroscopy was utilized to characterize the electronic characteristics of the transition metal atoms. Fig. 4a presents the Fe K-edge XANES of Fe foil, Fe2O3, MIL-101(Fe), bimetallic MIL-101(FeNi) and trimetallic MIL-101(FeNiTi). The pre-edge peak of MIL-101 is assigned to the 1s to 3d quadrupole transition of the octahedral Fe atom.43 The absorption edges of MIL-101(Fe), bimetallic MIL-101(FeNi) and trimetallic MIL-101(FeNiTi) are closer to that of the Fe2O3 reference and far away from that of Fe foil, suggesting that the Fe species in these samples exist in the cationic state (Fig. 4a). Moreover, k3-weighted Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectroscopy was conducted to explore the differences in atomic information. Fe K-edge FT-EXAFS spectra show that the Fe–O bond in bimetallic MIL-101(FeNi) and trimetallic MIL-101(FeNiTi) is longer than that in MIL-101(Fe) (Fig. 4b), suggesting that the octahedral geometry of the Fe sites is distorted after the introduction of Ni and Ti elements,44 which is also consistent with the XRD result. In addition, the Ni K-edge XANES spectra exhibit a slight shift towards higher energy (Fig. 4c), suggesting that Ni species with a higher valence state are formed due to the introduction of Ti atoms.45 The Fourier transforms of R-space oscillations show that only the Ni–O bond appears in MIL-101(FeNi) and MIL-101(FeNiTi) (Fig. S11†), implying that Ni exists in modified MIL-101 as single atoms through replacing the Fe atoms.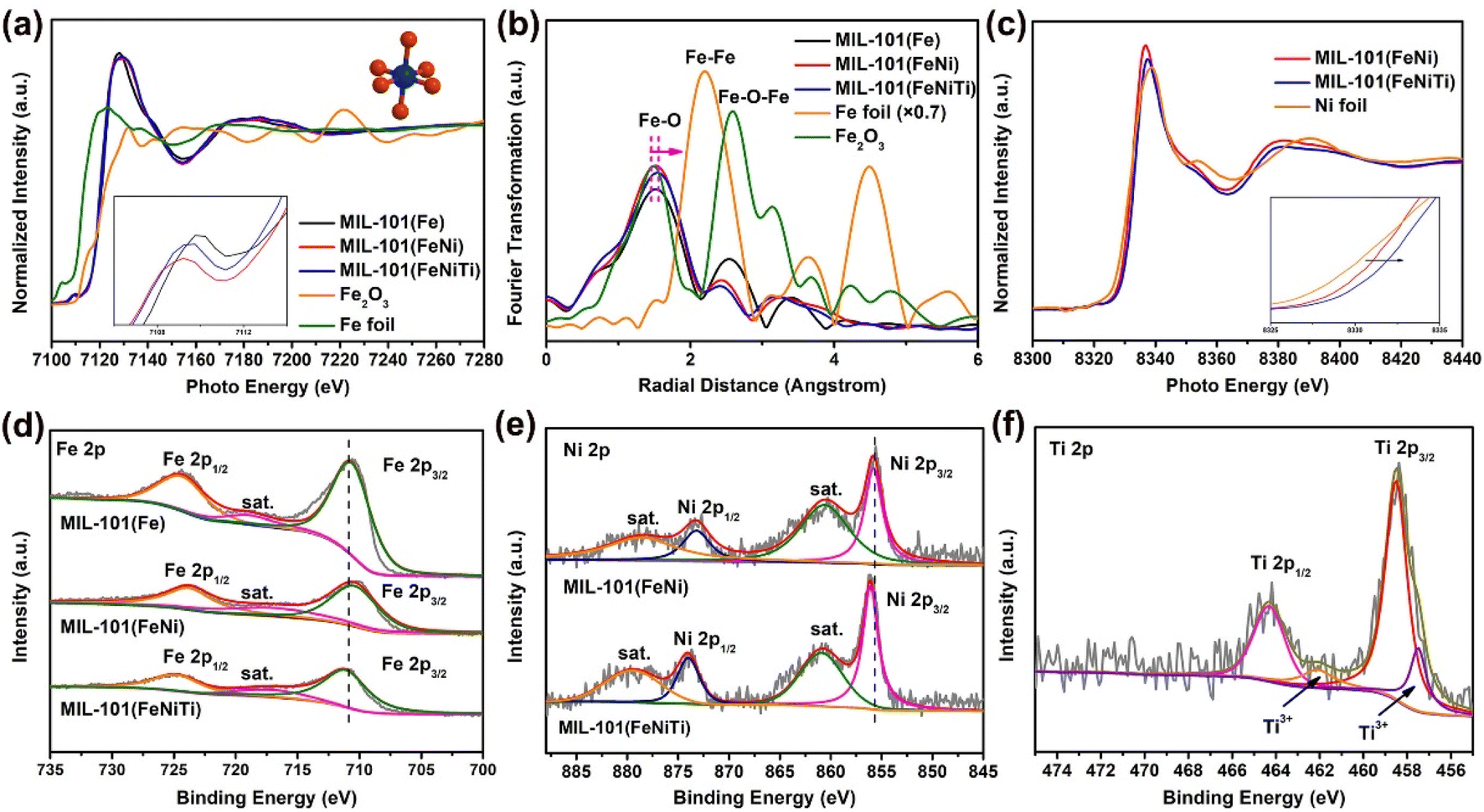 | ||
| Fig. 4 Electronic structure characterization of MIL-101(Fe), MIL-101(FeNi) and MIL-101(FeNiTi). (a) Fe K-edge XANES spectra of Fe foil, Fe2O3, MIL-101(Fe), bimetallic MIL-101(FeNi) and trimetallic MIL-101(FeNiTi); (b) Fourier transforms of R-space oscillations of Fe for the synthesized MOFs; (c) Ni K-edge XANES spectra of Ni foil, MIL-101(FeNi) and MIL-101(FeNiTi). XPS spectra of (d) Fe 2p, (e) Ni 2p and (f) Ti 2p for the prepared MOFs. | ||
X-ray photoelectron spectroscopy (XPS) was further applied to identify the electronic characteristics of different MIL-101 composites. In the Fe 2p XPS spectra (Fig. 4d), the signals of Fe3+ 2p3/2 and Fe3+ 2p1/2 are marked at 711.5 and 724.9 eV, respectively.46 Compared to MIL-101(Fe), the characteristic band of Fe3+ in MIL-101(FeNi) is slightly shifted towards lower energy, while that of MIL-101(FeNiTi) is shifted towards higher energy, which means that Fe atoms tend to act as electron acceptors and electron donors in MIL-101(FeNi) and MIL-101(FeNiTi), respectively. Additionally, the presence of Ni2+ is verified clearly by the high-resolution Ni 2p XPS spectra of MIL-101(FeNi) and MIL-101(FeNiTi) (Fig. 4e). The characteristic peaks at 855.6 and 872.9 eV and their respective shake-up satellites are attributed to Ni 2p3/2 and Ni 2p1/2, respectively.47 It is worth noting that the binding energy of the Ni 2p3/2 peak in MIL-101(FeNiTi) is positively shifted, suggesting that the electrons should be delocalized around the Ni sites after the introduction of the Ti dopant.48 In addition, the successful introduction of Ti4+ is verified by the Ti 2p data; a signal for Ti3+ is also observed in the bands located at 462.6 eV and 457.5 eV (Fig. 4f),49 demonstrating that the Ti dopant acts as an electron acceptor in MIL-101(FeNiTi). It is known that the spin state of transition metals is dependent on the electronic configuration and valence state.50 Based on the above results, the introduction of Ni and Ti causes distortion of the octahedral symmetric Fe–O sites, which may reduce the crystal-field splitting energy, thus leading to the transition from the low-spin state to the high-spin state of Fe3+.
Furthermore, density functional theory (DFT) calculations were also performed to investigate the spin transition of Fe3+ among the three kinds of MIL-101. The three models are displayed in Fig. S12.† As disclosed by the density of states (DOS) in Fig. 5a–c, the Fe ions in MIL-101(Fe) and MIL-101(FeNi) have d-band centers located at −0.775 eV and −1.159 eV, respectively, which are higher than for trimetallic MIL-101(FeNiTi) with a d-band center at −1.369 eV. A lower d band center is supposed to be less favorable for molecule adsorption.51 Moreover, the crystal-field splitting energy of the 3d orbitals was also calculated to understand the spin transition. As shown in Fig. 5d–f, the t2g and eg centers in MIL-101(Fe) are located at −1.039 and −0.378 eV with crystal-field splitting energy of 0.661 eV, while MIL-101(FeNi) and MIL-101(FeNiTi) have smaller crystal-field splittings of 0.297 and 0.147 eV, respectively. According to crystal-field theory,52 the Fe 3d electrons in trimetallic MIL-101(FeNiTi) preferentially occupy orbitals individually rather than in pairs, thus resulting in HS Fe atoms. The DFT calculation confirms that the crystal-field splitting energy of Fe atoms can be reduced through the introduction of aliovalent Ni and Ti ions into MIL-101(Fe), thus promoting the low-to-high spin state conversion of Fe3+.
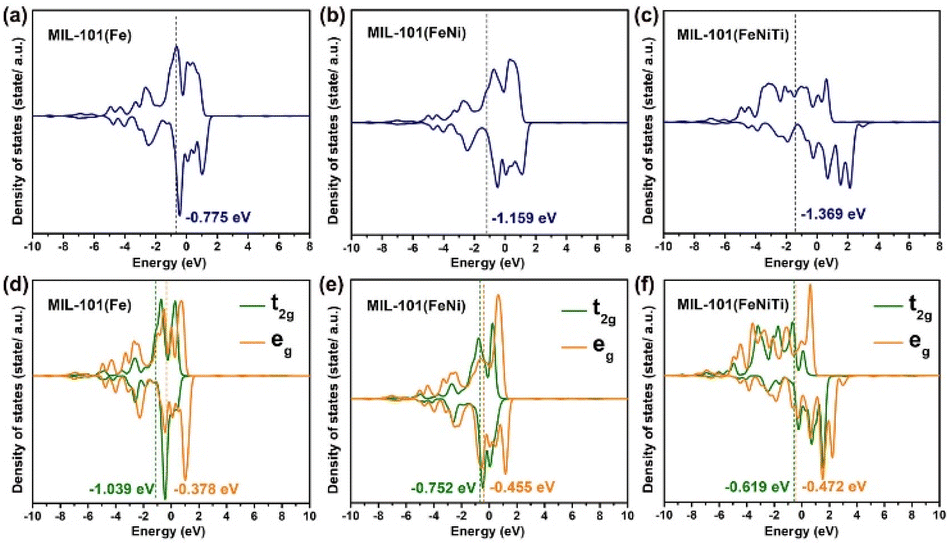 | ||
| Fig. 5 DFT calculation of the electronic characteristics of different MIL-101 materials. Projected DOS of the Fe 3d orbitals of (a) MIL-101(Fe), (b) MIL-101(FeNi) and (c) MIL-101(FeNiTi). Projected DOS of the t2g and eg orbitals of the Fe atoms in (d) MIL-101(Fe), (e) MIL-101(FeNi) and (f) MIL-101(FeNiTi). | ||
SERS mechanism of MIL-101(FeNiTi)
The possibility of resonance absorption for enhanced SERS activity was first definitively excluded (Fig. S13†) since there is no difference among the three kinds of MIL-101 material and no obvious resonance absorption peaks. The spin state of Fe3+ may influence the bonding strength between MIL-101 and the molecule since half-occupied (HS) and empty orbitals (LS) have different affinity to electrons.53 In light of this speculation, the adsorption energies of different MOF–MB systems were calculated by DFT calculations. As presented in Fig. 6a–c, the MB molecule is chemically adsorbed on the surfaces of MOF models via forming an Fe–S bond with the length in the range of 0.21–0.27 nm. The energies of MB adsorbed over MIL-101(Fe), MIL-101(FeNi) and MIL-101(FeNiTi) are −0.96, −1.46 and −2.16 eV, respectively (Table S3†). It turns out that the bonding strength between MB and MIL-101 follows the order of MIL-101(Fe) < MIL-101(FeNi) < MIL-101(FeNiTi), consistent with the ordering of the HS ratio. This result suggests that the metal–adsorbate interaction should be dependent on the spin state rather than the d-band center of Fe in MIL-101 since MIL-101(FeNiTi) has the lowest d band center but highest spin state.54 The enhanced bonding strength should benefit the interfacial electronic interaction between MB and MIL-101,55,56 which is critical to the SERS performance of MIL-101.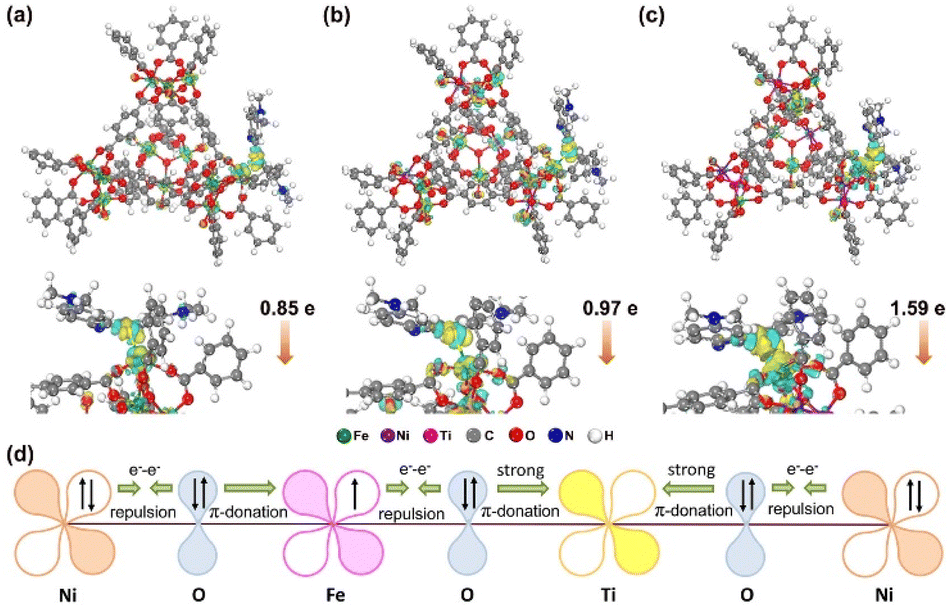 | ||
| Fig. 6 The DFT calculations of MB@MIL-101(Fe), MB@MIL-101(FeNi) and MB@MIL-101(FeNiTi). The difference in charge distributions for (a) MB@MIL-101(Fe), (b) MB@MIL-101(FeNi) and (c) MB@MIL-101(FeNiTi). The accumulation region for electron density is marked in yellow and the depletion region is marked in green. The colors of Fe, Ni, Ti, C, O, N and H atoms are green, purple, pink, gray, red, blue and white, respectively; (d) schematic representation of the electronic coupling among Fe, Ni and Ti in MIL-101(FeNiTi). | ||
The charge difference distribution of MIL-101 with adsorbed MB was further analyzed to understand the CT between them. The accumulation region (marked in yellow) for electron density is around the Fe atoms in the MIL-101 substrates, and the depletion region (marked in green) is near the S and C atoms in the aromatic ring. According to the Hirshfeld population analysis,57 0.85e and 0.97e are transferred from the MB molecule to MIL-101(Fe) and MIL-101(FeNi), respectively. While for trimetallic MIL-101(FeNiTi), the amount of charge transferred from MB increases to 1.59e. These results suggest that the charge transfer from the MB molecule is most efficiently promoted over trimetallic MIL-101(FeNiTi) with the completely HS state of Fe3+.
To explore the relationship between MIL-101(FeNiTi) containing high-spin Fe3+ and CT efficiency, the interaction among different transition metals was further discussed in light of the electronic characteristics of mixed-valent metal ions in MIL-101(FeNiTi). For MIL-101(Fe) with LS Fe3+, the dominant interaction between the fully occupied π-symmetry (t2g) d-orbitals of Fe3+ and the bridging oxygen (O2−, transition metal ions are connected through the bridging oxygen bridges) is electron–electron repulsion. For MIL-101(FeNi), Ni2+ with paired electrons in π-symmetry (t2g) d-orbitals interacts with O2−via electron–electron repulsion. However, because of the electronegativity difference between Fe3+ (1.651) and Ni2+ (1.367),58,59 the electrons would transfer from Ni to Fe sites (Fig. S14†), which also agrees well with the XANES and XPS results. The electron transfer would lead to electron delocalization in MIL-101(FeNi).60 As for MIL-101(FeNiTi) (Fig. 6d), electrons in Fe3+ would be repulsed by O2−, and the paired electrons in O2− interact with Ti4+via strong π-donation, resulting in partial electron transfer from Fe3+ to Ti4+. Meanwhile, via O2− as an intermediary, electrons in Ni2+ would transfer to d-orbitals of Ti4+ powered by strong π-donation. In this case, the electron transfer among mixed-valence metal ions with different filling degree of eg orbitals finally leads to electron delocalization in MIL-101(FeNiTi). It is suggested that the electron delocalization caused by the coupling between HS Fe, Ni and Ti ions would improve the conductivity of the SERS substrate,61 which is verified by the highest photocurrent and the minimum electrochemical impedance (Fig. S15†). The enhanced conductivity is supposed to promote CT.62,63 Therefore, benefitting from the formation of high-spin Fe3+, the strong bonding and promoted charge transfer between molecules and trimetallic MIL-101(FeNiTi) both contribute to the remarkable SERS activity.
Conclusions
In summary, the low-to-high spin conversion of Fe3+ in MIL-101 was tuned through the introduction of aliovalent Ni2+ and Ti4+ as a result of distortion of the octahedral symmetry. Compared with MIL-101(Fe) and bimetallic MIL-101(FeNi), the trimetallic MIL-101(FeNiTi) with completely HS Fe3+ exhibited a more distinct SERS activity with EF up to 6.1 × 106. Meanwhile, a prominent detection stability was revealed during long-term preservation under ambient conditions. The combination of electronic, magnetic and electrochemical analyses with DFT calculations demonstrated that the prominent SERS activity is attributed to the enhanced bonding strength and promoted charge transfer between molecules and MIL-101 with HS Fe3+. To our knowledge, to date, this is the first report on the in-depth investigation of the relationship between the spin state of transition metal-based substrates and their SERS activity. This development would open up a new avenue to rational design of distinguished non-noble metal-based SERS substrates.Data availability
The datasets generated and/or analyzed during the present study are available from the corresponding author upon reasonable request.Author contributions
Xinlu Zheng designed the experiments, analyzed data, performed theoretical calculations and wrote the manuscript. Xiao Wu and Letian Zhang contributed to sample fabrication and data acquisition. Jianjian Kang, Man Zhou and Yang Zhong assisted in the data interpretation. Jinlong Zhang contributed to the supervision and the theoretical understanding of the paper. Lingzhi Wang contributed to the supervision, the theoretical understanding of the paper and revising the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key R&D Program of China (2021YFC2103500), the National Natural Science Foundation of China (No. 21972040, 21822603, and 21773062), Shanghai Municipal Science and Technology (No. 21ZR1417900, 2018SHZDZX03, 17520711500, and 20DZ2250400), the Innovation Program of Shanghai Municipal Education Commission, the Program of Introducing Talents of Discipline to Universities (No. B16017 and B20031), and the Fundamental Research Funds for the Central Universities.Notes and references
- T. Chen, H. Wang, G. Chen, Y. Wang, Y. Feng, W. S. Teo, T. Wu and H. Chen, ACS Nano, 2010, 4, 3087–3094 CrossRef CAS PubMed.
- Y. Wang, C. Zhao, J. Wang, X. Luo, L. Xie, S. Zhan, J. Kim, X. Wang, X. Liu and Y. Ying, Sci. Adv., 2021, 7, eabe4553 CrossRef CAS PubMed.
- Y. Zeng, J.-Q. Ren, A.-G. Shen and J.-M. Hu, J. Am. Chem. Soc., 2018, 140, 10649–10652 CrossRef CAS PubMed.
- K. Liu, Y. Bai, L. Zhang, Z. Yang, Q. Fan, H. Zheng, Y. Yin and C. Gao, Nano Lett., 2016, 16, 3675–3681 CrossRef CAS PubMed.
- Z. Ye, C. Li, M. Celentano, M. Lindley, T. O’Reilly, A. J. Greer, Y. Huang, C. Hardacre, S. J. Haigh and Y. Xu, JACS Au, 2022, 2, 178–187 CrossRef CAS PubMed.
- Z. Yin, Y. Wang, C. Song, L. Zheng, N. Ma, X. Liu, S. Li, L. Lin, M. Li and Y. Xu, J. Am. Chem. Soc., 2018, 140, 864–867 CrossRef CAS PubMed.
- A. Li, X. Wang, L. Guo and S. Li, J. Phys. Chem. C, 2019, 123, 25394–25401 CrossRef CAS.
- X. Wang, G. Ma, A. Li, J. Yu, Z. Yang, J. Lin, A. Li, X. Han and L. Guo, Chem. Sci., 2018, 9, 4009–4015 RSC.
- K. Yuan, Q. Mei, X. Guo, Y. Xu, D. Yang, B. J. Sánchez, B. Sheng, C. Liu, Z. Hu and G. Yu, Chem. Sci., 2018, 9, 8781–8795 RSC.
- W. Lee, B.-H. Kang, H. Yang, M. Park, J. H. Kwak, T. Chung, Y. Jeong, B. K. Kim and K.-H. Jeong, Nat. Commun., 2021, 12, 1–10 CAS.
- X. Qin, Y. Si, D. Wang, Z. Wu, J. Li and Y. Yin, Anal. Chem., 2019, 91, 4529–4536 CrossRef CAS PubMed.
- S. Yang, J. Yao, Y. Quan, M. Hu, R. Su, M. Gao, D. Han and J. Yang, Light: Sci. Appl., 2020, 9, 1–7 CrossRef CAS PubMed.
- L. Yang, Y. Peng, Y. Yang, J. Liu, H. Huang, B. Yu, J. Zhao, Y. Lu, Z. Huang and Z. Li, Adv. Sci., 2019, 6, 1900310–1900319 CrossRef PubMed.
- H. Sun, S. Cong, Z. Zheng, Z. Wang, Z. Chen and Z. Zhao, J. Am. Chem. Soc., 2019, 141, 870–878 CrossRef CAS PubMed.
- E. Feng, T. Zheng, X. He, J. Chen and Y. Tian, Sci. Adv., 2018, 4, eaau3494 CrossRef CAS PubMed.
- B. Yang, S. Jin, S. Guo, Y. Park, L. Chen, B. Zhao and Y. M. Jung, ACS Omega, 2019, 4, 20101–20108 CrossRef CAS PubMed.
- J. Lin, W. Ren, A. Li, C. Yao, T. Chen, X. Ma, X. Wang and A. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 4204–4211 CrossRef CAS PubMed.
- J. Lin, J. Yu, O. Akakuru, X. Wang, B. Yuan, T. Chen, L. Guo and A. Wu, Chem. Sci., 2020, 11, 9414–9420 RSC.
- V. Rajput, R. K. Gupta and J. Prakash, J. Mater. Chem. C, 2022, 10, 73–95 RSC.
- D. Qi, L. Lu, L. Wang and J. Zhang, J. Am. Chem. Soc., 2014, 136, 9886–9889 CrossRef CAS PubMed.
- X. Zheng, W. Zhang, J. Zhang and L. Wang, Appl. Surf. Sci., 2021, 541, 148422–148430 CrossRef CAS.
- X. Tan, L. Wang, C. Cheng, X. Yan, B. Shen and J. Zhang, Chem. Commun., 2016, 52, 2893–2896 RSC.
- J. Lin, Y. Shang, X. Li, J. Yu, X. Wang and L. Guo, Adv. Mater., 2017, 29, 1604797–1604806 CrossRef PubMed.
- S. Cong, Y. Yuan, Z. Chen, J. Hou, M. Yang, Y. Su, Y. Zhang, L. Li, Q. Li and F. Geng, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- C. Gu, D. Li, S. Zeng, T. Jiang, X. Shen and H. Zhang, Nanoscale, 2021, 13, 5620–5651 RSC.
- G. Shen, L. Pan, R. Zhang, S. Sun, F. Hou, X. Zhang and J. J. Zou, Adv. Mater., 2020, 32, 1905988–1905997 CrossRef CAS PubMed.
- S. Chen, Z. Kang, X. Hu, X. Zhang, H. Wang, J. Xie, X. Zheng, W. Yan, B. Pan and Y. Xie, Adv. Mater., 2017, 29, 1701687–1701694 CrossRef PubMed.
- R. Gao, J. Wang, Z.-F. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang and C. Shi, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li and W. Yan, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu and Y. Jiang, Angew. Chem., 2015, 127, 8846–8851 CrossRef.
- Z. Lu, H. Wang, D. Kong, K. Yan, P.-C. Hsu, G. Zheng, H. Yao, Z. Liang, X. Sun and Y. Cui, Nat. Commun., 2014, 5, 1–7 CAS.
- K. M. Taylor-Pashow, J. Della Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261–14263 CrossRef CAS PubMed.
- F. Guo, S. Yang, Y. Liu, P. Wang, J. Huang and W.-Y. Sun, ACS Catal., 2019, 9, 8464–8470 CrossRef CAS.
- P. Xiong, H. Zhang, G. Li, C. Liao and G. Jiang, Sci. Total Environ., 2021, 797, 149179–149186 CrossRef CAS PubMed.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623–1805629 CrossRef PubMed.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C.-J. Yu, L. E. Darago, J. A. Mason and J.-O. Baeg, Nat. Mater., 2018, 17, 625–632 CrossRef CAS PubMed.
- M. Sathiya, G. Rousse, K. Ramesha, C. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau and W. Walker, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- L. Zhao, Y.-S. Meng, Q. Liu, O. Sato, Q. Shi, H. Oshio and T. Liu, Nat. Chem., 2021, 1–7 Search PubMed.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng and W. Chen, Adv. Mater., 2018, 30, 1803220–1803227 CrossRef PubMed.
- X. T. Wang, T. Ouyang, L. Wang, J. H. Zhong, T. Ma and Z. Q. Liu, Angew. Chem., 2019, 131, 13425–13430 CrossRef.
- G. Song, W. Gong, S. Cong and Z. Zhao, Angew. Chem., Int. Ed., 2021, 60, 5505–5511 CrossRef CAS PubMed.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- J. Kim, S. J. Kim, E. Jung, D. H. Mok, V. K. Paidi, J. Lee, H. S. Lee, Y. Jeoun, W. Ko and H. Shin, Adv. Func. Mater., 2022, 2110857–2110865 CrossRef CAS.
- J.-Y. Zhang, Y. Yan, B. Mei, R. Qi, T. He, Z. Wang, W. Fang, S. Zaman, Y. Su and S. Ding, Energy Environ. Sci., 2021, 14, 365–373 RSC.
- K. Fan, Y. Ji, H. Zou, J. Zhang, B. Zhu, H. Chen, Q. Daniel, Y. Luo, J. Yu and L. Sun, Angew. Chem., Int. Ed., 2017, 56, 3289–3293 CrossRef CAS PubMed.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li and Y. Luo, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu and E. Cortés, Appl. Catal., B, 2022, 121093–121102 CrossRef CAS.
- W. Zhao, K. Zhang, L. Wu, Q. Wang, D. Shang and Q. Zhong, J. Colloid Interf. Sci., 2021, 581, 76–83 CrossRef CAS PubMed.
- Y. Tong, J. Wu, P. Chen, H. Liu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2018, 140, 11165–11169 CrossRef CAS PubMed.
- A. Vojvodic and J. K. Nørskov, Science, 2011, 334, 1355–1356 CrossRef CAS PubMed.
- R. G. Burns and R. G. Burns, Mineralogical applications of crystal field theory, Cambridge University Press, 1993 Search PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- S. Bhattacharjee, U. V. Waghmare and S.-C. Lee, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- L. Tao, K. Chen, Z. Chen, C. Cong, C. Qiu, J. Chen, X. Wang, H. Chen, T. Yu and W. Xie, J. Am. Chem. Soc., 2018, 140, 8696–8704 CrossRef CAS PubMed.
- J. Seo, Y. Kim, J. Lee, E. Son, M.-H. Jung, Y.-M. Kim, H. Y. Jeong, G. Lee and H. Park, J. Mater. Chem. A, 2022, 10, 13298–13304 RSC.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- M. Ying, R. Tang, W. Yang, W. Liang, G. Yang, H. Pan, X. Liao and J. Huang, ACS Appl. Nano Mater., 2021, 4, 1967–1975 CrossRef CAS.
- H. Liu, M. Zha, Z. Liu, J. Tian, G. Hu and L. Feng, Chem. Commun., 2020, 56, 7889–7892 RSC.
- H. Li, J. Wang, R. Qi, Y. Hu, J. Zhang, H. Zhao, J. Zhang and Y. Zhao, Appl. Catal.,B, 2021, 285, 119778–1197787 CrossRef CAS.
- L. Yang and M. Dincă, Angew. Chem., Int. Ed., 2021, 60, 23784–23789 CrossRef CAS PubMed.
- R. Wang, C. Chen, Y. Zheng, H. Wang, J.-W. Liu and S.-H. Yu, Mater. Chem. Front., 2020, 4, 2881–2903 RSC.
- M. Pradhan, A. K. Sinha and T. Pal, Chem.–Eur. J., 2014, 20, 9111–9119 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03998b |
| This journal is © The Royal Society of Chemistry 2022 |