
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin-state crossover in photo-catalyzed nitrile dihydroboration via Mn-thiolate cooperation†
Matthew R.
Elsby‡
 a,
Changjin
Oh‡
bc,
Mina
Son
bc,
Scott Y. H.
Kim
a,
Mu-Hyun
Baik
*cb and
R. Tom
Baker
*a
a,
Changjin
Oh‡
bc,
Mina
Son
bc,
Scott Y. H.
Kim
a,
Mu-Hyun
Baik
*cb and
R. Tom
Baker
*a
aDepartment of Chemistry and Biomolecular Sciences and Centre for Catalysis Research and Innovation, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada. E-mail: rbaker@uottawa.ca
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 20th October 2022
Abstract
The role of S-donors in ligand-assisted catalysis using first-row metals has not been broadly investigated. Herein is described a combined experimental and computational mechanistic study of the dihydroboration of nitriles with pinacolborane (HBpin) catalyzed by the Mn(I) complex, Mn(κ3-SMeNS)(CO)3, that features thioether, imine, and thiolate donors. Mechanistic studies revealed that catalysis requires the presence of UV light to enter and remain in the catalytic cycle and evidence is presented for loss of two CO ligands. Stoichiometric reactions showed that HBpin reduces the imine N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the ligand backbone in the absence of nitrile, forming an inactive off-cycle by-product. DFT calculations showed that the bifunctional thiolate donor, coordinative flexibility of the SMeNS ligand, and access to an open-shell intermediate are all crucuial to accessing low-energy intermediates during catalysis.
C of the ligand backbone in the absence of nitrile, forming an inactive off-cycle by-product. DFT calculations showed that the bifunctional thiolate donor, coordinative flexibility of the SMeNS ligand, and access to an open-shell intermediate are all crucuial to accessing low-energy intermediates during catalysis.
Introduction
Homogeneous catalysts are important for carrying out highly selective synthesis of fine and commodity chemicals.1 Sustainable catalysis is a priority for green chemical processes,2 and the past decade has witnessed increased efforts towards the development of first-row metal catalysts to further this goal.3 The natural abundance4 and corresponding low-cost of the base metals areoften considered the main draws for their mainstream use. Nevertheless, these metals give access to diverse coordination geometries, odd–electron reaction pathways, and multiple spin states that could enable new catalytic pathways.5Metal–ligand cooperativity (MLC)6 is a valuable strategy that provides unique reactivity when first-row metals are combined with bifunctional ligands.7–10 Amongst the base metals, iron catalysts are the most developed,11 while the interest in manganese catalysts has increased exponentially in recent years.12 Since Beller's seminal report of Mn-catalyzed hydrogenation of carbonyls and nitriles,13 development of manganese catalysts which use an MLC approach has experienced tremendous growth.14–17 A shared motif amongst many reports of MLC-type Mn catalysts is the use of phosphine-based bifunctional ligands which often serve to stabilize the Mn centre and promote a low-spin ground state (Fig. 1A). To push sustainable catalysis forward, there has been interest in the study and development of sulfur-based pincer ligands for their air stability, economy, and facile syntheses, relative to P-derived ligand sets. Despite recent progress (Figure 1B),18 use of ligand frameworks composed of biomimetic donors such as sulfur or oxygen is underdeveloped, and more work is needed to allow for optimal catalyst design.
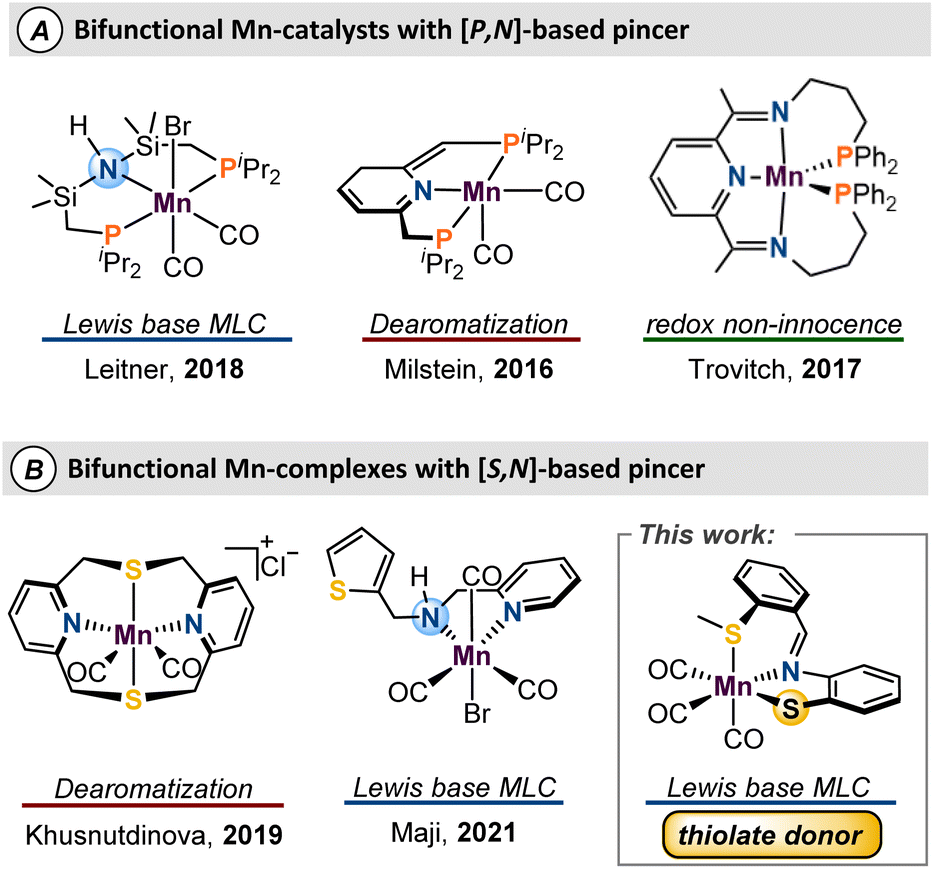 | ||
| Fig. 1 Notable manganese complexes featuring either P,N or S,N multidentate ligands. | ||
We have been investigating base-metal catalysts that leverage MLC with simple SNS ligands featuring either an amido or thiolate Lewis base donor.19 In a recent study, divalent bis(amido) complex, Mn(SMeNSMe)2, was shown to hydroborate carbonyls through a novel inner-sphere, metal hydride-free reaction pathway.20 While this work helped to highlight alternative reaction pathways enabled by sulfur-based pincer ligands, the MLC donor featured was the common amido. Overarchingly, the expansion of bifunctional Lewis base donors beyond nitrogen is sparse.21,22
In this work we use a Mn(I) platform with a [SMeNS] ligand which features thioether, imine, and thiolate donors to examine the ability of the latter to participate in MLC. While the use of bifunctional thiolate donors for MLC has been reported with Ru23 and Ir,24 their well-defined use with first-row metal catalysts is limited to select examples with Fe and Ni.25 Herein we describe a Mn(I)-thiolate complex which catalyses nitrile dihydroboration upon UV irradiation through an MLC mechanism. Detailed experimental and computational mechanistic studies demonstrate the importance of ligand flexibility, borane activation by the thiolate donor, and electronic flexibility of the base metal to access low-energy intermediates.
Results and discussion
Catalyst synthesis and nitrile dihydroboration
In previous work with mono-ligated Fe-[SNS] complexes, it was found that electron-rich co-ligands encouraged Caryl–S cleavage of the thioether donor.26 To avoid this deleterious side reaction during the design of a mono-ligated Mn-[SNS] complex, we prepared the tricarbonyl complex. Treatment of Mn(CO)5Br with one equiv. of LH and LiHMDS in toluene afforded Mn(SMeNS)(CO)3 (1) in 91% yield (Fig. 2). The thioether protons in the 1H NMR spectrum display a broadened singlet, likely due to thioether hemilability19 that allows for epimerization at sulfur. Observation of free CO in the 13C{1H} NMR spectra of solutions of 1 in CD3CN exposed to ambient light suggests lability of an ancillary carbonyl ligand as well,27 and UV-vis absorptions at 240 and 290 nm indicate that CO ligands may be labilized upon UV irradiation, potentially offering multiple opportunities to access unsaturated intermediates. Single crystal X-ray diffraction analysis revealed the expected pseudooctahedral geometry about Mn, where the [SMeNS] ligand is arranged in a facial manner. This is the first instance of our [SNS] ligand adopting this coordination pattern, suggesting potential for L to demonstrate coordinative fluxionality between fac- and mer-isomers that could also play a role in catalysis.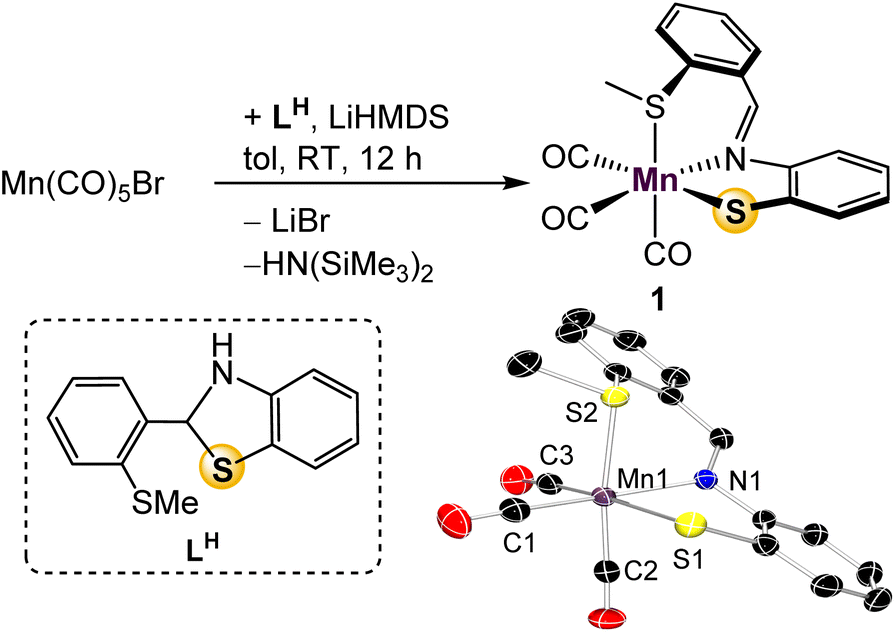 | ||
| Fig. 2 Synthesis and molecular depiction of 1. Thermal ellipsoids are set at 50% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å): Mn–N 2.0587(16), Mn–S(1) 2.3898(6), Mn–S(2) 2.3592(6), Mn–C(1) 1.797(2), Mn–C(2) 1.802(2), Mn–C(3) 1.805(2). | ||
To examine the bifunctional activity of the thiolate donor in 1, its affinity to perform bifunctional B–H activation was tested. The stoichiometric reaction of 1 and HBpin in CD3CN resulted in no color change, and 1H NMR analysis revealed no reaction. Heating the reaction mixture to 80 °C also gave no reactivity and led to decomposition, as evidenced by observation of free L in solution. Alternatively, upon irradiation with UV-light, a color change was observed from dark burgundy to light yellow. The crude 11B NMR spectrum displayed a signal at 24 ppm, diagnostic of a B–N species, and further analysis of the 1H NMR spectrum revealed signals indicative of the dihydroboration of the acetonitrile-d3 solvent.
To investigate the potential of this system for nitrile dihydroboration, the reaction conditions were optimized with benzonitrile as the model substrate, and 2.2 equiv. of HBpin were used to ensure complete reduction (Table 1). Attempts to thermally facilitate the dihydroboration of benzonitrile were unsuccessful (entry 1); only upon UV-irradiation of the reaction mixture was product formation observed. While 1 is most soluble in acetonitrile, performing the dihydroboration of benzonitrile in this solvent results in the reduction of both benzo- and acetonitrile (entry 2). Reaction in either benzene-d6 or toluene-d8 did not yield satisfactory conversion (Entries 3 and 4). Reaction in THF afforded complete conversion to the dihydroboration product in reasonable time frames with as little as 1 mol% of 1 (entries 5 and 6). Further control reactions were performed to ensure the coordination of deprotonated [SMeNS] to Mn was crucial to a successful reaction (see ESI†). Evaluating the optimized reaction conditions makes it clear that the rate is influenced by the presence of polar, coordinating solvents, suggesting a solvent-stabilized intermediate may be involved in the reaction pathway.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | mol% | Temp (°C) | hυ | Solvent | Time (h) | yield (%) |
| a Reaction conditions: nitrile (0.16 mmol), HBpin (0.34 mmol), 1 (see entry), THF (0.6 g) at room temperature with irradiation from UV-lamp. | ||||||
| 1 | 5 | 80 | ✗ | MeCN | 18 | 0 |
| 2 | 5 | 25 | ✓ | MeCN | 18 | 60 |
| 3 | 5 | 25 | ✓ | C6D6 | 18 | 54 |
| 4 | 5 | 25 | ✓ | Toluene | 18 | 62 |
| 5 | 1 | 25 | ✓ | THF | 2 | >99 |
| 6 | 1 | 25 | ✓ | THF | 6 | >99 |
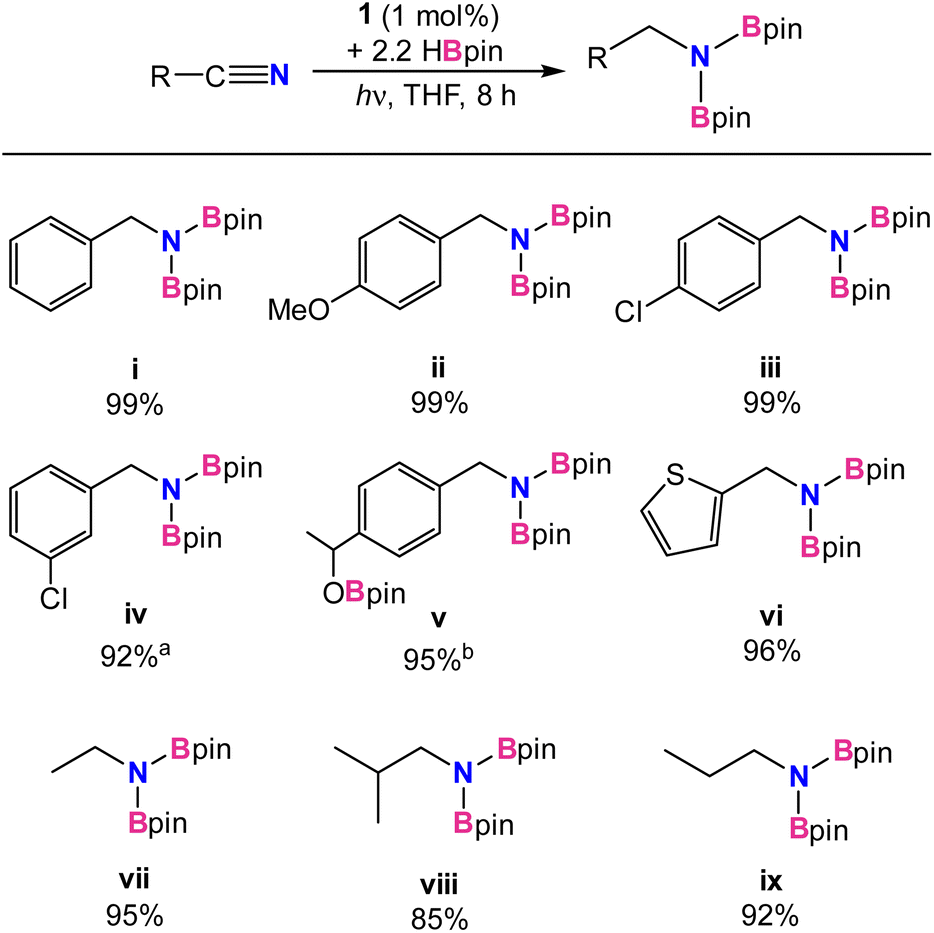
Under the optimized conditions, additional substrates for the nitrile dihydroboration reaction were investigated (Table 2). Greater than 90% NMR yields were observed for most substrates and isolated products were easily obtained as crystalline solids by hexane extraction of the dried reaction mixture. Performing the reduction with 0.25 g of benzonitrile gave the N,N-diborylbenzylamine product in 87% isolated yield. Catalysis was not significantly hindered for benzonitrile substituted with electron-donating or -withdrawing groups (ii and iii) or for one heterocycle example (vi). A functional group in more sterically compromising positions hindered catalysis, with 3-chlorobenzene requiring a 2.5% catalyst loading to reach 90% yield (iv). For p-acetyl-benzonitrile, addition of 3.3 equiv. of HBpin afforded the trihydroborated product (v), efficiently reducing both the carbonyl and nitrile functionalities. Using 1 equiv. of HBpin with the same substrate afforded the ketone hydroboration product exclusively in 5 min without the need for irradiation. While it was not unexpected for 1 to be an active catalyst for carbonyl hydroboration, this reaction occurs thermally at room temperature, indicating it likely follows an alternative pathway from nitrile reduction. Indeed, a reaction mixture of catalytic 1, p-acetyl benzonitrile, and 3 equiv. of HBpin which was kept in the dark at room temperature afforded the ketone reduced product with 2 equiv. of unreacted HBpin, confirming that ketone reduction takes place thermally without the need for light. Finally, for aliphatic nitriles (vii–ix) it was found that acetonitrile, propionitrile, and isobutylnitrile were all successfully converted, albeit in slightly poorer yields than aromatic substrates. Aromatic substrates containing alkenes, esters, and amides were also examined. High catalyst loadings and poor conversions (30–50%) for alkenes, as well as no reactivity for esters and amides deterred further investigation (see ESI†).
| a Reaction conditions: nitrile (0.16 mmol), HBpin (0.34 mmol), 1 (1 mol%), THF (0.6 g) at room temperature with irradiation from UV-lamp for 2–8 h. a Conducted with 2.5% loading of 1. b Conducted with 3.3 equiv of HBpin. Yields were determined from 1H NMR integrals with respect to internal standard mesitylene after workup. |
|---|
|
Photoexcitation and carbonyl ligand dissociation
The metal-pincer complex decorated with CO ligands is a widely used catalyst platform.28 Currently, many of these complexes use phosphine-based pincers and rely on elevated temperatures to kinetically access their respective reaction pathways. Despite the rich history of catalysis using photoinduced CO dissociation,29 use of UV irradiation in systems which use an MLC platform is rare. Photolysis may also provide potentially advantageous access to alternative reaction pathways involving open-shell intermediates. To further evaluate the capacity of UV-light in this system and understand the role of the bifunctional thiolate donor, a series of mechanistic experiments were performed. Initially, two parallel reaction mixtures of catalytic 1 with benzonitrile and 2.2 equiv of HBpin were irradiated until solution color change and product formation was observed. Subsequently, one of the samples was continually irradiated for a further 4 h, while the other was kept in the dark. Comparison of the two mixtures showed that the irradiated reaction went to completion, while catalysis in the “dark” mixture was halted, confirming that the persistent presence of light is needed to turn over the catalytic cycle. It is thus likely that dissociation of labile CO ligand(s) from 1 is the initial step, providing a vacant coordination site for substrate/solvent coordination. To gain insight into the number of CO ligands coordinated during catalysis, 13C-labeling studies were performed. The reaction of 2.2 equiv. of HBpin and 1 equiv. benzonitrile with a 10% loading of 13C-labelled 1* was monitored periodically by 13C{1H} NMR (Scheme 1A). The initial spectrum of 1* shows three distinct peaks in the M–CO region at 216, 220, and 222 ppm. Irradiation of the reaction mixture for 45 min initiated the expected color change and the onset of catalysis. The crude carbon NMR spectrum showed the disappearance of the three initial Mn–13CO signals and the emergence of a new very broad signal at 240 ppm (Fig. S7†). This single peak is indicative of either a single bound 13CO, or a highly symmetric intermediate with two coordinated 13CO's. Since the unsymmetrical nature of L likely precludes a high-symmetry environment, a species containing a single coordinated CO ligand is most likely. The broad signal at 240 ppm persisted throughout the reaction and remained detectable upon completion. Furthermore, observation of a new sharp signal at 184 ppm during catalysis was indicative of free 13CO in solution.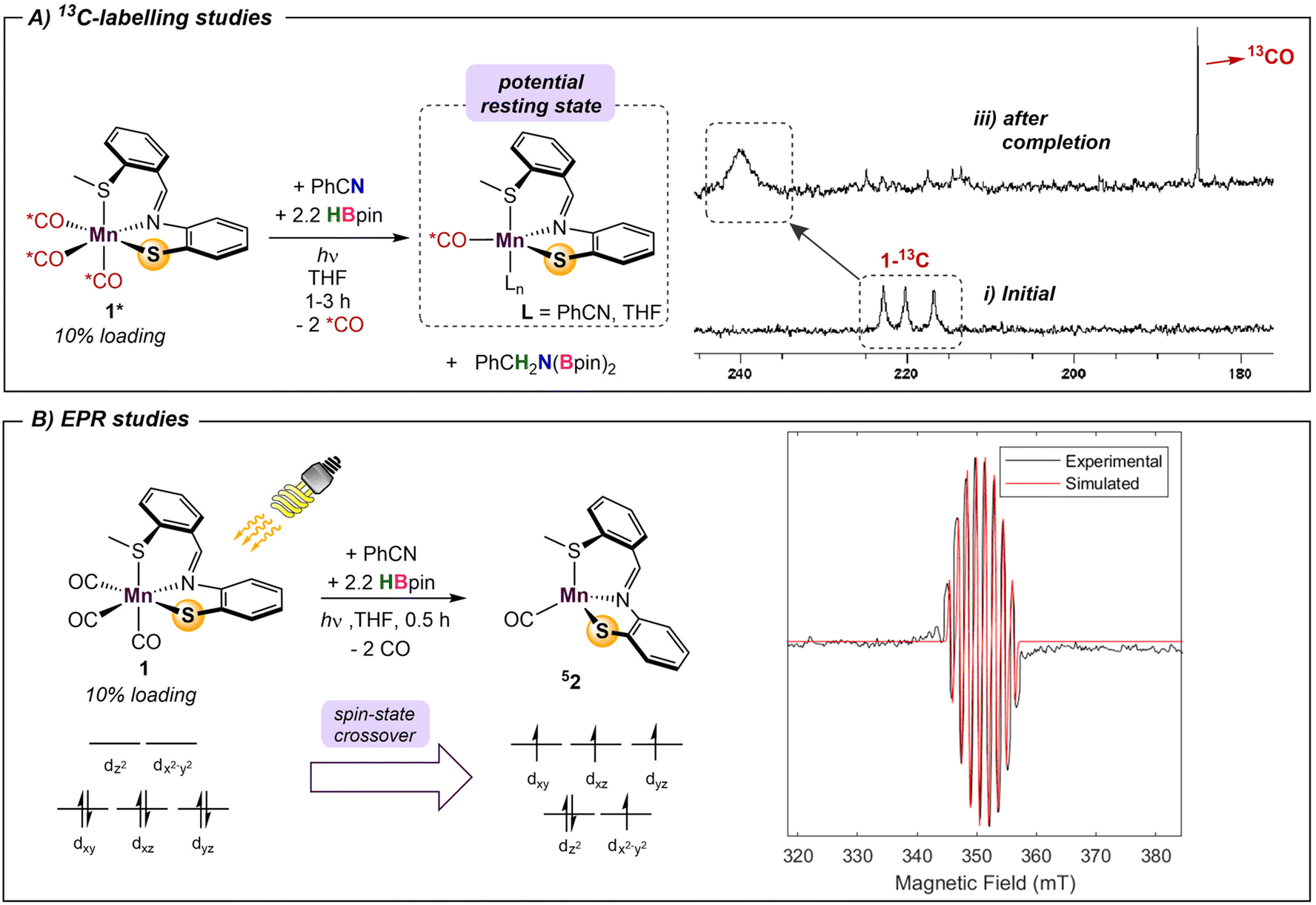 | ||
| Scheme 1 Spectroscopic and isotope-labelling experiments with 1. | ||
Time-dependent density functional theory (TD-DFT) calculations were employed to probe the photoexcitation. Geometry optimization and TD-DFT were carried out at the B3LYP-D3/6-31G**, LACVP level of theory. In accordance with 13C-labeling studies, a metal-to-ligand charge transfer excitation into a metal-CO antibonding orbital was observed around 5.06 eV, supporting the dissociation of two CO ligands.30 Natural transition orbitals (NTOs) corresponding to the major transition from metal- and ligand-based orbitals to an orbital comprising an out-of-phase combination between metal and two COs are depicted in Fig. 3. With the dissociation of two strong-field carbonyl ligands, the d6 Mn centre may gain access to a high-spin configuration with a singly-bound CO, as suggested by the very broad 13C NMR resonance.31 Indeed, DFT investigation into intermediates containing a single carbonyl ligand consistently showed that a high-spin quintet ground state is energetically favored over the low-spin singlet or intermediate-spin triplet states (Fig. S30†). Electron paramagnetic resonance studies were performed to experimentally probe this result. The ambient temperature EPR spectrum of a solution of HBpin, benzonitrile, and catalytic 1 gave no signal. Upon irradiation of light for 45 min, the isotropic EPR spectrum yielded a multiplet at g = 1.99 which, after modeling, was attributed to hyperfine coupling to 55Mn (42.5 G) and 14N (42.1 G) (Scheme 1B). This emergence of a paramagnetic signal supports the initial DFT results indicating that the Mn centre preferentially adopts a high-spin state upon photoactivation.
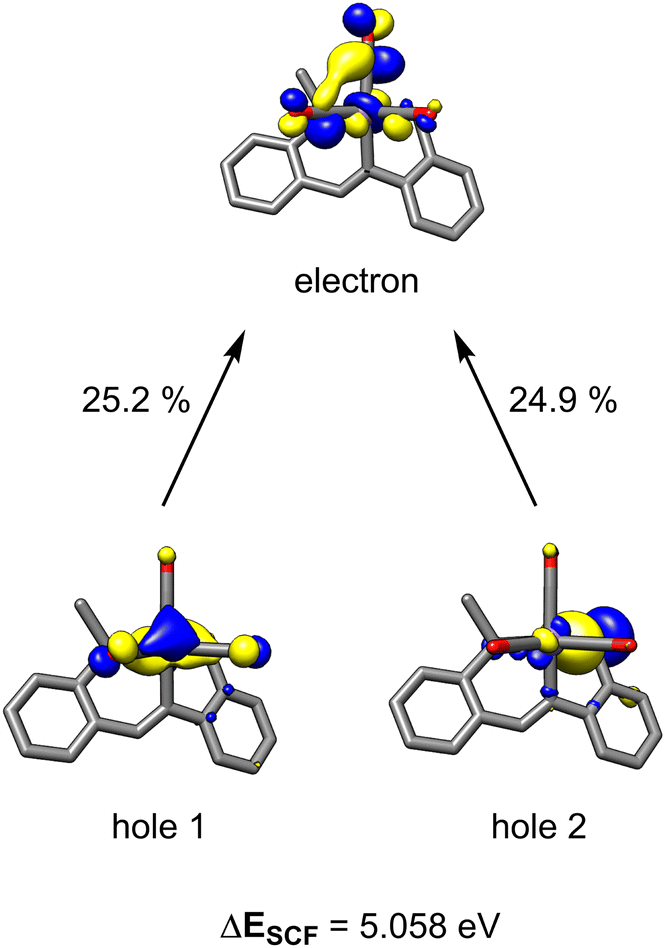 | ||
| Fig. 3 NTO pairs for the excitation responsible for CO dissociation. The top two contributing transitions are described. Other contributions are less than 15%. | ||
Catalytic cycle for nitrile dihydroboration
To probe the reaction's initial steps associated with the dihydroboration, a stoichiometric amount of 1 treated with HBpin in THF-d8 in the absence of nitrile was irradiated, giving a color change from dark yellow to red.32 The resultant 1H NMR spectrum showed the disappearance of original peaks associated with 1 and the emergence of a new species (Fig. S25†). Specifically, the disappearance of the imine C–H resonance at δ 8.81 paired with the growth of a new singlet at δ 4.85 was diagnostic for borylation of the C–N imine unit forming 9 (Scheme 2). While a small resonance was observed in the hydride region (δ −8.59), it was not of sufficient intensity to be considered as a viable reaction intermediate, similar to previous work by Leitner.15 Given the literature precedence for imine hydroboration33 in addition to a previous report of the activity of a ligand backbone for H2 activation,34 the observed hydroboration of the imine backbone in the SMeNS ligand is not unexpected. Additionally, the hydroborated ligand was previously isolated and characterized in our lab,35 and its presence in this reaction mixture was confirmed by comparison of 1H and 11B NMR data. It is of note that this intermediate is preferentially formed over Mn–H derived from traditional bifunctional B–H activation by 41.2 kcal mol−1 (Fig. S29†). To further investigate 9 as a catalytic intermediate, 1 equiv. of benzonitrile and an additional equiv. of HBpin were added to the reaction mixture. Curiously, no product conversion was observed upon heating or irradiation. The lack of reactivity of 9 suggests it is an off-cycle product or an unproductive species that does not form under catalytic conditions. This is supported by DFT calculations showing that formation of 9 is exergonic by −17.1 kcal mol−1, confirming it to be a thermodynamic sink. In addition, while direct insertion of HBpin to the ligand imine moiety in the absence of nitrile is feasible with a barrier of 31.4 kcal mol−1 (Fig. S36† in ESI), the proposed pathway traversing 4-TS in Fig. 4 is energetically favored in the presence of nitrile. This suggests that the initial presence of both HBpin and nitrile are required in solution to enter the productive catalytic cycle. Finally, re-examining the crude catalysis solution by 1H NMR after product workup showed persistence of the imine C–H resonance, indicating that ligand imine hydroboration is not favored under catalytic conditions.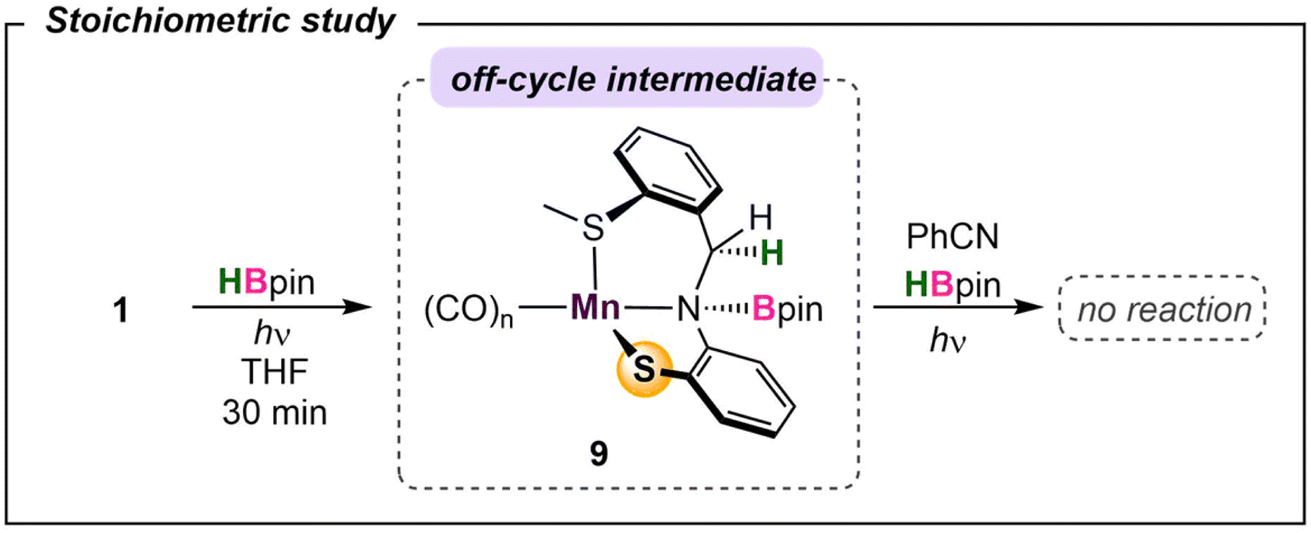 | ||
| Scheme 2 Off-pathway intermediate in the absence of nitrile. | ||
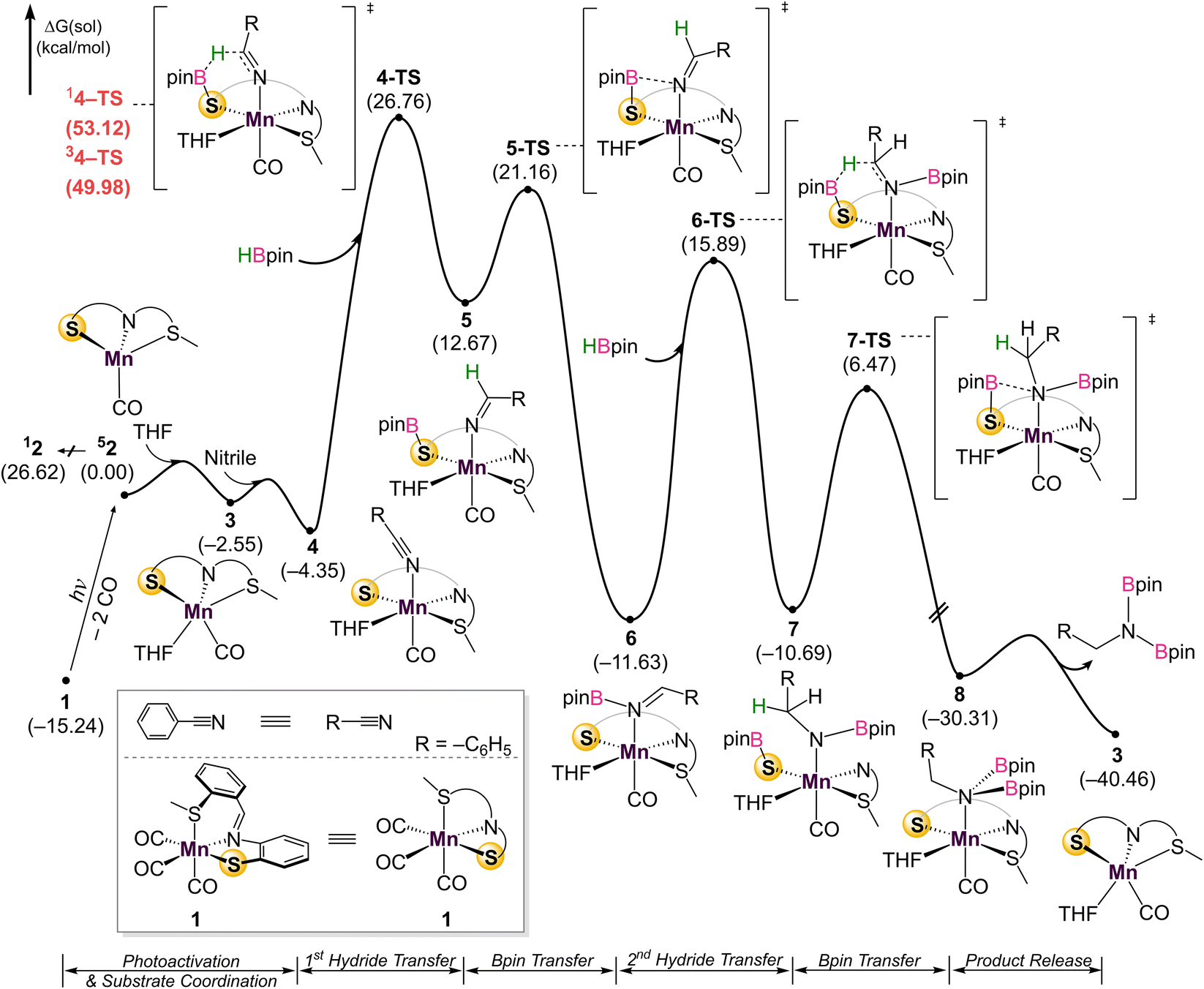 | ||
| Fig. 4 DFT-calculated energy profile for the dihydroboration of benzonitrile with catalyst 1. | ||
Given the challenges associated with monitoring the reaction in real-time, DFT calculations on the overall mechanism were carried out at the B3LYP-D3/cc-pVTZ(-f)/LACV3P//B3LYP-D3/6-31G**/LACVP level of theory36 with a self-consistent reaction field (ε = 7.6 for THF) approach to account for solvation (Fig. 4).37 Initial photoactivation and carbonyl dissociation forms the monocarbonyl species 2. With the loss of two carbonyls, which act as strong-field ligands, a high-spin configuration is favored. Thus, the high-spin quintet intermediate 52 is preferred over the low-spin species 12 by 26.6 kcal mol−1. While the intermediate-spin triplet 32, located 4.4 kcal mol−1 higher than 12, is relatively close to the ground quintet state, the preference for high-spin species is retained throughout the catalytic cycle. A detailed comparison of different spin states is given in the ESI (Fig. S30)†.
Coordination of the solvent THF to form 3 is followed by nitrile coordination to the metal centre, leading to stable intermediate 4. The calculations confirm experimental results that excluded significant participation of Mn–H, with HBpin instead coordinatingto the bifunctional thiolate sulfurand allowing for direct hydride transfer to the coordinated nitrile through 4-TS to form 5. An alternate pathway featuring the direct coordination of HBpin in lieu of solvent to form a Mn–S–B–O metallacycle was also considered, inspired by previous work (Fig. S31†).20 This pathway has a barrier of 41.5 kcal mol−1 due to the penalty of forming a sterically hindered 4-membered metallacycle, indicating that a solvent molecule must be incorporated. Hemilability of the thioether group was also interrogated (Fig. S32†). As the thioether arm dissociates, the phenyl linker interacts with a THF molecule. Two transition states featuring the simple cleavage of thioether and η2-coordination of imine were captured and the corresponding barriers are 41.5 and 38.1 kcal mol−1, respectively. Notably higher barriers with the labile thioether group suggest that the SNS ligand likely remains tridentate during reaction. This first hydride transfer through 4-TS represents the turnover limiting step with a 31.1 kcal mol−1 barrier, which looks too high at first sight for a room–temperature reaction. Extensive efforts did not indicate any technical problems and no alternative transition state with a lower energy could be located (See ESI† for details). Taking into account that the reaction does not proceed without continuous UV irradiation, we reason that the Mn-complex obtains additional energy from the Mn–CO bond cleavage process, as the excitation energy is dissipated locally through heat and molecular vibrations. Alternatively, if rebinding of the CO does not occur, the Mn-catalyst may absorb light and undergo non-radiative decay. In either case, the local vibrational temperature of the Mn-catalyst will be much higher than what is expected for an undisturbed ground state system. In other words, UV irradiation not only cleaves the Mn–CO bond, but also provides additional energy during hydroboration to overcome the reaction barrier.
Substitution of THF with a weakly coordinating solvent, benzene, gives a greater hydride transfer barrier of 34.0 kcal mol−1 which coincides well with the observed conversions in Table 1 (Fig. S34†). Closer inspection of the hydride transfer in 4-TS shows that a transient metallo-imine species is generated, with the CO ligand trans to the anionic iminato moiety to electronically stabilize the transition state as in structure 5. For the trans effect to successfully stabilize the intermediate, the SNS ligand must be arranged in a mer-configuration. Indeed, a comparison between mer- and fac-conformations shows the former is favored by 7.4 kcal mol−1, indicating that the coordinative flexibility of the SNS ligand is crucial for rearrangement to the lowest energy conformer (Fig. S35†). The resulting intermediate 5 then undergoes sequential Bpin transfer to generate a monoborylimine in 6 and complete the first reduction cycle. Evidence for the monoborylimine product was never observed experimentally, and it was found that steric hindrance is responsible for the selective hydride transfer to the monoborylimine over the imine of the ligand framework. In the absence of THF, the ligand imine is preferentially reduced (Fig. S36 and S37† in ESI), supporting the kinetic preference for reduction of the monoboryl-imine. Finally, undergoing a similar mechanism involving sequential hydride and Bpin transfer through 7 and 7-TS, the second reduction cycle furnishes intermediate 8 which releases product diborylamine, as depicted in the proposed full mechanism for both reduction cycles (Fig. 5).
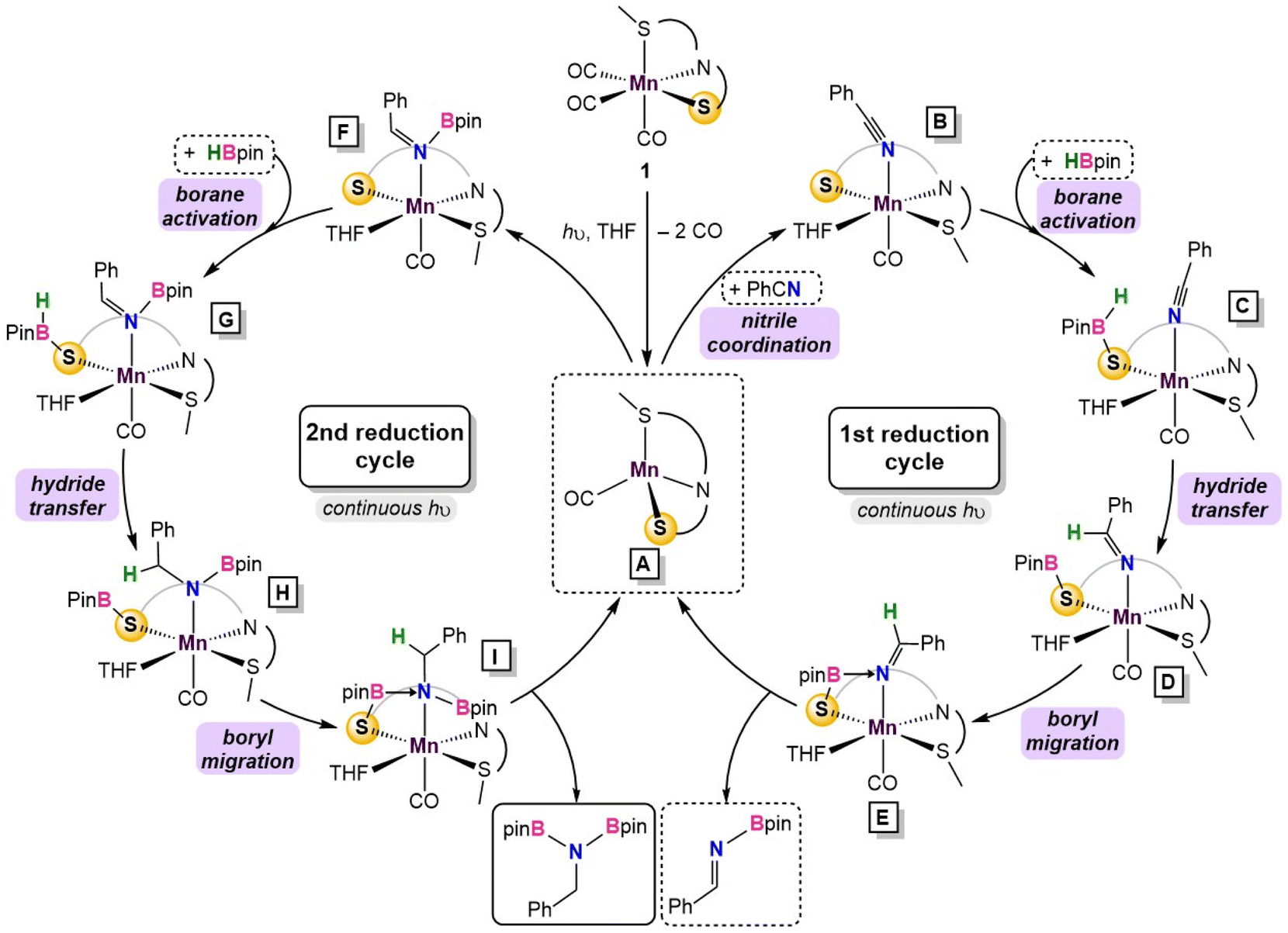 | ||
| Fig. 5 Summary of the catalytic cycle for the photodissociation and two borylation steps involved in the reaction. | ||
Conclusions
Catalytic dihydroboration of nitriles is an effective strategy to access synthetically valuable N,N,-diborylamine precursors38 and has been previously reported in systems using Mo,39 Th,40 Zn,41 Mg,42 and Ru43 catalysts with catalyst loadings between 1 and 10% and temperatures between 22 and 80 °C. Previous reports with first-row transition metal complexes are limited to Ni,44 Co45 and a single Mn example46 which required elevated reaction temperatures.46 In this work, a Mn(I) complex featuring an SNS-thiolate ligand was prepared to study the bifunctional activity of less studied thiolate donors. While Mn(κ3-SMeNS)(CO)3 complex (1) was an active catalyst for carbonyl hydroboration at room temperature, reduction of nitrile substrates required continuous photolysis to access additional coordination sites on the Mn centre. 13C-labeling studies revealed a Mn-species likely featuring one coordinated CO ligand, in good agreement with TD-DFT calculations. EPR studies combined with DFT calculations indicated that the Mn-centre undergoes a spin state change to preferentially adopt the high-spin configuration during catalysis. This feature is a benefit of first-row metals,5 whereby embracing the electronic flexibility of the metal allows for access to lower energy intermediates. Experimental mechanistic studies and calculations suggest the catalytically active species incorporating a single CO ligand is formed under UV light, and upon traversing two reduction cycles, diborylamine is released as the product (Fig. 5). The bifunctional SMeNS ligand is crucial to the success of the reaction in three aspects: (i) the weak field donors comprising the SNS ligand enable Mn to access a low-energy, high-spin intermediate, (ii) the thiolate donor allows for an MLC pathway for borane activation and Bpin transfer (5 and 5-TS in Fig. 5), and (iii) the coordinative flexibility of the SMeNS ligand enables facile rearrangement between fac- and mer-isomers which is integral to stabilization of the turnover-limiting hydride transfer step (4-TS in Fig. 4). The presence of coordinating solvent THF in lieu of non-coordinating solvent was also determined to be crucial for access to the sterically relaxed transition state, mitigating the need to form a sterically disfavored metallacycle intermediate.The use of synthetically accessible ligands with biomimetic N-, O- and S-donors, in combination with first-row metals, is vital to meet the growing demand for more sustainable catalyst options. Although photolysis for CO loss is a well-established paradigm, its necessity in this work to access high-spin reaction intermediates highlights how the unique electronic flexibility of the base-metals can decrease activation barriers.47 Whereas our previously reported divalent Mn catalyst was shown to operate via an amido donor,20 this work uses a thiolate donor coordinated to Mn(I). This shows that this alternate inner-sphere, hydride-free reaction pathway does not discriminate on the basis of the bifunctional donor or even the metal d-electron count; it is instead likely a consequence of the weaker donors on the SMeNS ligand vs. P-containing pincer ligands. The combination of a first-row metal and a thiolate donor in MLC catalysis remains rare,25 and this is the first instance of a Mn catalyst using a bifunctional thiolate. As more phosphine-free ligand variants emerge,48 it will be critical to gain understanding of their associated reactivity patterns to enable optimal designs of sustainable and high-performing first-row metal catalysts.
Data availability
All data are included in the ESI.†Author contributions
MRE and CO contributed equally to this work. MRE performed all experimental work associated with synthesis, catalysis, and mechanistic studies and contributed to writing the manuscript. CO and MS performed all computational work. SYHK assisted with EPR experiments and catalyst stability studies. RTB and MHB provided supervision, funding, ideation and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSERC (Discovery Grant 2019-05959) for generous financial support and the University of Ottawa, Canada Foundation for Innovation, and the Ontario Ministry of Economic Development and Innovation for essential infrastructure. M.-H. B. acknowledges financial support from the Institute for Basic Science in Korea (IBS-R010-A1). M. R. E. acknowledges a graduate scholarship from the province of Ontario and we thank Inorganic Chemistry Exchange undergrad Keshia Gaudet for initial catalyst synthesis studies.References
- (a) P. W. N. M. Van Leeuwen, Homogeneous Catalysis: Understanding the Art, Springer, 2006 Search PubMed; (b) G. P. Chiusoli and P. M. Maitlis, Metal-Catalysis in Industrial Organic Processes, Royal Society of Chemistry, 2019 Search PubMed; (c) B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied Homogeneous Catalysis With Organometallic Compounds: A Comprehensive Handbook in Four Volumes, John Wiley & Sons, 2017 CrossRef.
- (a) P. T. Anastas and R. H. Crabtree, Green Catalysis: Homogeneous Catalysis, John Wiley & Sons, 2014 Search PubMed; (b) J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 CrossRef CAS.
- (a) R. M. Bullock, Catalysis Without Precious Metals, John Wiley & Sons, 2011 Search PubMed; (b) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed; (c) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin and S. D. Minteer, Science, 2020, 369, abc3183 CrossRef PubMed.
- K. H. Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef CAS.
- P. L. Holland, Chem, 2017, 2, 443–444 CAS.
- (a) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- (a) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (b) M. Fritz and S. Schneider, in The Periodic Table II: Catalytic, Materials, Biological and Medical Applications, ed. D. M. P. Mingos, 2019, pp. 1–36 Search PubMed; (c) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2018, 119, 2681–2751 CrossRef PubMed; (d) J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS; (e) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed; (f) R. M. Bullock and M. L. Helm, Acc. Chem. Res., 2015, 48, 2017–2026 CrossRef CAS PubMed.
- (a) S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS; (b) N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS; (c) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed; (d) R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350–6354 CrossRef CAS.
- (a) A. Mikhailine, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS PubMed; (b) A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS PubMed; (c) L. De Luca, A. Passera and A. Mezzetti, J. Am. Chem. Soc., 2019, 141, 2545–2556 CrossRef PubMed; (d) R. Bigler, R. Huber, M. Stöckli and A. Mezzetti, ACS Catal., 2016, 6, 6455–6464 CrossRef CAS; (e) M. Pang, J.-Y. Chen, S. Zhang, R.-Z. Liao, C.-H. Tung and W. Wang, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- (a) M. Pang, C. Wu, X. Zhuang, F. Zhang, M. Su, Q. Tong, C.-H. Tung and W. Wang, Organometallics, 2018, 37, 1462–1467 CrossRef CAS; (b) C. Ghosh, S. Kim, M. R. Mena, J.-H. Kim, R. Pal, C. L. Rock, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2019, 141, 15327–15337 CrossRef CAS PubMed; (c) M. R. Elsby and R. T. Baker, Chem. Commun., 2019, 55, 13574–13577 RSC.
- (a) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed; (b) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed; (c) T. Zell and R. Langer, in Homogeneous Hydrogenation with Non-Precious Catalysts, ed. J. F. Teichert, Wiley-VCH, 2020, pp. 15–38 Search PubMed.
- (a) R. J. Trovitch, Acc. Chem. Res., 2017, 50, 2842–2852 CrossRef CAS PubMed; (b) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (c) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- (a) S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed; (b) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed; (c) N. A. Espinosa‐Jalapa, A. Nerush, L. J. Shimon, G. Leitus, L. Avram, Y. Ben‐David and D. Milstein, Chem. - Eur. J., 2017, 23, 5934–5938 CrossRef PubMed; (d) F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed; (e) F. Freitag, T. Irrgang and R. Kempe, J. Am. Chem. Soc., 2019, 141, 11677–11685 CrossRef CAS PubMed; (f) K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS; (g) N. V. Kulkarni, W. W. Brennessel and W. D. Jones, ACS Catal., 2018, 8, 997–1002 CrossRef CAS; (h) S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed; (i) A. Kumar, T. Janes, N. A. Espinosa‐Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed.
- C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- (a) U. K. Das, A. Kumar, Y. Ben-David, M. A. Iron and D. Milstein, J. Am. Chem. Soc., 2019, 141, 12962–12966 CrossRef CAS PubMed; (b) U. K. Das, Y. Ben-David, G. Leitus, Y. Diskin-Posner and D. Milstein, ACS Catal., 2019, 9, 479–484 CrossRef CAS; (c) J. Masdemont, J. A. Luque-Urrutia, M. Gimferrer, D. Milstein and A. Poater, ACS Catal., 2019, 9, 1662–1669 CrossRef CAS; (d) Y.-Q. Zou, S. Chakraborty, A. Nerush, D. Oren, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2018, 8, 8014–8019 CrossRef CAS PubMed.
- (a) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef CAS PubMed; (b) T. K. Mukhopadhyay, C. L. Rock, M. Hong, D. C. Ashley, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2017, 139, 4901–4915 CrossRef CAS PubMed; (c) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Chem. Sci., 2018, 9, 7673–7680 RSC.
- (a) R. van Putten, J. Benschop, V. J. de Munck, M. Weber, C. Müller, G. A. Filonenko and E. A. Pidko, ChemCatChem, 2019, 11, 5232 CrossRef CAS PubMed; (b) A. Sarbajna, P. H. Patil, M. H. Dinh, O. Gladkovskaya, R. R. Fayzullin, S. Lapointe, E. Khaskin and J. R. Khusnutdinova, Chem. Commun., 2019, 55, 3282–3285 RSC; (c) K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- (a) U. K. Das, S. L. Daifuku, S. I. Gorelsky, I. Korobkov, M. L. Neidig, J. J. Le Roy, M. Murugesu and R. T. Baker, Inorg. Chem., 2016, 55, 987–997 CrossRef CAS PubMed; (b) U. K. Das, S. L. Daifuku, T. E. Iannuzzi, S. I. Gorelsky, I. Korobkov, B. Gabidullin, M. L. Neidig and R. T. Baker, Inorg. Chem., 2017, 56, 13766–13776 CrossRef CAS PubMed; (c) M. R. Elsby, S. Y. Kim, S. N. Steinmann and R. T. Baker, Dalton Trans., 2021, 50, 14542–14546 RSC.
- M. R. Elsby, M. Son, C. Oh, J. Martin, M.-H. Baik and R. T. Baker, ACS Catal., 2021, 11, 9043–9051 CrossRef CAS.
- (a) A. Dubey, S. W. Rahaman, R. R. Fayzullin and J. R. Khusnutdinova, ChemCatChem, 2019, 11, 3844–3852 CrossRef CAS; (b) J. Liu, J.-Y. Chen, M. Jia, B. Ming, J. Jia, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2019, 9, 3849–3857 CrossRef CAS.
- (a) A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS; (b) K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC.
- L. Omann, C. D. F. Königs, H. F. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed.
- K. D. Hesp, R. McDonald, M. J. Ferguson and M. Stradiotto, J. Am. Chem. Soc., 2008, 130, 16394–16406 CrossRef CAS.
- (a) H. Song, K. Ye, P. Geng, X. Han, R. Liao, C.-H. Tung and W. Wang, ACS Catal., 2017, 7, 7709–7717 CrossRef CAS; (b) F. Zhang, H. Song, X. Zhuang, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2017, 139, 17775–17778 CrossRef CAS PubMed; (c) X. Yu, M. Pang, S. Zhang, X. Hu, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2018, 140, 11454–11463 CrossRef CAS.
- M. R. Elsby, K. Ghostine, U. K. Das, B. M. Gabidullin and R. T. Baker, Organometallics, 2019, 38, 3844–3851 CrossRef CAS.
- Two samples of 1 were prepared in CD3CN. One sample heated for 18 h at 70 °C in darkness showed an unchanged 1H NMR spectrum. The other sample which was exposed to ambient light for 18 h changed color and showed broadened 1H NMR resonances.
- (a) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC; (b) M. E. O'Reilly and A. S. Veige, Chem. Soc. Rev., 2014, 43, 6325–6369 RSC; (c) M. Vogt and R. Langer, Eur. J. Inorg. Chem., 2020, 2020, 3885–3898 CrossRef CAS.
- (a) M. Wrighton, Chem. Rev., 1974, 74, 401–430 CrossRef CAS; (b) M. Wrighton, G. S. Hammond and H. B. Gray, J. Organomet. Chem., 1974, 70, 283–301 CrossRef CAS; (c) M. A. Schroeder and M. S. Wrighton, J. Am. Chem. Soc., 1976, 98, 551–558 CrossRef CAS; (d) M. S. Wrighton, D. Ginley, M. Schroeder and D. Morse, Pure Appl. Chem., 1975, 41, 671–697 CrossRef CAS; (e) P. L. Bogdan, P. J. Sullivan, T. A. Donovan Jr and J. D. Atwood, J. Organomet. Chem., 1984, 269, c51–c54 CrossRef CAS.
- (a) For an example of concerted loss of two CO ligands see: P. T. Snee, C. K. Payne, S. D. Mebane, K. T. Kotz and C. B. Harris, J. Am. Chem. Soc., 2001, 123, 6909–6915 CrossRef CAS; (b) For examples of multiple CO loss in N-ligated Mn carbonyl complexes see: M. A. Gonzalez, M. A. Yim, S. Cheng, A. Moyes, A. J. Hobbs and P. K. Mascharak, Inorg. Chem., 2012, 51, 601–608; (c) I. Chakraborty, S. J. Carrington and P. K. Mascharak, Acc. Chem. Res., 2014, 47, 2603–2611 CrossRef CAS PubMed.
- While we cannot rule out signal broadening by the 55Mn quadrupole (I = 5/2), the 13C NMR line widths at half height for unsymmetrical 1 average ca. 30 Hz vs. 225 Hz for the signal at 240 ppm in Scheme 1.
- The possibility of initial nitrile coordination was also investigated. Irradiating a solution of 1 in acetonitrile-d3 afforded no color change and the 1H NMR spectrum showed no indication of a new species. Additionally, addition of stoichiometric benzonitrile to a THF solution of 1 gave no reactivity and resulted in decomposition upon heating or irradiation.
- (a) C. C. Chong and R. Kinjo, ACS Catal., 2015, 5, 3238–3259 CrossRef CAS; (b) A. Kaithal, B. Chatterjee and C. Gunanathan, J. Org. Chem., 2016, 81, 11153–11161 CrossRef CAS PubMed; (c) X. Wang and X. Xu, RSC Adv., 2021, 11, 1128–1133 RSC.
- A. Friedrich, M. Drees, M. Käss, E. Herdtweck and S. Schneider, Inorg. Chem., 2010, 49, 5482–5494 CrossRef CAS PubMed.
- S. Ataie, S. Hogeterp, J. S. Ovens and R. T. Baker, Chem. Commun., 2022, 58, 3795–3798 RSC.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed; (c) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (d) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS; (e) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (f) A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS; (g) T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef; (h) D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- B. Marten, K. Kim, C. Cortis, R. A. Friesner, R. B. Murphy, M. N. Ringnalda, D. Sitkoff and B. Honig, J. Phys. Chem., 1996, 100, 11775–11788 CrossRef CAS.
- D. Hayrapetyan and A. Y. Khalimon, Asian J. Chem., 2020, 15, 2575–2587 CrossRef CAS PubMed.
- (a) A. Y. Khalimon, P. M. Farha and G. I. Nikonov, Dalton Trans., 2015, 44, 18945–18956 RSC; (b) A. Y. Khalimon, P. Farha, L. G. Kuzmina and G. I. Nikonov, Chem. Commun., 2012, 48, 455–457 RSC.
- S. Saha and M. S. Eisen, ACS Catal., 2019, 9, 5947–5956 CrossRef CAS.
- S. Das, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 16812–16818 RSC.
- (a) C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC; (b) J. Li, M. Luo, X. Sheng, H. Hua, W. Yao, S. A. Pullarkat, L. Xu and M. Ma, Org. Chem. Front., 2018, 5, 3538–3547 RSC.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- G. Nakamura, Y. Nakajima, K. Matsumoto, V. Srinivas and S. Shimada, Catal.: Sci. Technol., 2017, 7, 3196–3199 RSC.
- (a) A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS; (b) H. Ben-Daat, C. L. Rock, M. Flores, T. L. Groy, A. C. Bowman and R. J. Trovitch, Chem. Commun., 2017, 53, 7333–7336 RSC.
- T. T. Nguyen, J.-H. Kim, S. Kim, C. Oh, M. Flores, T. L. Groy, M.-H. Baik and R. J. Trovitch, Chem. Commun., 2020, 56, 3959–3962 RSC.
- P. Ma and H. Chen, ACS Catal., 2019, 9, 1962–1972 CrossRef CAS.
- (a) A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS; (b) A. Léval, A. Agapova, C. Steinlechner, E. Alberico, H. Junge and M. Beller, Green Chem., 2020, 22, 913–920 RSC; (c) S. Coufourier, Q. Gaignard Gaillard, J.-F. Lohier, A. Poater, S. Gaillard and J.-L. Renaud, ACS Catal., 2020, 10, 2108–2116 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and catalysis details; NMR spectra; X-ray data; details of computational chemistry and XYZ coordinates. CCDC 2060584–2060585. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04339d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |