
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular dearomative [4 + 2] cycloaddition of naphthalenes via visible-light energy-transfer-catalysis†
Pramod
Rai‡
,
Kakoli
Maji‡
,
Sayan K.
Jana‡
and
Biplab
Maji
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur 741246, WB, India. E-mail: bm@iiserkol.ac.in
First published on 5th October 2022
Abstract
The dearomative cycloaddition reaction serves as a blueprint for creating sp3-rich three-dimensional molecular topology from flat-aromatic compounds. However, severe reactivity and selectivity issues make this process arduous. Herein, we describe visible-light energy-transfer catalysis for the intermolecular dearomative [4 + 2] cycloaddition reaction of feedstock naphthalene molecules with vinyl benzenes. Tolerating a wide range of functional groups, structurally diverse 2-acyl naphthalenes and styrenes could easily be converted to a diverse range of bicyclo[2.2.2]octa-2,5-diene scaffolds in high yields and moderate endo-selectivities. The late-stage modification of the derivatives of pharmaceutical agents further demonstrated the broad potentiality of this methodology. The efficacy of the introduced methods was further highlighted by the post-synthetic diversification of the products. Furthermore, photoluminescence, electrochemical, kinetic, control experiments, and density-functional theory calculations support energy-transfer catalysis.
Introduction
Constructing sp3-rich three-dimensional (3D) scaffolds from two-dimensional (2D) molecules is highly challenging yet significantly impacts organic synthesis and drug discovery programs.1 The [4 + 2] cycloaddition reaction in which two new σ-bonds, and one π-bond are formed in a 3D six-membered ring topology from two simple unsaturated reaction components, diene and dienophile, has been recognized as a powerful tool for this purpose (Fig. 1a).2,3 In fact, this thermally allowed process has been a fundamental reaction type demonstrating its molecular complexity generating power for many years.4 In this context, polycyclic aromatic hydrocarbons such as naphthalenes also contain alternating double bonds. Besides, they are abundant and inexpensive feedstock chemicals.5 However, these 2D molecules displayed limited application in 3D complexity generating cycloaddition reactions due to severe challenges associated with breaking the increased stabilization conferred by aromaticity (resonance energy = 80.3 kcal mol−1) and selectivity (Fig. 1b and c).6 A typical thermal [4 + 2] cycloaddition with naphthalenes required harsh reaction conditions (high temperature up to 210 °C, pressure up to 103 atm),7 specially designed reaction conditions,8 or reactive dienophile9 to overcome the high kinetic barrier (Fig. 1c). However, since the free energy is often positive, the reverse reaction is preferred resulting in lower product yields (Fig. 1d, blue curve).7–9 Photochemistry provides alternative strategies for achieving challenging chemical processes in this context.10 However, since most organic molecules are incapable of absorbing visible light efficiently, direct high-energy ultraviolet (UV) light irradiation is required. Indeed, the UV-light mediated [4 + 2] cycloaddition of naphthalene is known.9b,11 However, their utility in organic synthesis is minimal due to the requirement of specific arenophiles, meager product yields, and unpredictable side reactions conferred by UV light (Fig. 1c). Eliminating UV irradiation should ideally broaden the synthetic applicability of this process with enriched structural diversity. The recent renaissance of visible-light photocatalysis provides a new space for dearomative [4 + 2] cycloaddition reaction via sensitization induced energy transfer (EnT) catalysis12 or direct visible-light excitation of the dienophile in some cases.13 Conceptually, a EnT process can selectively excite a ground state of a polycyclic hydrocarbon by using a photosensitizer to a higher triplet state (naphthalenes exhibit ETs of 54–60 kcal mol−1),12d lowering the kinetic barriers significantly compared to thermal processes (Fig. 1d, black curve). Furthermore, milder reaction conditions and substantially higher ET of the dearomatized product prevent the reverse reaction resulting in higher product yields.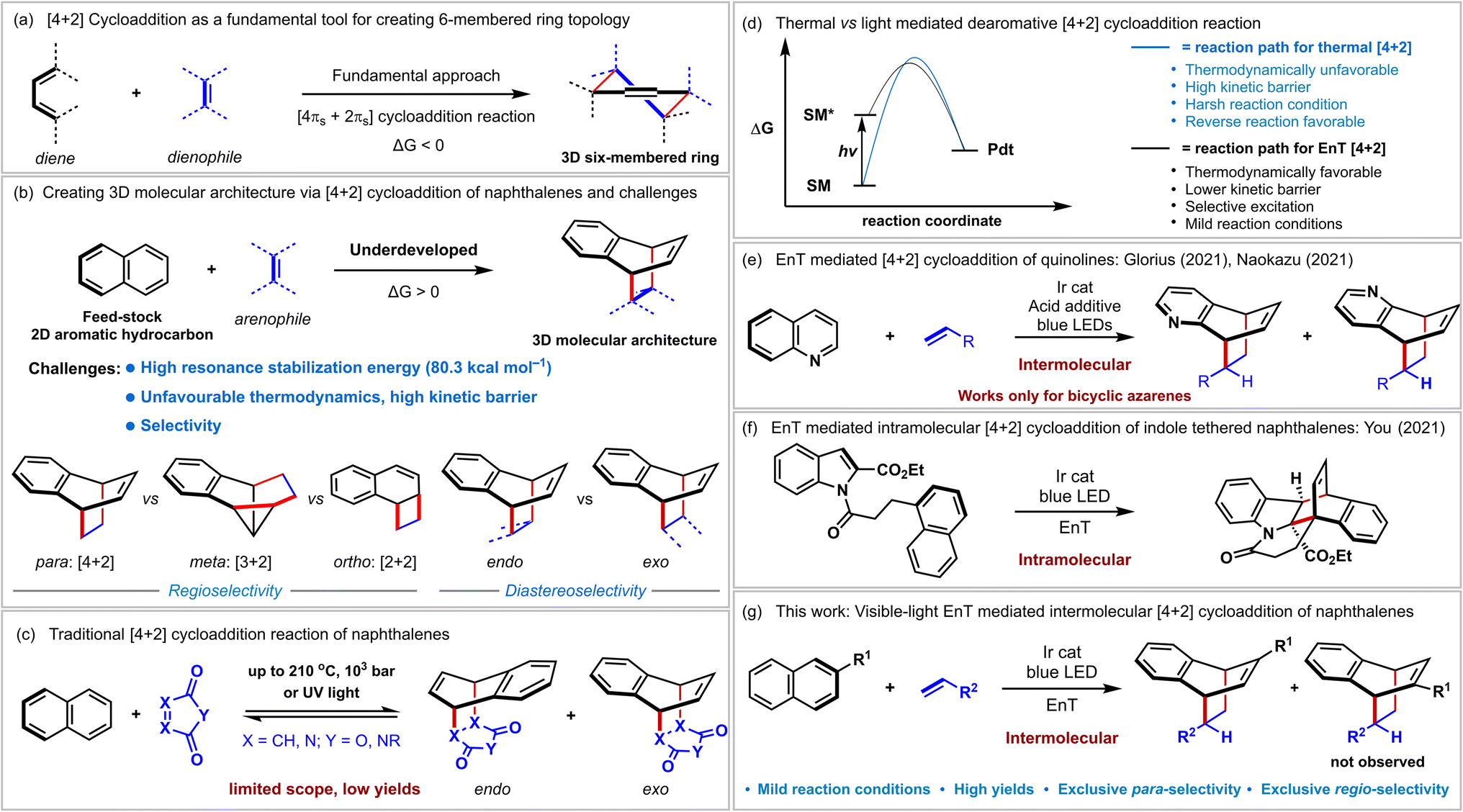 | ||
| Fig. 1 (a and b) Creating molecular complexity via [4 + 2] cycloaddition reaction and their challenges. (c) Traditional [4 + 2] cycloaddition reaction with naphthalenes. (d) Thermal vs. light-mediated cycloaddition reaction. (e and f) EnT Mediated [4 + 2] cycloaddition reaction of bicyclic azaarenes and indole tethered naphthalenes. (g) This work: visible-light EnT mediated intermolecular [4 + 2] cycloaddition reaction of naphthalenes. SM = starting material. Pdt = product. | ||
Recently, Glorius and coworkers disclosed the sole report on intermolecular dearomative [4 + 2] cycloaddition of bicyclic azaarenes (Fig. 1e) via visible-light EnT catalysis.12c A stoichiometric Brønsted acid additive was shown to play a vital role in increasing the reactivity of quinolines' triplet state toward olefins.12b You reported intramolecular cycloaddition of indole tethered naphthalenes (Fig. 1f).14 The intramolecular [2 + 2] cycloaddition of 1-naphthol derivatives and [4 + 2] cycloaddition of pyridines via visible-light EnT catalysis were recently developed by Glorius.12a,15 The intramolecularity prepaid the crucial entropic requirements for the last three reactions.
Besides, various groups demonstrated the application of the EnT process in diverse chemical transformations.16 However, to the best of our knowledge, visible-light-induced intermolecular dearomative [4 + 2] cycloaddition reactions of aromatic hydrocarbons with simple alkenes have not been documented yet. Herein, we report our initial results on EnT mediated dearomative [4 + 2] cycloaddition of naphthalenes with styrenes to access bicyclo[2.2.2]octa-2,5-diene scaffolds (Fig. 1g).
Results and discussion
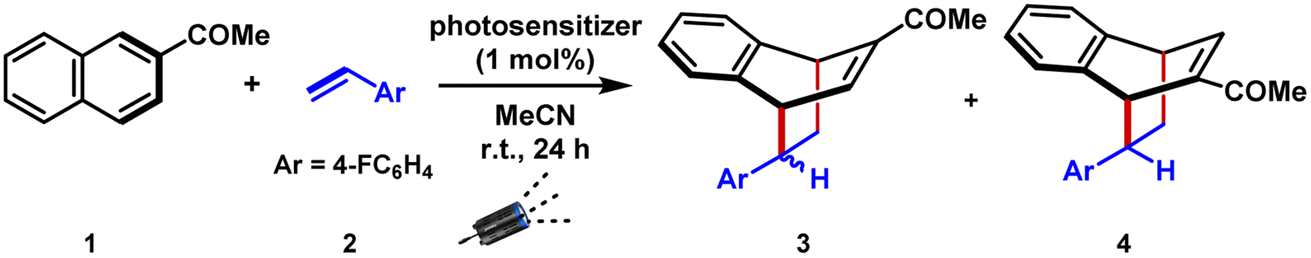
We commenced the visible-light mediated dearomative [4 + 2] cycloaddition reaction using 2-acetyl naphthalene 1 and 1-fluoro-4-vinyl benzene 2 as the model substrates (Table 1 and S1–S6†). Pleasingly, the irradiation of blue light-emitting diodes (λmax = 427 nm) in the presence of commercially available photosensitizer Ir[(dFCF3ppy)2dtbbpy]PF6 (PC1, 1 mol%) in acetonitrile solution of 1 and 2 (in stoichiometric ratio) at room temperature resulted in the formation of the desired [4 + 2] cycloadduct 3 in 98% yield in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, favoring endo (entry 1). Notably, the regioisomeric product 4 resulting from the syn attack of the alkene to the 6-membered aromatic ring was not observed by the 1H nuclear magnetic resonance analysis of the crude reaction mixture. The ortho and meta cycloaddition products (Fig. 1b) were also undetected in a measurable amount. We then followed the kinetics of the reaction via1H NMR spectroscopy (Fig. S18†).
:1 ratio, favoring endo (entry 1). Notably, the regioisomeric product 4 resulting from the syn attack of the alkene to the 6-membered aromatic ring was not observed by the 1H nuclear magnetic resonance analysis of the crude reaction mixture. The ortho and meta cycloaddition products (Fig. 1b) were also undetected in a measurable amount. We then followed the kinetics of the reaction via1H NMR spectroscopy (Fig. S18†).
|
|
||||
|---|---|---|---|---|
| Entry | Photosensitizer (PC) | E T (kcal mol−1), hνmax (LED) | Yield ofb 3 % |
endo:exob |
|
a Reaction condition: 1 (0.1 mmol), 2 (0.12 mmol), sensitizer (1 mol%), CH3CN (1 mL), blue LED irradiation under N2 at rt., 24 h.
b Yield and endo:exo ratio of 3 were determined by 1H NMR analysis by using trimethoxy benzene as an internal standard. Isolated yield in the parenthesis. Triplet energy ET from ref. 16d.
c In the dark.
d In the dark at 150 °C.
e 5 mol% sensitizer is used.
|
||||
| 1 | Ir[(dFCF3ppy)2dtbbpy]PF6 (PC1) | 61.8, 427 | 98 (90) | 2:1 |
| 2 | — | 61.8, 427 | <5 | — |
| 3c | PC1 | — | <5 | — |
| 4d | PC1 | — | <5 | — |
| 5e | Xanthone (PC2) | 74, 370 | 51 | 1.3:1 |
| 6 | Ir(ppy)3 (PC3) | 58.1, 427 | 25 | 2:1 |
| 7 | Ir[(ppy)2bpy]PF6 (PC4) | 53.1, 427 | 30 | 2:1 |
| 8e | 4CzIPN (PC5) | 53, 427 | 50 | 1.5:1 |
| 9 | Ir[(ppy)2dtbbpy]PF6 (PC6) | 49.2, 427 | 45 | 2:1 |
The rate of formation of 3 was found to be almost equal to the disappearance of 1 (Fig. S19†). This supports the direct formation of [4 + 2] cycloadduct 3 without the intermediacy of an [2 + 2] adduct, as previously observed for the intramolecular reaction.14
Product 3 was computed to be thermodynamically uphill (Fig. S24†). However, it can be purified by flash chromatography on silica gel and was found to be stable when heated at 60 °C for 24 h. The structure of endo-3 was confirmed by single-crystal X-ray crystallography.17 Control experiments confirm that the light source and photosensitizer were necessary for this [4 + 2] cycloaddition reaction, and their absence led to no product formation (entries 2–3). As anticipated, the reaction does not provide any product when heated at 150 °C for 24 h (entry 4).
We then stride forward to gain more insights into the EnT mediated [4 + 2] cycloaddition reaction (Fig. 2). The cyclic voltammetry analysis suggested that an electron transfer from the excited PC1 (E1/2Ir(III)*/Ir(IV) = −0.89 V vs. SCE) to either 1 or 2 (reduction potentials E1/2 = −1.81 V and −1.42 V vs. SCE, respectively) is not thermodynamically feasible (Fig. 2a). Similarly, an oxidative quenching of the excited PC1 (E1/2Ir(III)*/Ir(II) = 1.21 V vs. SCE) can also be ruled out. However, the Stern–Volmer analysis shows that 2-acetyl naphthalene 1 efficiently quenches the luminescence emission of the excited photosensitizer (Stern–Volmer quenching constant kq = 1.6 ×104 M−1 s−1) (Fig. 2b and c). Vinyl benzene 2 quenched the excited PC1 at a lower rate (Fig. 2c). We then computed the S0–T1 gap for 1 and 2 using B3LYP/6-311+G(2d,p) level of density functional theory (Fig. S20†). The calculated triplet energies ET = 55.1 kcal mol−1 for 1 and 57.9 kcal mol−1 for 2 are in good agreement with the previously reported values.16w Accordingly, the triplet energy transfer EnT from the photoexcited PC1 (ET = 61.8 kcal mol−1)18 to substrate 1 is exergonic and more likely.
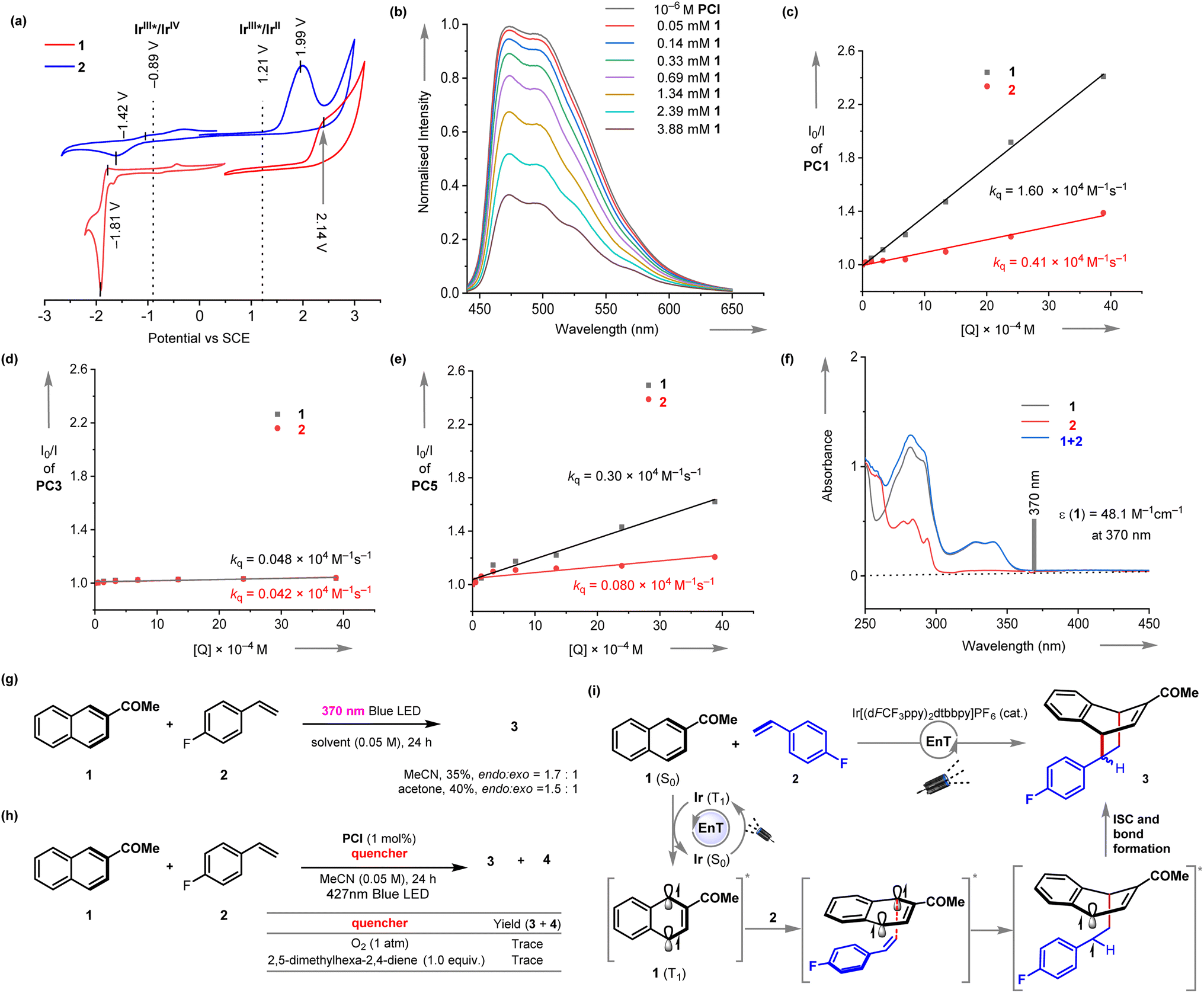 | ||
| Fig. 2 Mechanistic studies and proposed mechanism. (a) Cyclic voltammetry studies. (b) Decrease in luminescence intensity of PC1 upon addition of 1. The Stern–Volmer plot of the luminescence quenching for (c) PC1, (d) PC3, (e) PC5. (f) UV-vis studies. (g) Direct excitation of 1 in the absence of photosensitizer. (h) Reaction inhibition by external ET quencher. (i) Proposed mechanism. | ||
The reaction was also found to be amenable to some triplet-energy sensitizers, albeit at lesser efficiencies (Table 1, entries 5–9). However, a direct correlation between the ETs of the catalysts and product yields could not be drawn.16h,19 To gain more insight into the interaction of other photosensitizers with substrates 1 and 2 and their efficiencies in catalyzing the EnT mediated cycloaddition reaction, we have performed the luminescence quenching experiments with PC3 and PC5, which gave a moderate to low yield of 3. We have found both 1 and 2 do not interact with the excited PC3 at an appreciable rate (Fig. 2d). The luminescence intensity of excited PC5 is moderately quenched by increasing the concentration of 1 (kq = 0.30 × 104 M−1 s−1) (Fig. 2e). However, 2 does not communicate with the excited PC5. Further quenching studies are incorporated in ESI.† In addition, it was confirmed that an electron donor–acceptor complex between 1 and 2 does not occur, as evidenced by the lack of a change in the UV/vis spectrum (Fig. 2f). The absorption spectrum of 2 possesses small residual absorption at 370 nm (molar absorptivity ε = 48.1 M−1 cm−1 at 370 nm). However, direct irradiation using 370 nm Kessil LEDs (emission spectral window of ∼360 to 410 nm) only gave a 35% yield of 3 (endo:exo = 1.7:1) (Fig. 2g). A similar observation was made in the acetone solvent. These results additionally suggest that the photochemical dearomative cycloaddition reaction of 1 is more efficiently triggered upon energy transfer from the excited PC1. Additionally, complete inhibition of the dearomative [4 + 2] cycloaddition reaction under well-known ET quenchers, including oxygen and 2,5-dimethylhexa-2,4-diene, further supported the EnT process (Fig. 2h).
Based on the observations outlined above and the literature report, it is proposed that the photosensitizer, upon visible-light excitation produces its long-lived triplet excited state (Fig. 2i). The naphthalene 1 is then activated via EnT to afford triplet intermediate 1 (T1). The latter is then engaged in [4 + 2] cycloaddition with styrene 2 to yield the cycloadduct 3via radical capture, intersystem crossing (ISC), and bond formation.
The orbital coefficients of the relevant frontier molecular orbitals were examined to understand the origin of regioselectivity (see Page S74†). The preferential formation of 3 can approximately be conceptualized considering the maximum interaction between the largest orbital components in the triplet intermediate 1 (T1) and the lowest unoccupied molecular orbital of 2. However, a detailed computational elaboration is necessary.
Finally, we explored the generality and synthetic utility of the EnT mediated dearomative [4 + 2] cycloaddition reaction (Fig. 3). First, the scalability of our protocol was tested for a 1.0 mmol scale reaction between 1 and 2 that provided 0.25 g (84% yield) of 3 with the same endo selectivity. Then, the scope of the arenophile was investigated. Halogenated (F, Cl, Br) styrenes were excellent coupling partners, yielding bicyclo[2.2.2]octa-2,5-diene scaffolds 3–8 in high yields and selectivities. The electronic nature of the substituents does not perturb the reaction. Vinyl benzenes with both electron-withdrawing (CF3) and electron-donating substituents (OMe, OAc, Me, SMe) at different positions of the aryl ring participated in this reaction in equal efficiencies. The adducts 9–16 were obtained in 25–80% yields with a similar endo/exo ratio. Interestingly, vinyl naphthalene could also be utilized as an arenophile, and the adduct 17 was isolated in 39% yield. It indicated selective energy transfer to 1. Notably, vinyl biphenyls containing diverse electronic substituents, heteroarenes, and 4-vinyl pyridines were compatible with this reaction, and the dearomative cycloadducts 18–20 were obtained in moderate to good yields. However, α- and β-methyl styrene fails to deliver the product in synthetically useful yields under these conditions. Pleasingly, the reaction was compatible with styrene derivatives of complex molecules, including gemfibrozil, clofibric acid, fenbufen, ketoprofen, and oleic acid. The adducts 21–25 were isolated in 64–80% yields and moderate endo selectivities. It highlighted the functional group compatibility and synthetic utility of the process for late-stage diversification of these molecules.
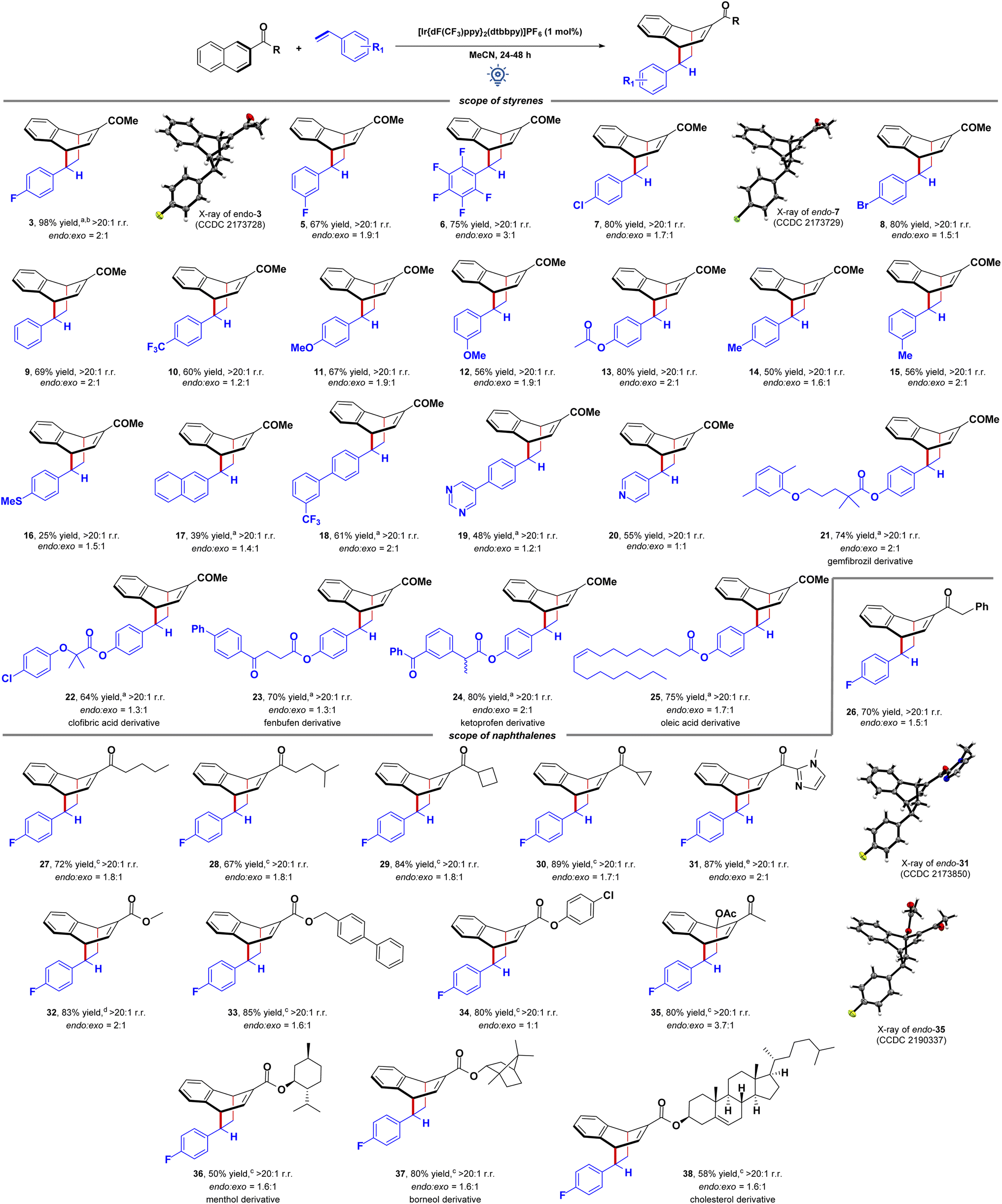 | ||
| Fig. 3 Synthetic utility of the EnT mediated [4 + 2] cycloaddition reaction. Reaction conditions: Table 1, entry 1. Reaction performed in 2 mL MeCN. Endo/exo ratio of products was determined by 1H NMR analysis using 1,3,5-trimethoxy benzene as an internal standard. Combined yields are given. Isolated yields of each isomer are provided in the ESI.† CCDC information ref. 17 [a] reaction performed in 1 mL MeCN. [b] 0.246 g, 84% yield, >20:1 rr., endo:exo = 1.6:1, when performed in 1 mmol scale. [c] Reaction carried out for 48 h. [d] Reaction carried out in MeOH solvent. rr. = regioisomeric ratio. | ||
The scope of naphthalene derivatives was then tested. Acyl naphthalenes with diverse alkyl chains participate in this EnT mediated [4 + 2] cycloaddition reaction, delivering the adducts 26–28 in high yields and selectivities. Naphthalenes containing cyclic alkyl groups, including cyclobutane 29 and cyclopropane 30, were also viable reaction partners. A CC-bond cleavage in a strained cyclopropane ring was not observed. In comparison, Brown recently discovered EnT mediated CC-bond cleavage in bicyclo[1.1.0]butyl naphthyl ketone for [2π + 2σ] cycloaddition reactions with styrenes.19a Severe ring strain in the caged molecule can be reasoned responsible. N-Methyl imidazole bearing keto naphthalene performed well in this reaction 31. The structures of both the endo- and exo-diastereomers were assigned by single-crystal X-ray crystallography.17 Interestingly, 2-carboxyalkyl naphthalenes also smoothly undergo visible-light mediated dearomative cycloaddition reaction providing high 80–85% yields of the cycloadducts 32–34, albeit in a moderate endo/exo ratio. Interestingly, 1,2-disubstituted naphthalene derivative also undergoes the dearomative cycloaddition providing the desired product 35 in high yield, and the structure of the endo-diastereomer was assigned by single-crystal X-ray crystallography.17 The reaction was also compatible with the naphthalene derivatives of (−)-menthol 36, (−)-borneol 37, and cholesterol 38. It again highlights the utility of the visible-light-driven dearomatization reaction.
To further demonstrate the synthetic application of the [4 + 2] dearomative cycloaddition reaction, we have performed the derivatization of the cycloadduct endo-3 to obtain complex 3D molecular architecture (Fig. 4). The palladium-catalyzed reduction of the α,β-unsaturated double bond of endo-3 in the presence of diphenyl silane gave the saturated product 39 in 52% yield in >20:1 dr. NOE spectroscopy confirmed the stereochemical assignment. Furthermore, a Michael addition of N-Boc-L-cysteine methyl ester delivered the adduct 40 in 56% yield and 2:1 dr. Notably, the Corey–Chaykovsky cyclopropanation of endo-3 offered the cyclopropane 41 in 63% yield and >99:1 dr. Similarly, the epoxidation of endo-3 in the presence of H2O2/NaOH delivered the epoxide 42 in 70% isolated yield as a single diastereomer.
 | ||
| Fig. 4 Post-synthetic application of [4 + 2] cycloaddition products, dr. = diastereoselective ratio. | ||
Conclusions
In conclusion, a new strategy for intermolecular dearomative [4 + 2] cycloaddition reaction of abundant aromatic hydrocarbon naphthalene was developed. The reaction operates via visible-light energy transfer utilizing a commercially available sensitizer. Diverse bicyclo[2.2.2]octa-2,5-diene scaffolds were synthesized in high yields. The reaction can be scaled-up and is amicable for late-stage modification of various complex molecules. The ease of post-synthetic diversifications adds to the utility of the introduced method. Photoluminescence, electrochemical, kinetic, and control experiments helped understand the features of this visible-energy transfer catalysis. We anticipate that the protocol will further illuminate the EnT catalysis for designing complex molecular scaffolds. Further research exploring the reaction scope and mechanisms is currently underway in this laboratory.Data availability
The ESI† includes all experimental details, including optimization of the synthetic method, synthesis and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products, crystallography, and computation details are included as well.Author contributions
P. R., K. M., S. K. J. and B. M. conceived and designed the project. P. R., K. M., S. K. J. performed the experiment and contributed equally to this work. P. R., K. M., S. K. J. and B. M. wrote the manuscript. B. M. acquired the funding and directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. R., K. M., and S. K. J. acknowledge DST-INSPIRE, IISER Kolkata, and PMRF for PhD fellowships, respectively. BM thanks SERB (Grant No. CRG/2019/001232) for financial support. The authors thank Prof. A. K. Roy (IISER K) for the helpful discussions. Dedicated to Prof. Herbert Mayr on the occasion of his 75th birthday.References
- (a) R. S. Vardanyan and V. J. Hruby, in Synthesis of Essential Drugs, Elsevier, 2006 Search PubMed; (b) L. F. Raveglia and G. A. Giardina, Future Med. Chem., 2009, 1, 1019–1023 CrossRef CAS PubMed; (c) K. C. Nicolaou, D. J. Edmonds, A. Li and G. S. Tria, Angew. Chem., Int. Ed., 2007, 46, 3942–3945 CrossRef CAS PubMed; (d) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- (a) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Elsevier Science, 2013 Search PubMed; (b) Y. Hayashi, in Cycloaddition Reactions in Organic Synthesis, ed. S. Kobayashi and K. A. Jørgensen, Wiley-VCH, 2001 Search PubMed; (c) L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418–14420 CrossRef CAS PubMed; (d) S. N. Semenov, L. Belding, B. J. Cafferty, M. P. S. Mousavi, A. M. Finogenova, R. S. Cruz, E. V. Skorb and G. M. Whitesides, J. Am. Chem. Soc., 2018, 140, 10221–10232 CrossRef CAS PubMed; (e) C. Zheng and S.-L. You, Chem, 2016, 1, 830–857 CrossRef CAS.
- (a) N. Chopin, H. Gérard, I. Chataigner and S. R. Piettre, J. Org. Chem., 2009, 74, 1237–1246 CrossRef CAS PubMed; (b) M. Essers, C. Mück-Lichtenfeld and G. Haufe, J. Org. Chem., 2002, 67, 4715–4721 CrossRef CAS PubMed.
- (a) W. Xiao, Q.-Q. Yang, Z. Chen, Q. Ouyang, W. Du and Y.-C. Chen, Org. Lett., 2018, 20, 236–239 CrossRef CAS PubMed; (b) S. G. Bartko, P. J. Hamzik, L. Espindola, C. Gomez and R. L. Danheiser, J. Org. Chem., 2020, 85, 548–563 CrossRef CAS PubMed; (c) Q. Lu, X. Huang, G. Song, C.-M. Sun, J. P. Jasinski, A. C. Keeley and W. Zhang, ACS Comb. Sci., 2013, 15, 350–355 CrossRef CAS PubMed.
- G. Collin, H. Höke and H. Greim, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2003 Search PubMed.
- (a) C. W. Bird, Tetrahedron, 1996, 52, 9945–9952 CrossRef CAS; (b) R. Remy and C. G. Bochet, Chem. Rev., 2016, 116, 9816–9849 CrossRef CAS PubMed.
- W. H. Jones, D. Mangold and H. Plieninger, Tetrahedron, 1962, 18, 267–272 CrossRef CAS.
- (a) K. i. Takeda, S. Hagishita, M. Sugiura, K. Kitahonoki, I. Ban, S. Miyazaki and K. Kuriyama, Tetrahedron, 1970, 26, 1435–1451 CrossRef CAS; (b) Y. Fang, T. Murase and M. Fujita, Chem. Lett., 2015, 44, 1095–1097 CrossRef CAS; (c) T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed.
- (a) V. D. Kiselev, E. A. Kashaeva, L. N. Potapova and G. G. Iskhakova, Russ. Chem. Bull., 2004, 53, 51–54 CrossRef CAS; (b) G. W. Breton and K. A. Newton, J. Org. Chem., 2000, 65, 2863–2869 CrossRef CAS PubMed; (c) K. Hagiwara, M. Iwatsu, D. Urabe and M. Inoue, Heterocycles, 2015, 90, 659–672 CrossRef CAS; (d) M. C. Kloetzel and H. L. Herzog, J. Am. Chem. Soc., 1950, 72, 1991–1995 CrossRef CAS.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- (a) D. Döpp and H. R. Memarian, Chem. Ber., 1990, 123, 315–319 CrossRef; (b) K. Kishikawa, S. Akimoto, S. Kohmoto, M. Yamamoto and K. Yamada, J. Chem. Soc., Perkin Trans. 1, 1997, 77–84, 10.1039/A601710J.
- (a) J. Ma, F. Strieth-Kalthoff, T. Dalton, M. Freitag, J. L. Schwarz, K. Bergander, C. Daniliuc and F. Glorius, Chem, 2019, 5, 2854–2864 CrossRef CAS; (b) T. Morofuji, S. Nagai, Y. Chitose, M. Abe and N. Kano, Org. Lett., 2021, 23, 6257–6261 CrossRef CAS PubMed; (c) J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 CrossRef CAS PubMed; (d) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- (a) E. H. Southgate, J. Pospech, J. Fu, D. R. Holycross and D. Sarlah, Nat. Chem., 2016, 8, 922–928 CrossRef CAS PubMed; (b) L. W. Hernandez, J. Pospech, U. Klöckner, T. W. Bingham and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 15656–15659 CrossRef CAS PubMed.
- M. Zhu, H. Xu, X. Zhang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2021, 60, 7036–7040 CrossRef CAS PubMed.
- M. J. James, J. L. Schwarz, F. Strieth-Kalthoff, B. Wibbeling and F. Glorius, J. Am. Chem. Soc., 2018, 140, 8624–8628 CrossRef CAS PubMed.
- (a) A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed; (b) K. Singh, S. J. Staig and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef CAS PubMed; (c) Q. Cheng, J. Chen, S. Lin and T. Ritter, J. Am. Chem. Soc., 2020, 142, 17287–17293 CrossRef CAS PubMed; (d) X. Zhang and T. Rovis, J. Am. Chem. Soc., 2021, 143, 21211–21217 CrossRef CAS PubMed; (e) A. Tröster, A. Bauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 3538–3541 CrossRef PubMed; (f) K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186–17192 CrossRef CAS PubMed; (g) Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (h) A. Singh, C. J. Fennell and J. D. Weaver, Chem. Sci., 2016, 7, 6796–6802 RSC; (i) T. Patra, M. Das, C. G. Daniliuc and F. Glorius, Nat. Catal., 2021, 4, 54–61 CrossRef CAS; (j) T. Patra, P. Bellotti, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 3172–3177 CrossRef CAS PubMed; (k) R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 477–482, DOI:10.1038/s41586-022-04636-x; (l) M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed; (m) J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625–13634 CrossRef CAS PubMed; (n) Z. D. Miller, B. J. Lee and T. P. Yoon, Angew. Chem., Int. Ed., 2017, 56, 11891–11895 CrossRef CAS; (o) M. Plaza, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 12785–12788 CrossRef CAS PubMed; (p) J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef; (q) J. J. Molloy, M. Schäfer, M. Wienhold, T. Morack, C. G. Daniliuc and R. Gilmour, Science, 2020, 369, 302–306 CrossRef CAS PubMed; (r) C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678–687 CrossRef CAS; (s) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed; (t) X. Jiang, E. Li, J. Chen and Y. Huang, Chem. Commun., 2021, 57, 729–732 RSC; (u) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (v) L. Wu, C. Huang, B. P. Emery, A. C. Sedgwick, S. D. Bull, X.-P. He, H. Tian, J. Yoon, J. L. Sessler and T. D. James, Chem. Soc. Rev., 2020, 49, 5110–5139 RSC; (w) T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed; (x) M. Huang, L. Zhang, T. Pan and S. Luo, Science, 2022, 375, 869–874 CrossRef CAS PubMed; (y) F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CrossRef CAS; (z) E. R. Wearing, D. E. Blackmun, M. R. Becker and C. S. Schindler, J. Am. Chem. Soc., 2021, 143, 16235–16242 CrossRef CAS; (a a) M. R. Becker, E. R. Wearing and C. S. Schindler, Nat. Chem., 2020, 12, 898–905 CrossRef CAS PubMed; (a b) Z. Zhang, D. Yi, M. Zhang, J. Wei, J. Lu, L. Yang, J. Wang, N. Hao, X. Pan, S. Zhang, S. Wei and Q. Fu, ACS Catal., 2020, 10, 10149–10156 CrossRef CAS.
- CCDC-2173728 (endo-3), CCDC-2173729 (endo-7), CCDC-2173850 (endo-31), CCDC-2173723 (exo-31), CCDC-2190337 (endo-35) contain the supplementary crystallographic data.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- (a) R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed; (b) F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jakob and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 827–831 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, crystallography, computation and characterization details (PDF). CCDC 2173728, 2173729, 2173850, 2173723 and 2190337. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04005k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |