
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium–peptide oxidative addition complexes for bioconjugation†
Anthony J.
Rojas‡
 ab,
Justin M.
Wolfe‡
a,
Heemal H.
Dhanjee
a,
Ivan
Buslov
a,
Nicholas L.
Truex
a,
Richard Y.
Liu
c,
Walter
Massefski
a,
Bradley L.
Pentelute
*adef and
Stephen L.
Buchwald
*a
ab,
Justin M.
Wolfe‡
a,
Heemal H.
Dhanjee
a,
Ivan
Buslov
a,
Nicholas L.
Truex
a,
Richard Y.
Liu
c,
Walter
Massefski
a,
Bradley L.
Pentelute
*adef and
Stephen L.
Buchwald
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: blp@mit.edu; sbuchwal@mit.edu
bDepartment of Chemistry and Biochemistry, Kennesaw State University, 1000 Chastain Road NW, Kennesaw, GA 30144, USA
cDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
dThe Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, 500 Main Street, Cambridge, MA 02142, USA
eCenter for Environmental Health Sciences, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA
fBroad Institute of MIT and Harvard, 415 Main Street, Cambridge, MA 02142, USA
First published on 19th September 2022
Abstract
The synthesis of palladium oxidative addition complexes derived from unprotected peptides is described. Incorporation of 4-halophenylalanine into a peptide during solid phase peptide synthesis allows for subsequent oxidative addition at this position upon treatment with a palladium precursor and suitable ligand. The resulting palladium–peptide complexes are solid, storable, water-soluble, and easily purified via high-performance liquid chromatography. These complexes react with thiols in aqueous buffer, offering an efficient method for bioconjugation. Using this strategy, peptides can be functionalized with small molecules to prepare modified aryl thioether side-chains at low micromolar concentrations. Additionally, peptide–peptide and peptide–protein ligations are demonstrated under dilute aqueous conditions.
Introduction
Reactions that modify complex biomolecules enable researchers to synthesize molecularly defined conjugates, probe structure–function relationships, and develop new therapeutics.1,2 In particular, chemoselective reactions are desirable for bioconjugation because their ability to target specific functional groups in the presence of many others obviates the need for protecting groups or extraneous synthetic manipulations during the synthesis of large biomolecules.3 Bioconjugation reactions must also satisfy a number of strict criteria to be considered practical: the reaction should be effective at low concentrations,4 proceed in aqueous solutions, generate kinetically stable linkages, and produce, at most, minor side products.5,6 Additionally, bioconjugation reactions are typically conducted at low micromolar concentrations and therefore should have kinetic rate constants large enough to achieve near-quantitative conversions on experimental timescales.7Many of the widely used bioconjugation reactions could be improved in one or more of these regards.1,9 For instance, thiol–maleimide conjugation reactions, while effective, are known to generate chemically unstable linkages.10 Moreover, even relatively fast reactions such as Staudinger reactions11 and oxime ligations12 generally lose effectiveness at low micromolar concentrations and copper-catalysed azide–alkyne cycloadditions13 can lead to undesired disulfide bond formation in the presence of free thiols.7 One approach that achieves high reaction rates is the pericyclic reaction between tetrazines and strained alkenes (e.g., trans-cyclooctenes) but the performance of the reaction depends on the nature of the tetrazine component.14
Recently, transition metal-mediated transformations have emerged as effective tools in bioconjugation.15–21 Transition metals are widely used in organic synthesis for highly selective and efficient transformations. Drawing from extensive research in this context, we have been able to develop bioconjugation reactions that occur rapidly, exhibit excellent chemoselectivity and proceed under the mild conditions necessary for use on biomolecules.22 Recently, our laboratories reported palladium oxidative addition complexes that rapidly and chemoselectively modify cysteine,8 a naturally occurring amino acid (Fig. 1A). With appropriate ligand modifications, this reaction could take place under fully aqueous conditions while maintaining exclusive selectivity for reaction with cysteine over other nucleophilic amino acids such as lysine.23 Other groups have also utilized transition metals for the modification of cysteine, with great success.24,25
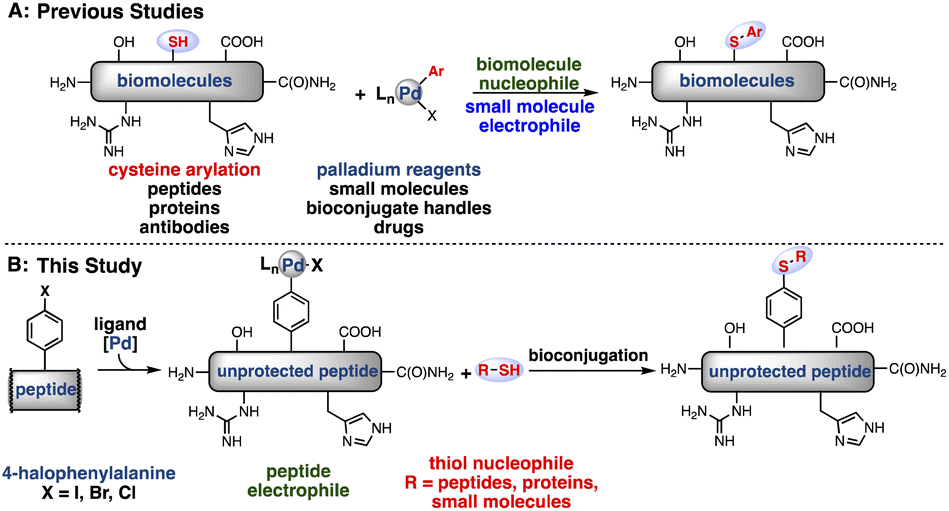 | ||
| Fig. 1 Strategies for bioconjugation mediated by palladium oxidative addition complexes. (A) Previous reports of cysteine S-arylation with palladium-small molecule oxidative addition complexes.7 (B) This report of peptide bioconjugation achieved through the generation of palladium–peptide oxidative addition complexes. | ||
As an alternative to small molecule-bearing palladium reagents that functionalize nucleophilic peptides, a complementary approach was envisioned wherein a peptide-bound palladium reagent could engage a diverse array of external nucleophiles (Fig. 1B). We have thus far observed inefficient reactivity when attempting to carry out an oxidative addition step directly on biomolecules under aqueous, buffered conditions. We reasoned that direct oxidative addition on the aryl-halide containing peptides in organic solvent would permit formation of a palladium–peptide oxidative addition complex (OAC). Isolation of this Pd-peptide-OAC as a lyophilized solid would then allow conjugation to a wide array of partners in aqueous conditions, including small molecules, peptides, or proteins, via the synthesis of a single palladium–peptide complex. Thus, by separating the oxidative addition and conjugation steps by first isolating the Pd-peptide-OAC, we would enable the formation and use of Pd-peptide-OACs for bioconjugation.
Results and discussion
To synthesize a palladium–peptide reagent, we first incorporated 4-bromophenylalanine into the primary sequence of a peptide during solid-phase peptide synthesis. Next, the purified unprotected peptide was treated with a water-soluble ligand (sSPhos) and (1,5-COD)Pd(CH2TMS)2 in dimethylsulfoxide (DMSO) (Fig. 2A). Surprisingly, our first attempt afforded the corresponding palladium–peptide oxidative addition complex in sufficient yield. Moreover, the synthesis did not require specialized air-free Schlenk techniques or the use of an inert-atmosphere glovebox. We note that the reagents for Pd-peptide-OAC formation are all commercially available and easily handled on the bench. Additional experiments revealed that the reaction could proceed using peptides containing 4-iodophenylalanine and 4-chlorophenylalanine.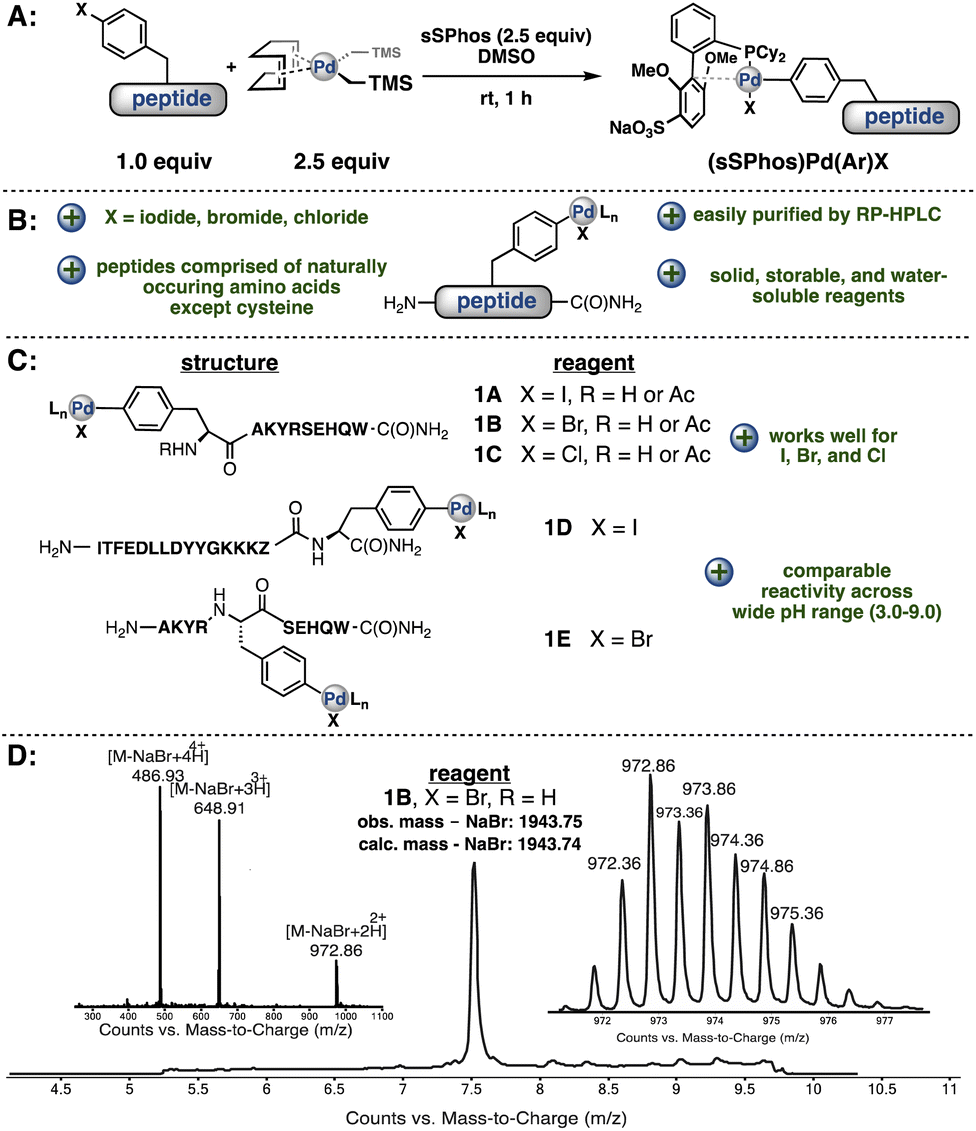 | ||
| Fig. 2 Synthesis and characterization of palladium–peptide oxidative addition complexes is facile and robust. (A) General procedure for the generation of palladium–peptide complexes. (B) Palladium–peptide complex formation is efficient for a variety of peptides containing 4-halophenylalanines. (C) Palladium–peptide oxidative addition complexes are successfully synthesized regardless of the position of the 4-halophenylalanine in the sequence. In sequence 1D, 6-aminohexanoic acid is denoted as Z. (D) LC-MS analysis of palladium–peptide oxidative addition complex 1B. | ||
The resulting palladium oxidative addition complexes are stable as lyophilized solids stored on the bench and amenable to chromatography. When left under ambient conditions, these lyophilized solids showed no observable decomposition after 12 months (Fig. 2D, ESI†). The complexes can be purified via reverse-phase high-performance liquid chromatography (RP-HPLC) using water/acetonitrile mobile phases containing 0.1% (v/v) trifluoroacetic acid. para-Halophenylalanine was incorporated in the middle of our model peptides or at the N- or C-terminus and, in each case, successful Pd-peptide-OAC formation occurred to give sufficient quantities of material after purification for further evaluation in cross-coupling experiments (Fig. 2C). More importantly, the presence of other nucleophilic or basic residues (except cysteine) were tolerated in the oxidative addition process.
Based on well-characterized Pro-Pro-Xaa-type peptides from the Wennemers group,36 we investigated the solution-state conformation of a tripeptide containing C-terminal 4-bromophenylalanine (H-Pro-Pro-Phe(4-Br)-NH2, 2A, Fig. 3) and the corresponding Pd complex supported by the SPhos ligand (H-Pro-Pro-Phe(4-Br)–NH2–OAC, 2B). A combination of COSY, TOCSY, HSQC, and HMBC allowed for the assignment of proton and carbon resonances of peptide 2A. Quantitative analysis of nuclear Overhauser effects (NOEs) obtained from ROESY spectra of 2A and 2B provided NOE-derived H–H distances. We then performed computational studies and compared the resulting conformers with the NMR spectroscopic data. The conformations that we inferred from ROESY data were comparable with the energy-minimized conformations obtained from DFT calculations (ESI†). We sought to gain insight into the potential change of ground-state conformation of tripeptide 2A upon insertion of SPhos-Pd into the C(aryl)–Br bond. The conformations of 2A and 2B are each consistent with their respective NMR data, and indicate that the conformation of the OAC does not perturb the conformation of the parent peptide.
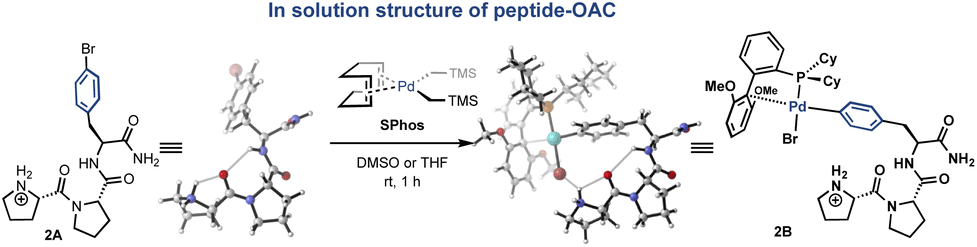 | ||
| Fig. 3 Conformational analysis using both 2D-NMR and DFT calculations suggests that the solution-state structure of the peptidic backbone is largely unperturbed by OAC formation. A representative optimized structure is shown for the Pd-peptide-OAC (right) derived from the p-bromophenylalanine containing peptide (left). | ||
We then sought to apply these complexes toward late-stage side-chain functionalization, a potentially powerful strategy to rapidly diversify unprotected peptides. To evaluate the feasibility of this method, the oxidative addition complex, 1B, was mixed with 5.0 equivalents of a small-molecule thiol in low micromolar concentration in aqueous tris(hydroxymethyl)aminomethane (Tris, pH 7.5) buffer at 37 °C. After 30 min, we observed the expected site-selective modification of the aryl side-chain in excellent conversions using eight distinct thiols (Scheme 1). Moreover, the reactivity was maintained across a wide range of aqueous buffered conditions (pH 3.0–9.0, see ESI† for details).
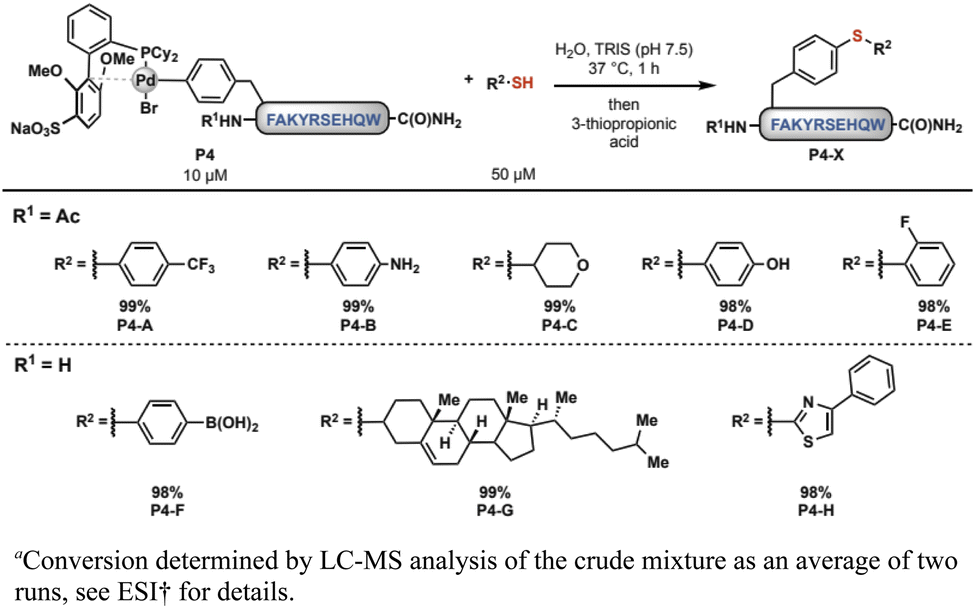 | ||
| Scheme 1 Palladium–peptide reagents react efficiently with small molecule thiols.a | ||
In addition to ligation with small molecules, we envisioned that these palladium–peptide complexes might be useful in ligation with other peptides. Peptide–peptide conjugates are important both for studying the function of biomolecules and developing new biotherapeutics for disease treatment.26 Among others, leading chemical methods for peptide ligation include enzyme-mediated ligation27 and protein-templated ligation.27,28 However, many ligation reactions are limited by producing relatively labile bonds and development of chemical ligation strategies that produce stable linkages has been a longstanding ambition.29 We reasoned that our palladium–peptide reagents might allow for rapid, irreversible ligation with a variety of cysteine-containing peptides. While the resulting linkages will not be native to proteins that occur in nature, proteins have been shown to retain their function even with the introduction of non-native backbones.30
Using automated fast-flow peptide synthesis,31 three peptides (P1, P2, and P3) were prepared containing a single cysteine residue at different locations along the primary amino acid sequence. The peptides were dissolved independently in aqueous Tris (pH 7.5) buffer containing oxidative addition complex 1D. After 6 h at 37 °C, 3-thiopropionic acid was added to the mixture to quench the reaction. Analysis of the reaction mixtures by LC-MS indicated that all the peptide ligations proceeded with both high conversion and selectivity under these conditions (Scheme 2). Scaling up this method, we were able to isolate purified ligated peptides following RP-HPLC in good yield (76% isolated yield, see ESI†).
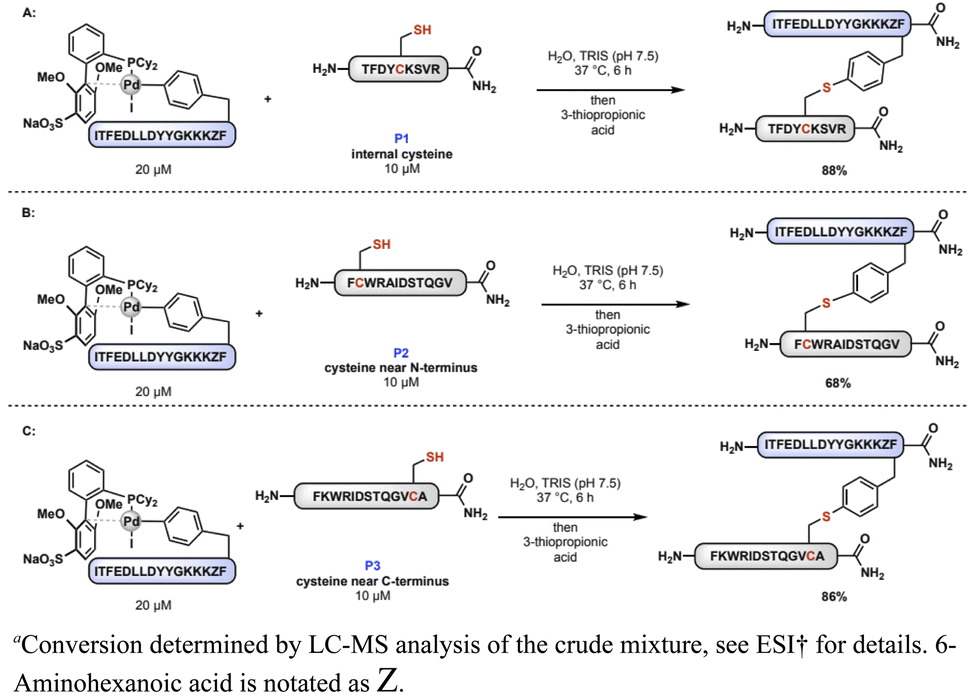 | ||
| Scheme 2 Palladium–peptide reagent enables peptide–peptide ligation. (A) Peptide ligation with internal cysteine, (B) peptide ligation with cysteine near N-terminus, and (C) peptide ligation with cysteine near C-terminus.a | ||
Given these results, we envisioned that another class of suitable nucleophiles would be cysteine-containing proteins. This kind of peptide–protein conjugation could potentially allow site-specific attachment of therapeutic peptides to antibodies or other large proteins containing cysteine, a strategy of significant interest for the development of targeted therapeutics or vaccines.32,33 Furthermore, the ability to conjugate a peptide to the side-chains of proteins under physiologically relevant conditions in dilute aqueous solutions would be an advancement for transition metal-based reagents. An additional advantage of this method is that the aryl thioether bond is known to exhibit high chemical stability.8
As a model substrate, the protein protective antigen (PA)34,35 from Bacillus anthracis was modified to contain a single internal cysteine (ESI† for details). In aqueous buffer containing His (pH 7.0), PA (2 μM) was incubated with 10 equivalents of palladium–peptide complex 1B, itself prepared as a solution in DMSO. After 18 hours at 37 °C, conjugation of the peptide to the protein was observed to occur with 90% conversion (Scheme 3, see ESI† for details). Subjecting wild type PA (lacking cysteine) to conjugation conditions led to no protein modification (see ESI† for details), confirming that the reaction is specific to cysteine residues.
 | ||
| Scheme 3 Ligation of a peptide to anthrax toxin protective antigen proceeds in one hour at low micromolar concentrations.a | ||
Conclusions
In conclusion, palladium–peptide oxidative addition complexes were synthesized through the introduction of 4-halophenylalanine into peptides during SPPS. These complexes react efficiently with small molecule thiols, as well as cysteine-containing peptides and proteins. The ligation reactions proceed at low concentrations under physiologically relevant conditions. We have yet to investigate reactions at higher concentrations (in the millimolar range) and this is a topic of future research. These transformations represent a bioconjugation approach complementary to current methods. We anticipate applications in the development of targeted therapeutics, the investigation of structure–function relationships, and the design of unconventional, branched polypeptide structures.Data availability
All data generated during this study are available either in the main text or ESI.†Author contributions
A. J. R., J. M. W., H. H. D., B. L. P. and S. L. B. designed the research; A. J. R., J. M. W., H. H. D., I. B., N. L. T., R. Y. L. and W. M. performed the experiments; A. J. R., J. M. W., H. H. D., I. B., R. Y. L., W. M., B. L. P. and S. L. B. analyzed the data; A. J. R., J. M. W. and H. H. D. generated the figures; A. J. R., J. M. W. and H. H. D. wrote the manuscript with input from all of the authors.Conflicts of interest
The authors declare the following competing financial interest(s): B. L. P. is a co-founder and/or member of the scientific advisory board of several companies focusing on the development of protein and peptide therapeutics. MIT has obtained or has filed patents on some of the ligands that are described in the paper from which S. L. B. and former co-workers receive royalty payments.Acknowledgements
Financial support for this work was provided by the National Institutes of Health (grants R01 GM046059 to S. L. B., R35 GM122483 to S. L. B., and R01 GM110535 to B. L. P.) and the Sontag Foundation Distinguished Scientist Award (to B. L. P.). A. J. R. and J. M. W. gratefully acknowledge support from the National Science Foundation Graduate Research Fellowship under grant no. 1122374. H. H. D. acknowledges support from an N. I. H. postdoctoral fellowship (grant F32 GM131592). I. B. is grateful to the Swiss National Science Foundation for generous financial support through an Early Postdoctoral Mobility Fellowship. The Varian 300 spectrometer used for portions of this work was purchased with funds from the NSF (grant CHE-9808061). We thank Sigma Aldrich for the generous donation of sSPhos. We thank Alexander Loftis for providing the protective antigen protein used in these studies. We thank Dr Andy Thomas for assistance in the preparation of this manuscript.Notes and references
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, CA, 3rd edn, 2013 Search PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 1–14 CrossRef PubMed.
- I. S. Carrico, Chem. Soc. Rev., 2008, 37, 1423–1431 RSC.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- C. S. Mckay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- F. Saito, H. Noda and J. W. Bode, ACS Chem. Biol., 2015, 10, 1026–1033 CrossRef CAS.
- E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute and S. L. Buchwald, Nature, 2015, 526, 687–691 CrossRef CAS PubMed.
- N. Justine, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef PubMed.
- S. D. Fontaine, R. Reid, L. Robinson, G. W. Ashley and D. V. Santi, Bioconjugate Chem., 2015, 26, 145–152 CrossRef CAS PubMed.
- E. Saxon, J. I. Armstrong and C. R. Bertozzi, Org. Lett., 2000, 2, 2141–2143 CrossRef CAS PubMed.
- S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem. - Eur. J., 2014, 20, 34–41 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- W. Zhao, H. G. Lee, S. L. Buchwald and J. M. Hooker, J. Am. Chem. Soc., 2017, 139, 7152–7155 CrossRef CAS PubMed.
- S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T. Iavarone, J. A. Wells, F. D. Toste and C. J. Chang, Science, 2017, 602, 597–602 CrossRef.
- A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576–1580 CrossRef CAS PubMed.
- S. D. Tilley and M. B. Francis, J. Am. Chem. Soc., 2006, 128, 1080–1081 CrossRef CAS.
- J. Ohata, S. C. Martin and Z. T. Ball, Angew. Chem., 2019, 131, 6238–6264 CrossRef.
- P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed.
- J. Rodríguez and M. Martínez-Calvo, Chem. – Eur. J., 2020, 26, 9792–9813 CrossRef PubMed.
- J. M. Chalker, in Chemoselective and Bioorthogonal Ligation Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 231–270 Search PubMed.
- A. J. Rojas, B. L. Pentelute and S. L. Buchwald, Org. Lett., 2017, 19, 4263–4266 CrossRef CAS PubMed.
- M. A. Waddington, X. Zheng, J. M. Stauber, E. Hakim Moully, H. R. Montgomery, L. M. A. Saleh, P. Král and A. M. Spokoyny, J. Am. Chem. Soc., 2021, 143, 8661–8668 CrossRef CAS PubMed.
- T. Schlatzer, J. Kriegesmann, H. Schröder, M. Trobe, C. Lembacher-Fadum, S. Santner, A. V. Kravchuk, C. F. W. Becker and R. Breinbauer, J. Am. Chem. Soc., 2019, 141, 14931–14937 CrossRef CAS.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, J. Am. Chem. Soc., 2017, 139, 7416–7427 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS.
- N. Brauckhoff, G. Hahne, J. T. Yeh and T. N. Grossmann, Angew. Chem., Int. Ed., 2014, 53, 4337–4340 CrossRef CAS.
- L. P. Miranda and P. F. Alewood, Biopolymers, 2000, 55, 217–226 CrossRef CAS.
- W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed.
- A. J. Mijalis, D. A. Thomas III, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, Nat. Chem. Biol., 2017, 13, 464–466 CrossRef CAS.
- K. Kafi, D. J. Betting, R. E. Yamada, M. Bacica, K. K. Steward and J. M. Timmerman, Mol. Immunol., 2009, 46, 448–456 CrossRef CAS PubMed.
- K. Temming, D. L. Meyer, R. Zabinski, P. D. Senter, K. Poelstra, G. Molema and R. J. Kok, Mol. Pharm., 2007, 4, 686–694 CrossRef CAS.
- S. F. Little, S. H. Leppla and E. Corat, Infect. Immun., 1988, 56, 1807–1813 CrossRef CAS.
- J. A. T. Young and R. J. Collier, Annu. Rev. Biochem., 2007, 76, 243–265 CrossRef CAS PubMed.
- C. Rigling, J. K. Kisunzu, J. Duschmalé, D. Häussinger, M. Wiesner, M.-O. Ebert and H. Wennemers, Conformational properties of a peptidic catalyst: insights from NMR spectroscopic studies, J. Am. Chem. Soc., 2018, 140, 10829–10838 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04074c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |