
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChain transfer agents for the catalytic ring opening metathesis polymerization of norbornenes†‡
Indradip
Mandal
 ,
Ankita
Mandal
,
Md Atiur
Rahman
and
Andreas F. M.
Kilbinger
*
,
Ankita
Mandal
,
Md Atiur
Rahman
and
Andreas F. M.
Kilbinger
*
Department of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.kilbinger@unifr.ch
First published on 13th October 2022
Abstract
Here, we present a detailed study of the metathesis activity of conjugated 1,3 diene derivatives in ring opening metathesis polymerization (ROMP) using Grubbs' 3rd generation catalyst (G3). A comprehensive screening of those derivatives revealed that monosubstituted 1,3 dienes show similar reactivities towards G3-alkylidenes as norbornene derivatives. Therefore, they represent perfect candidates for chain transfer agents in a kinetically controlled catalytic ROMP. This unprecedented reactivity allowed us to catalytically synthesize mono-end-functional poly(norborneneimide)s on the gram scale. Much more complex architectures such as star-shaped polymers could also be synthesized catalytically for the very first time via ROMP. This inexpensive and greener route to produce telechelic ROMP polymers was further utilized to synthesize ROMP block copolymers using bifunctional ROMP and ATRP/NCL initiators. Finally, the regioselective reaction of G3 with 1,3 diene derivatives was also exploited in the synthesis of a ROMP-PEG diblock copolymer initiated from a PEG macroinitiator.
Introduction
Over the last few decades, ring opening metathesis polymerization (ROMP) has emerged as a powerful polymerization technique to produce highly functional and diverse materials.1 Typical metathesis initiators for ROMP are based on ruthenium2 and molybdenum3 complexes. Among these, commercially available ruthenium-based complexes discovered by Grubbs' and coworkers are often preferred due to their robustness and excellent tolerance toward many functional groups.4 Grubbs' 2nd generation catalyst (G2) and Grubbs' 3rd generation catalyst (G3) are frequently used for polymerizations.5–7 The mechanism of ROMP involves a chain-growth polymerization where a strained cyclic olefin (monomer) is converted to a polymeric species.8 The high functional group tolerance of Grubbs' catalysts allowed the synthesis of telechelic ROMP polymers9–15 that has great importance in many disciplines of chemistry.Based on the choice of monomers, ROMP, in general, can either be catalytic or stoichiometric in the ruthenium complex (Scheme 1). For example, polymerization of cyclooctene and its derivatives or unsubstituted norbornene can be carried out with catalytic amounts of ruthenium carbene complexes using irreversible symmetrical chain transfer agents (CTAs) under thermodynamically controlled conditions.16–19 This method heavily relies on “backbiting” to produce homotelechelic polymers. Even though it reduces the loading of costly ruthenium catalysts, this method is restricted to very few monomers.
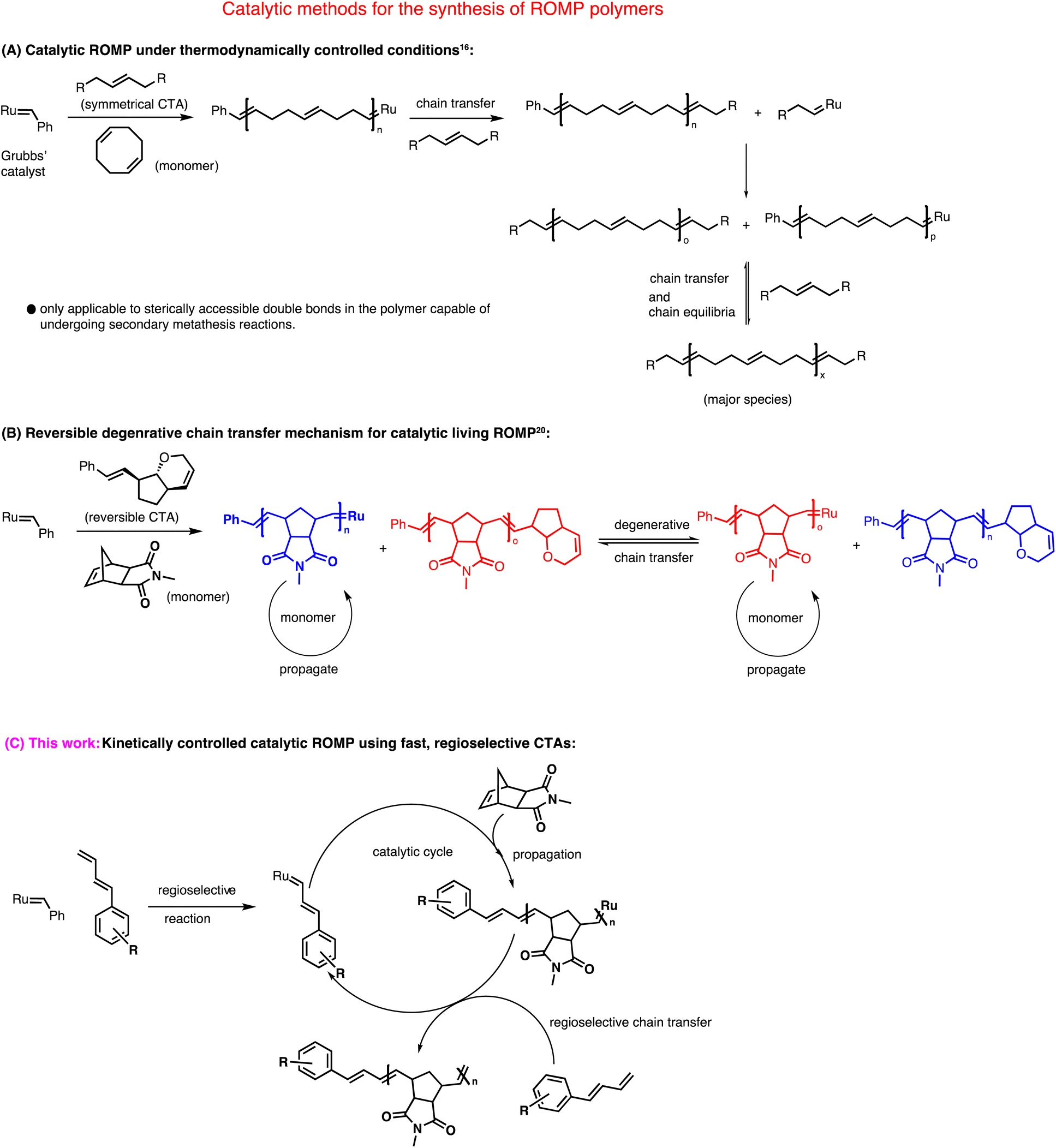 | ||
| Scheme 1 Mechanistic illustration of three different approaches for the catalytic synthesis of ROMP polymers. | ||
On the other hand, in a typical living ROMP where norbornene imide-based monomers are often used, equimolar amounts of metathesis catalyst are required with respect to the number of polymer chains formed, making the whole process non-catalytic. Our group recently addressed this issue utilizing a degenerative chain-transfer mechanism that used only catalytic amounts of ruthenium complex.20,21 Although this new method is mechanistically unique; it is limited in terms of functional and readily available chain transfer agents. Pulsed monomer addition is another technique in ROMP to obtain telechelic polymers using a sub-stoichiometric amount of metal complex.22–25 Lastly, a theoretically straightforward approach to synthesizing ROMP polymers is based on a kinetically controlled chain transfer mechanism. Katayama et al. reported true heterotelechelic ROMP polymers using functional vinyl ethers, vinyl thioethers, and vinyl acetates that undergo regioselective chain transfer.26–28 Only norbornene was used as a monomer to give heterotelechelic poly(norbornene) in a kinetically controlled catalytic process. Therefore, this method is limited in terms of monomer scope.
Here, we report a new kinetically controlled chain transfer ROMP mechanism. We developed a new class of chain transfer agents containing monosubstituted 1,3 dienes to catalytically produce mono-end-functional poly(norborneneimide)s, bi, tri, and tetrafunctionally initiated linear and three and four-arm star polymers for the very first time in metathesis polymerization.
Results and discussion
Mono-end functional poly(norborneneimide)s via catalytic ROMP
Grubbs' Ru complexes react with electron-rich double bonds such as vinyl ethers in a regioselective manner.29,30 We hypothesized that monosubstituted 1,3 diene compounds containing an electron-rich as well as the sterically accessible conjugated double bond (Scheme S1‡) would exhibit a similar fast, regioselective cross-metathesis reaction with G3. Therefore, a functional monosubstituted 1,3- diene (CTA1, Fig. 1) was synthesized in a few straightforward steps (see ESI‡).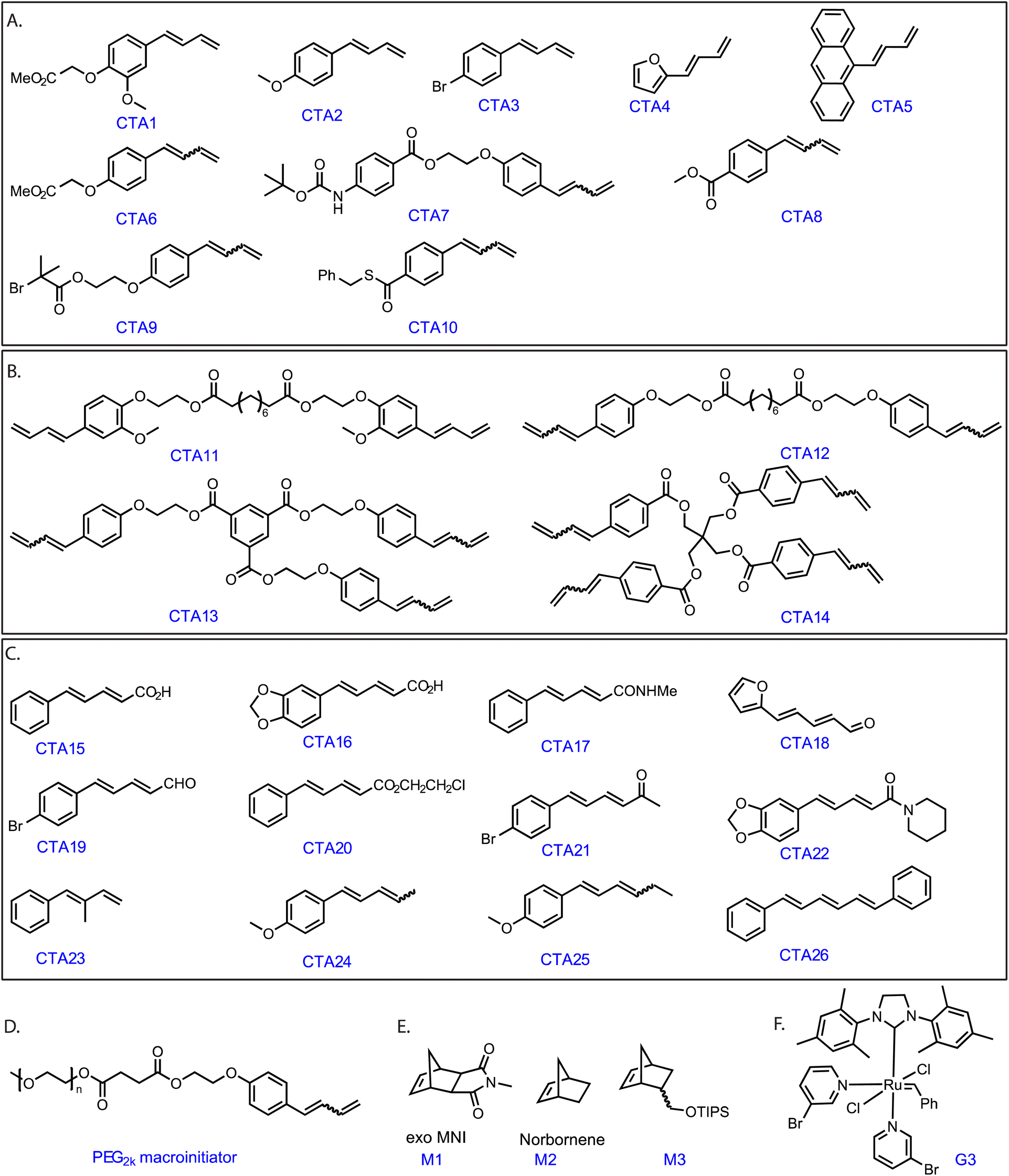 | ||
| Fig. 1 Chemical structures of compounds investigated in this study: (A) monosubstituted, functional chain transfer agents (CTAs) (CTA1–20). (B) Bi, tri, and tetra functional CTAs (CTA11–14). (C) Disubstituted CTAs (CTA15–26). (D) poly(ethylene glycol) based macroinitiator (PEG2k macroinitiator). (E) Structures of monomers. (F) Structure of Grubbs' 3rd generation catalyst (G3). | ||
To demonstrate our concept, G3 (1 equiv.) and 1 equiv. of CTA1 was dissolved in dichloromethane-d2, added to an NMR tube and the reaction was followed by 1H NMR spectroscopy (see Fig. S1‡). Within the first measurement (<10 min), the signals of the terminal double-bond (CTA1) (5.15 ppm and 5.32 ppm) disappeared along with the generation of new peaks at 5.25 ppm and 5.75 ppm, which corresponded to the formation of styrene.
Moreover, looking at the ruthenium alkylidene region, we observed the formation of a new doublet at 18.56 ppm. As proposed, the ruthenium complex reacted regioselectively, forming an electronically stable conjugated carbene (thus, producing styrene) rather than the methylidene complex. The fact that only 1 equiv. CTA1 could fully pre-functionalize commercial G3 benzylidene indicated an unexpectedly high reactivity of the terminal conjugated double bond. Additionally, an end-capping experiment was performed where a G3-benzylidene initiated poly (exo-N-methyl norbornene imide, M1) (M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G3 = 20) was end-capped with 1.2 equiv. of CTA1 in an NMR tube. To our expectation, the complete disappearance of the G3-alkylidene signal (at 18.45 ppm) and the formation of a doublet at 18.56 ppm and a doublet of doublet at 8.40 ppm were observed immediately, suggesting the formation of only one type of conjugated Ru complex (Fig. 2). This regioselective chain transfer was further confirmed by isotopically resolved MALDI-ToF MS analysis of the precipitated polymer (P1), showing a single set of mass distributions with expected polymer end groups: phenyl on one end (from G3) and methylene on the other (from CTA1, mono-isotopic mass: 4106.70 vs. observed mass: 4106.61, Fig. S3‡). Analysis of P1 by size exclusion chromatography (SEC) showed a single, monomodal peak indicating absence of any chain end coupling products (Fig. S208‡). The exceptionally high metathesis reactivity of CTA1 prompted us to investigate a catalytic ring opening metathesis polymerization that exploits a kinetically controlled chain transfer process. Next, we performed a one-pot polymerization using a ratio of G3:CTA1:M1 = 1:40:400. SEC analysis of the resulting polymer revealed a monomodal distribution with the molecular weight very close to the M1/CTA1 ratio (P2, Mn, M1/CTA1 = 2.0 kDa vs. Mn, SEC (CHCl3) = 2.6 kDa) and dispersity of 1.77. This suggested a catalytic polymerization involving a very efficient chain transfer to CTA1. In this mechanism, a propagating polymer chain reacts regioselectively with a CTA to regenerate the initiating species while terminating the polymer chain irreversibly (Fig. 3A). For this mechanism to work optimally, the chain transfer and monomer propagation rate constants should be identical. Moreover, isotopically resolved MALDI-ToF MS analysis confirmed the polymer structure with both CTA1 end groups: a functional aromatic ring and a methylene unit (Fig. 3B). The polymerization was repeated, varying the M1 to CTA1 ratio (P3–5) (Table S1‡). The resulting polymers showed a linear dependence between M1/CTA1, and the number average molecular weight (Mn) determined by SEC (Fig. 3C) indicating that excellent control over molecular weight could be obtained by this method. Next, a methoxy substituted CTA2 was employed, and the G3 to CTA2 ratio was lowered to 1:300. A polymer with reasonable control over molecular weight (P6, Mn, M1/CTA2 = 1.9 kDa vs. Mn, SEC (CHCl3) = 2.5 kDa) was obtained in high yield (91%). To the best of our knowledge, this is the lowest Ru-complex to CTA ratio ever employed to synthesize norbornene imide-based metathesis polymers. Then, M1 was polymerized using CTA3 (P7, G3:CTA3 = 1:100, Mn, SEC (CHCl3) = 3.9 kDa), CTA3 (P8,G3:CTA3 = 1:50, Mn, SEC (CHCl3) = 44.0 kDa), CTA4 (P9, G3:CTA4 = 1:100, Mn, SEC (CHCl3) = 4.4 kDa), and CTA5 (P10, G3:CTA5 = 1:200, Mn, SEC (CHCl3) = 2.3 kDa) (Table S1‡). The purified polymers were fully characterized using NMR spectroscopy and MALDI-ToF mass spectrometry. P10 draws special attention here due to the ease at which our method introduced a fluorescent group (anthracene) to a polymer chain end. It is worth mentioning that there have been many attempts in ROMP to synthesize precisely labeled metathesis polymers, in particular, fluorescent labels, due to their applications in bioimaging, drug delivery, etc.31,32
:G3 = 20) was end-capped with 1.2 equiv. of CTA1 in an NMR tube. To our expectation, the complete disappearance of the G3-alkylidene signal (at 18.45 ppm) and the formation of a doublet at 18.56 ppm and a doublet of doublet at 8.40 ppm were observed immediately, suggesting the formation of only one type of conjugated Ru complex (Fig. 2). This regioselective chain transfer was further confirmed by isotopically resolved MALDI-ToF MS analysis of the precipitated polymer (P1), showing a single set of mass distributions with expected polymer end groups: phenyl on one end (from G3) and methylene on the other (from CTA1, mono-isotopic mass: 4106.70 vs. observed mass: 4106.61, Fig. S3‡). Analysis of P1 by size exclusion chromatography (SEC) showed a single, monomodal peak indicating absence of any chain end coupling products (Fig. S208‡). The exceptionally high metathesis reactivity of CTA1 prompted us to investigate a catalytic ring opening metathesis polymerization that exploits a kinetically controlled chain transfer process. Next, we performed a one-pot polymerization using a ratio of G3:CTA1:M1 = 1:40:400. SEC analysis of the resulting polymer revealed a monomodal distribution with the molecular weight very close to the M1/CTA1 ratio (P2, Mn, M1/CTA1 = 2.0 kDa vs. Mn, SEC (CHCl3) = 2.6 kDa) and dispersity of 1.77. This suggested a catalytic polymerization involving a very efficient chain transfer to CTA1. In this mechanism, a propagating polymer chain reacts regioselectively with a CTA to regenerate the initiating species while terminating the polymer chain irreversibly (Fig. 3A). For this mechanism to work optimally, the chain transfer and monomer propagation rate constants should be identical. Moreover, isotopically resolved MALDI-ToF MS analysis confirmed the polymer structure with both CTA1 end groups: a functional aromatic ring and a methylene unit (Fig. 3B). The polymerization was repeated, varying the M1 to CTA1 ratio (P3–5) (Table S1‡). The resulting polymers showed a linear dependence between M1/CTA1, and the number average molecular weight (Mn) determined by SEC (Fig. 3C) indicating that excellent control over molecular weight could be obtained by this method. Next, a methoxy substituted CTA2 was employed, and the G3 to CTA2 ratio was lowered to 1:300. A polymer with reasonable control over molecular weight (P6, Mn, M1/CTA2 = 1.9 kDa vs. Mn, SEC (CHCl3) = 2.5 kDa) was obtained in high yield (91%). To the best of our knowledge, this is the lowest Ru-complex to CTA ratio ever employed to synthesize norbornene imide-based metathesis polymers. Then, M1 was polymerized using CTA3 (P7, G3:CTA3 = 1:100, Mn, SEC (CHCl3) = 3.9 kDa), CTA3 (P8,G3:CTA3 = 1:50, Mn, SEC (CHCl3) = 44.0 kDa), CTA4 (P9, G3:CTA4 = 1:100, Mn, SEC (CHCl3) = 4.4 kDa), and CTA5 (P10, G3:CTA5 = 1:200, Mn, SEC (CHCl3) = 2.3 kDa) (Table S1‡). The purified polymers were fully characterized using NMR spectroscopy and MALDI-ToF mass spectrometry. P10 draws special attention here due to the ease at which our method introduced a fluorescent group (anthracene) to a polymer chain end. It is worth mentioning that there have been many attempts in ROMP to synthesize precisely labeled metathesis polymers, in particular, fluorescent labels, due to their applications in bioimaging, drug delivery, etc.31,32
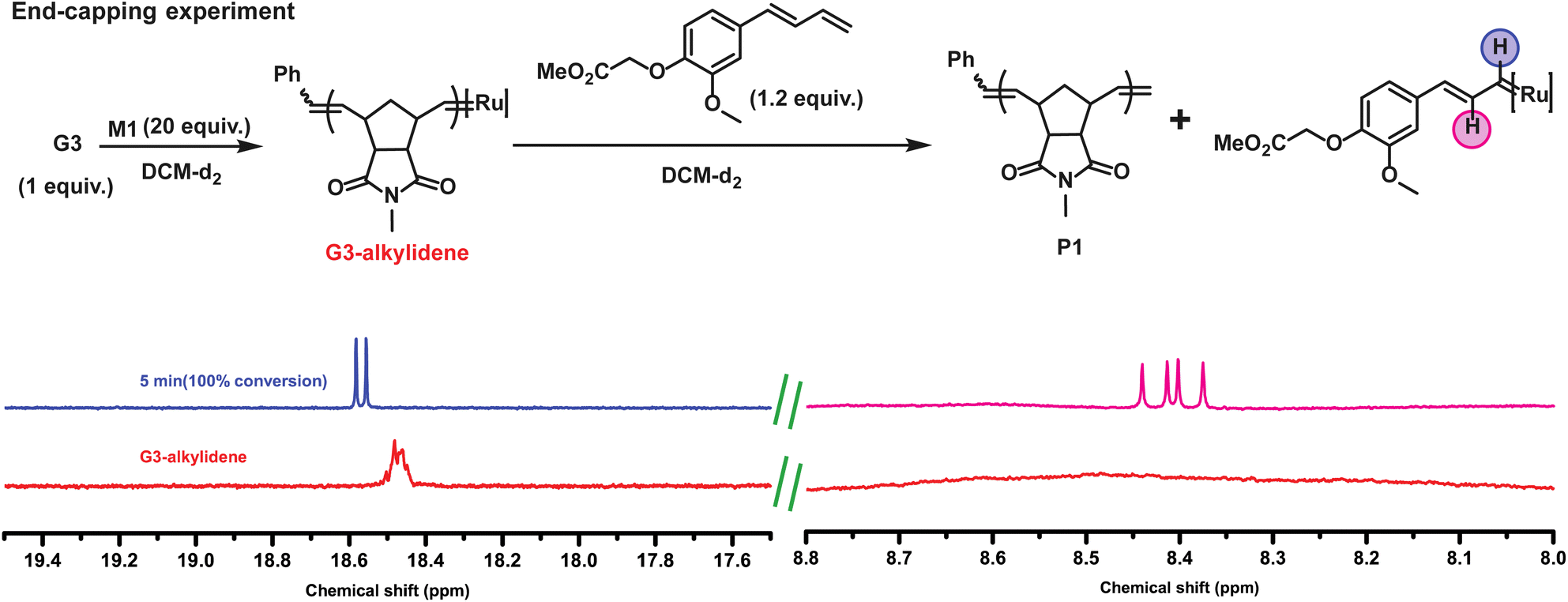 | ||
| Fig. 2 1H NMR spectra (CD2Cl2, 400 MHz) showing the propagating G3-alkylidene complex (at 18.45 ppm, red line) and its reaction with only 1.2 equiv. of CTA1 after 5 min (doublet at 18.56 ppm, in blue and doublet of doublet at 8.40 ppm, in magenta). | ||
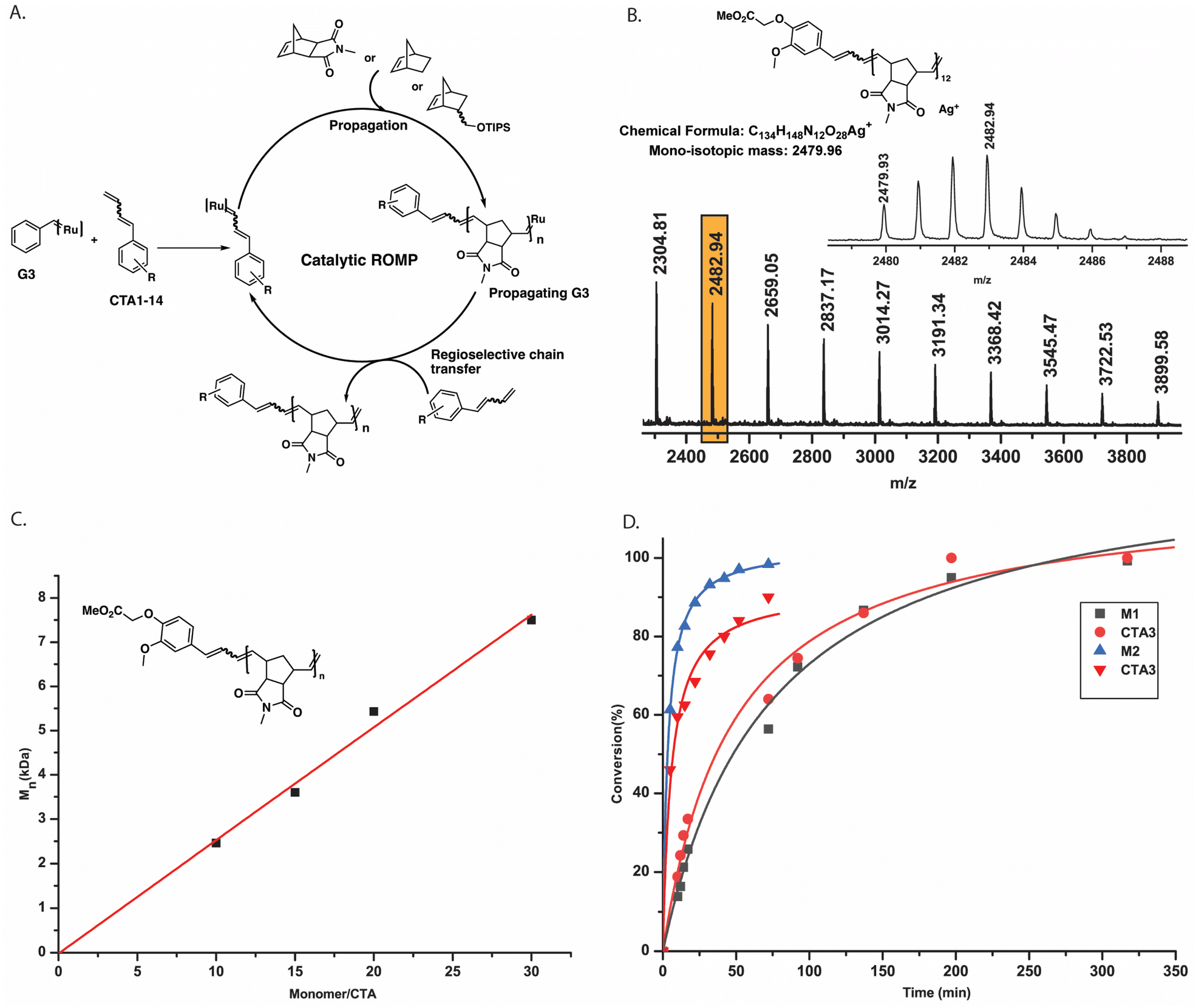 | ||
| Fig. 3 Mechanistic details for the proposed kinetically controlled mechanism. (A) Mechanism of catalytic ROMP. G3 catalyst reacts with CTA regioselectively to give a functional catalyst which, upon introduction of monomer (M1 was used in the catalytic cycle as an example), produces a propagating G3. As the rate of chain transfer to the CTA is fast, the propagating G3 reacts regioselectivity to give back the functional catalyst, which closes the catalytic cycle. (B) Isotopically resolved MALDI-ToF mass spectrum (DCTB, AgTFA) of the polymer P2 matching the proposed end groups that resulted from CTA1. (C) A plot of the number average molecular weight (Mn) versus monomer to CTA ratio showing a linear correlation. Polymerizations were carried out with M1, CTA1, and G3 under standard degassed conditions. Mn was measured using SEC in chloroform. (D) A plot of monomers (M1 and M2, 1000 equiv.) and CTA3 (50 equiv.) conversion vs. time determined by 1H NMR spectroscopy (CDCl3, 300 MHz) showing both monomer and CTA were consumed at a similar rate during the polymerization, thus, suggesting a kinetically controlled mechanism. | ||
To elucidate the proposed kinetically controlled mechanism in detail, a 1H NMR kinetics experiment was performed taking G3 (1 equiv.), CTA3 (50 equiv.) and M1 (1000 equiv.) in CDCl3. 3-bromopyridine was added as an additive to slow down the polymerization, and thus, conversion of M1 and CTA3 were followed over time. A plot of conversion vs. time for M1 and CTA3 showed that both components of the polymerization mixture were consumed almost at the same rate during the polymerization (over 317 min) (Fig. 3D and S6‡). This strongly suggests a kinetically controlled mechanism for this type of CTA. To further elucidate the mechanism detailed kinetic study was performed and the rate of consumption of both monomer (M1) and CTA (CTA3) (see Fig. S263 and S264‡) were determined. Gratifyingly, rate constants obtained for both monomer consumption (kM1 = 0.01531 min−1) and CTA3 consumption (kCTA3 = 0.01434 min−1) are of a similar magnitude which was predicted for a kinetically controlled chain transfer mechanism.
Moreover, the effect of CTA concentration on the kinetics of polymerization was also exploited using 1H NMR spectroscopy. As expected for a chain transfer chain growth polymerization, rate of the reaction was inversely proportional to the concentration of the CTA (see Fig. S272 and Table S6‡).
In CTA1–5, the diene double bond was necessarily trans configured as their synthesis was based on trans-cinnamaldehyde derivatives. Only very few such derivatives are commercially available and thus limit the introduction of functionality in our CTAs. However, we envisioned that a CTA with a mixture of cis and trans double bonds could be synthesized easily from an aromatic aldehyde using allyl triphenylphosphonium bromide in a Wittig reaction. Thus, CTA6–10 were prepared. A polymer end-capping experiment, as described above, was performed using CTA6, and the reaction was followed by 1H NMR spectroscopy (Fig. S11‡). Although all the propagating poly(M1) alkylidene signals vanished immediately, several doublet peaks were observed in the ruthenium alkylidene region, presumably, due to the coordination of the cis double bond of CTA6 to the ruthenium complex. Nonetheless, a clean MALDI-ToF mass spectrum (Fig. S12‡) with methylene terminated polymer chains confirmed the expected regioselectivity. Furthermore, M1 was polymerized using CTA6 (P11, G3:CTA6 = 1:100, Mn, SEC (CHCl3) = 6.5 kDa), CTA8 (P12, G3:CTA8 = 1:20, Mn, SEC (CHCl3) = 3.4 kDa), CTA9 (P13, G3:CTA9 = 1:100, Mn, SEC (CHCl3) = 7.9 kDa), and CTA10 (P14, G3:CTA10 = 1:100, Mn, SEC (CHCl3) = 5.0 kDa) (Table S1‡). All showed distinct end groups in the 1H NMR spectra and isotopically resolved MALDI ToF MS signals matching the two expected end groups (see ESI‡).
Norbornene (M2) is a highly strained monomer that has been studied immensely in ROMP. Norbornene propagates more rapidly than norborneneimide derivatives and is, therefore, more difficult to polymerize catalytically under kinetic control. Here, we have taken into account the very high reactivity of our CTAs and synthesized monotelechelic poly (norbornene) using a catalytic amount of Grubbs' initiator. For example, CTA2 (P15, G3:CTA2 = 1:100, Mn, SEC (CHCl3) = 7.2 kDa), CTA5 (P16, G3:CTA5 = 1:100, Mn, SEC (CHCl3) = 8.8 kDa), CTA7 (P17, G3:CTA7 = 1:100, Mn, SEC (CHCl3) = 8.8 kDa), CTA9 (P18, G3:CTA9 = 1:100, Mn, SEC (CHCl3) = 8.2 kDa), and CTA10 (P19, G3:CTA10 = 1:170, Mn, SEC (CHCl3) = 7.0 kDa) (Table S1‡) were employed to obtain poly(norbornene)s with excellent end group fidelity as observed by both 1H NMR spectroscopy and MALDI-ToF mass spectrometry. The molecular weights of the poly(norbornene)s measured by SEC using CHCl3 as eluent showed more than twice the theoretical value (see Table S2‡). However, monodisperse poly(styrene) samples were used as calibrants for SEC measurements, which have a different hydrodynamic radius compared to polydisperse poly(norbornene)s, and therefore a discrepancy in the observed molecular weights can be expected as Grubbs' and coworkers have already reported.19
To demonstrate similar kinetic controlled mechanism works for M2, G3 (1 equiv.), CTA3 (50 equiv.) M2 (1000 equiv.) and 3-bromopyridine (60 equiv.) were mixed in CDCl3 in an NMR tube. The consumption of M2 and CTA3 were followed over time. Since M2 has a higher propagation rate than M1, full consumption of M2 was observed within 70 min and the conversion of CTA3 was almost at 90% (Fig. 3D, Fig. S7‡). This, again, implied a kinetically controlled chain transfer mechanism even for M2.
To obtain insight into the structure–property relationships of monosubstituted 1,3 diene derivatives for catalytic ROMP, Hammett studies were performed using CTA2, CTA3 and CTA8 (Fig. S275‡). A negative reaction parameter (ρ = −0.88) indicated electron rich CTAs have higher reactivity and thus should be the choice of CTA for catalytic ROMP.
Branched ROMP polymers
Encouraged by the unusual reactivity of our regioselective CTAs, we synthesized CTA11 carrying a long spacer between the two reacting sites. Then, a 1H NMR experiment was performed to prove the pre-functionalization of CTA11 on both sides (Fig. S8‡). Bifunctional CTA12 was employed for the catalytic polymerization of both, M1 and M2 to obtain P20 (G3:CTA12 = 1:50, Mn, SEC (CHCl3) = 6.8 kDa) and P24 (G3:CTA12 = 1:100, Mn, SEC (CHCl3) = 7.7 kDa) with a good control over the molecular weights (Table S2‡). Since we could not rely on MALDI-ToF MS analyses to confirm the bidirectional growth of the polymers, additional support for the expected polymeric species was given by 1H NMR spectroscopic data. There, we observed that upon polymerization, the conjugated double bonds of the initiator (CTA) shifted completely into the expected signals for the chain ends of the polymer (see ESI‡).
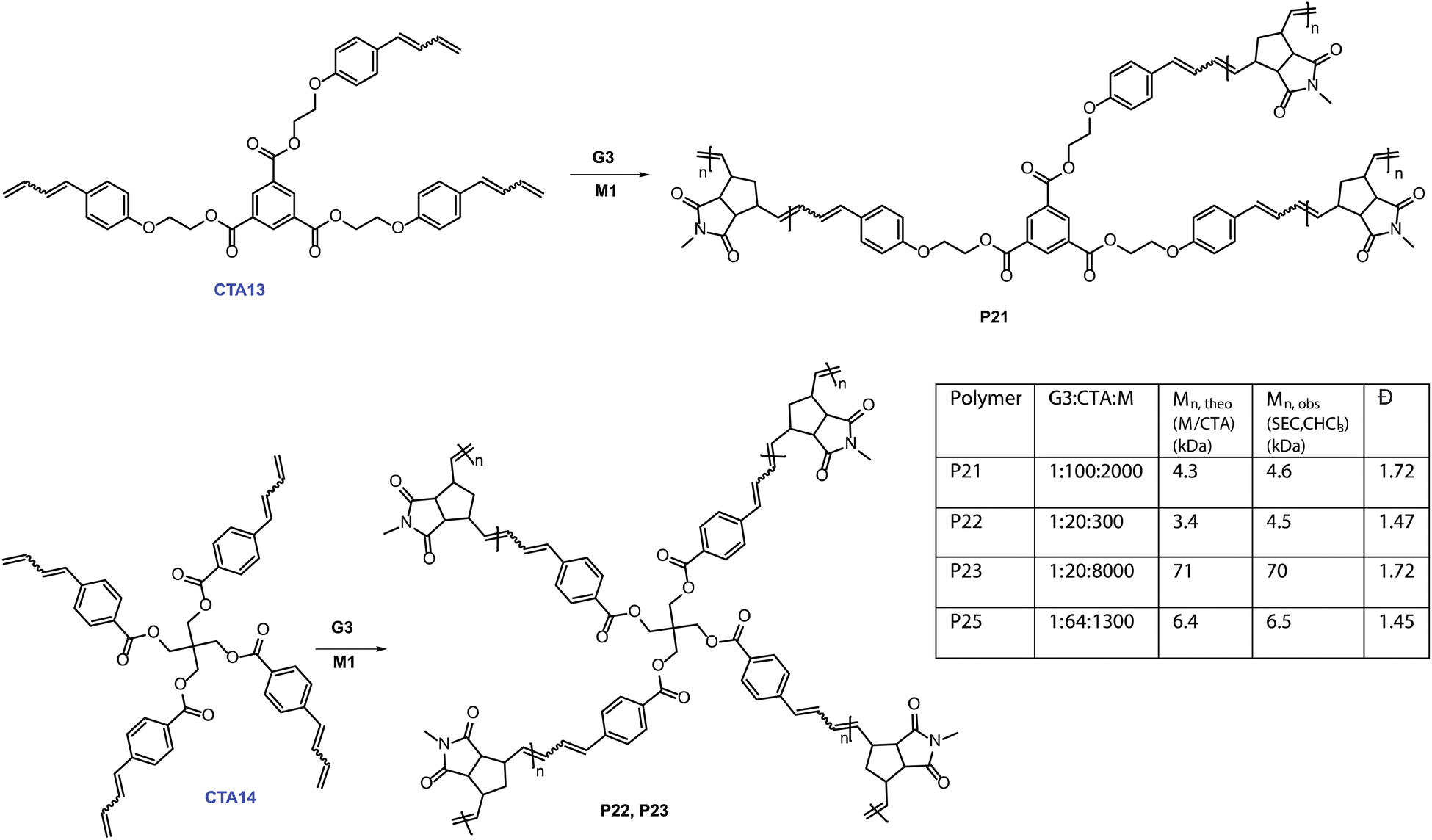 | ||
| Fig. 4 Catalytic synthesis of three-arm and four-arm star polymers. Right inset: polymerization results for the synthesis of star-shaped polymers. M = Monomer, Đ = dispersity. | ||
We first synthesized trifunctional CTA13. Then, a 100 times catalytic polymerization using M1 (M1:CTA13 = 1:100)produced tri-arm star polymer, P21, with the molecular weight (4.6 kDa) controlled by the monomer (M1) to CTA13 ratio. We then expanded our investigation further into catalytically synthesizing four-arm star polymers. CTA14 was synthesized in three straightforward steps. A 1H NMR tube polymerization employing CTA14 and M1 was performed to show full consumption of both CTA14 and M1, suggesting a tetra functional growth (Fig. S10‡). In addition, SEC analysis of the resulting polymer P22 provided a monomodal distribution with controlled Mn, SEC (CHCl3) = 4.5 kDa (Mn, M1/CTA14 = 3.4 kDa). To further demonstrate the applicability of this method, a higher molecular weight tetra-arm star polymer P23 (Mn, SEC (CHCl3) = 70 kDa) was also synthesized. Finally, M3 was employed to obtain a four-arm functional star polymer (P25, G3:CTA14 = 1:64, Mn, SEC (CHCl3) = 6.5 kDa). A combination of SEC and 1H NMR spectroscopy confirmed the expected star-shaped structure of the polymer. This straightforward route to making homo-arm star polymers is unique and highly efficient. To the best of our knowledge, this is the first report of synthesizing such complex polymeric architectures using a catalytic amount of ruthenium complex in metathesis chemistry.
A major drawback of our catalytic method is that molecular weight distribution or dispersity is relatively broad (1.45–2.10) due to a mechanistic necessity of a kinetically controlled catalytic polymerization. Nevertheless, the end-group fidelity of polymer chain ends is always very high. Besides, recently it has been documented that both high and low dispersity polymers exhibit distinctive properties. In many cases, high dispersity polymeric materials are shown to be beneficial in different applications, including polymeric self-assembly, polymer blends' processability, and in different rheological properties.43,44
Interestingly, two sets of doublets were observed when (1E,3E,5E)-1,6-diphenylhexa-1,3,5-triene (CTA26) (see Fig. S37‡) was introduced to check the regioselectivity towards G3. Moreover, the end-capping experiment proved that non-regioselective chain transfer was involved in the case of sterically accessible triene derivatives (see Fig. S38 and S39‡).
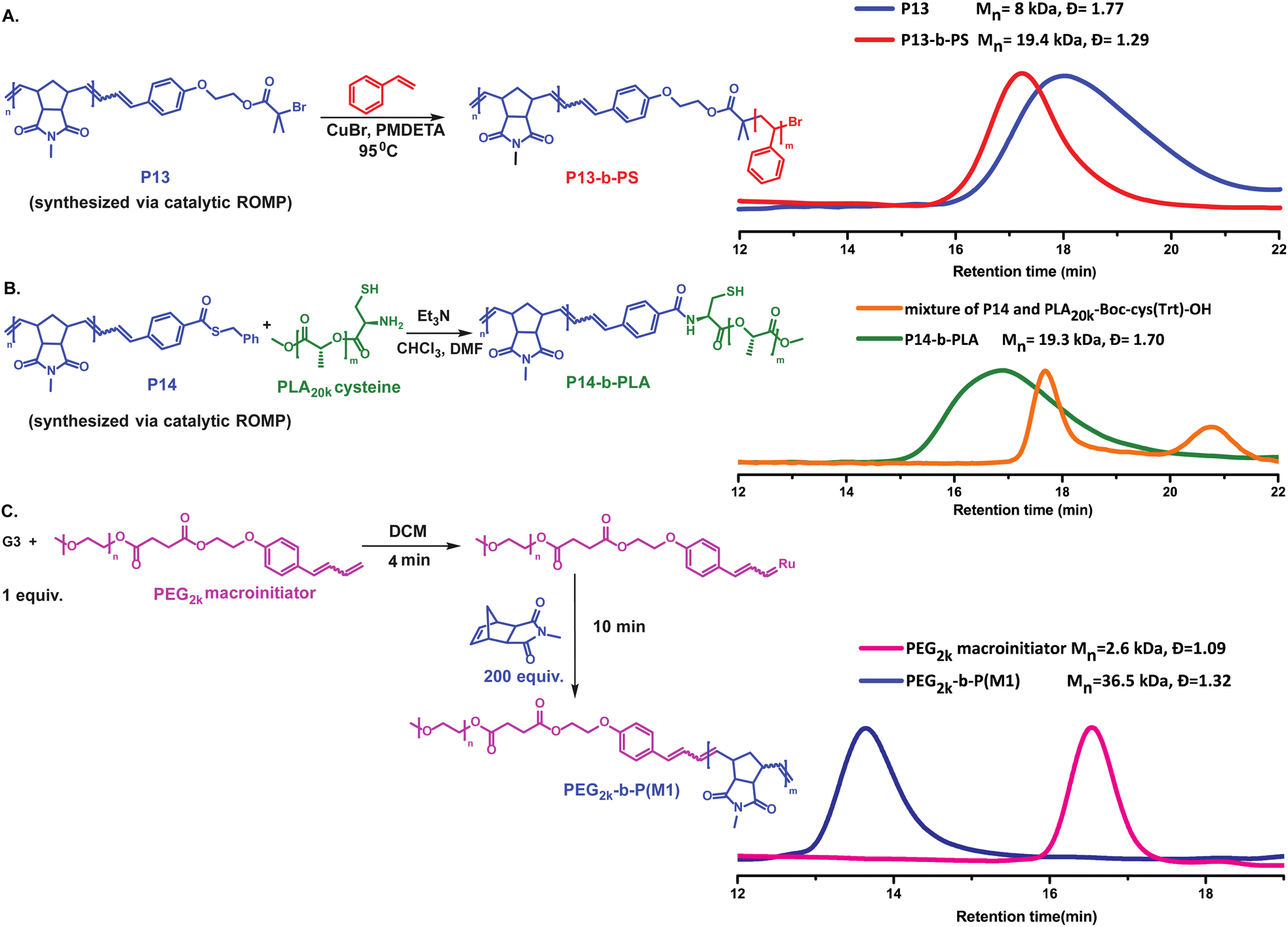 | ||
| Fig. 5 Synthesis of block copolymers. (A) Synthesis of P13-b-PS diblock copolymer via catalytic ROMP followed by ATRP and SEC (CHCl3) characterization. (B) NCL synthesis of the diblock copolymer P14-b-PLA and SEC (CHCl3) characterization. (C) Synthesis of diblock copolymer PEG2k-b-P(M1)via macroinitiation from a poly(ethylene glycol) macroinitiator (PEG2k macroinitiator) and SEC (DMF) characterization. | ||
Conclusions
In conclusion, we have successfully designed a simple yet efficient synthetic method to prepare ROMP polymers catalytically in a one-pot approach using a new type of chain transfer agents. The total polymerization time is typically less than fifteen minutes which emphasizes the ultrafast kinetics of the reported CTAs. Various substituents are introduced selectively at one terminus of the polymer chain producing monotelechelic polymers in a catalytic fashion. The catalyst to CTA ratio studied in this report was as low as 1 to 300, meaning a 300-fold saving of costly and toxic ruthenium carbene complex. Complex polymeric architectures such as homo-arm star polymers have been synthesized catalytically using a core-first approach for the first time in metathesis-based polymerization. Telechelic polymers obtained via our method were further employed to produce block copolymers using orthogonal chemistry like ATRP and NCL. Moreover, a macroinitiation approach was introduced to prepare ROMP block polymers.The development of catalytic ring opening metathesis polymerization methods is an ongoing effort. It reduces the cost of expensive metathesis-based catalysts and offers considerable potential for synthesizing functional ROMP polymers for biomedical or materials uses where lower contaminations of toxic ruthenium metal are critical. The current environmentally friendly approach can be used to synthesize end functional homopolymers, block copolymers, and other complex architectures that could lead to new applications in industrial and materials chemistry.
Data availability
We have included all data in the ESI section.‡Author contributions
I. M and A. F. M. K designed the experiments. A. M synthesized PLA20k-cysteine and P31-b-PLA. M. A. R performed a few MALDI-ToF MS analyses. I. M carried out the rest of the syntheses, analyses, and kinetic studies. I. M and A. F.M. K. wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. F. M. K, I. M, A. M, M. A. R thanks Swiss National Science Foundation, National Center of Competence in Research (NCCR Bio-inspired Materials) and the Fribourg Center for Nanomaterials (FriMat) for support. We also acknowledge Dr Krzysztof Piech, Rahul Giri and Subrata Patra for their kind help during GC-MS measurements and analyses.Notes and references
- S. Sutthasupa, M. Shiotsuki and F. Sanda, Polym. J., 2010, 42, 905–915 CrossRef CAS.
- Y. Chen, M. M. Abdellatif and K. Nomura, Tetrahedron, 2018, 74, 619–643 CrossRef CAS.
- M. R. Buchmeiser, Chem. - Eur. J., 2018, 24, 14295–14301 CrossRef CAS PubMed.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- M. S. Sandford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6554 CrossRef PubMed.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed.
- T. L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- J. B. Matson and R. H. Grubbs, Macromolecules, 2010, 43, 213–221 CrossRef CAS PubMed.
- S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537–546 CrossRef CAS PubMed.
- A. F. M. Kilbinger, Synlett, 2019, 30, 2051–2057 CrossRef CAS.
- S. Pal, F. Lucarini, A. Ruggi and A. F. M. Kilbinger, J. Am. Chem. Soc., 2018, 140, 3181–3185 CrossRef CAS PubMed.
- B. R. Elling and Y. Xia, ACS Macro Lett., 2018, 7, 656–661 CrossRef CAS PubMed.
- L. Fu, T. Zhang, G. Fu and W. R. Gutekunst, J. Am. Chem. Soc., 2018, 140, 12181–12188 CrossRef CAS PubMed.
- P. Liu, M. Yasir, A. Ruggi and A. F. M. Kilbinger, Angew. Chem., 2018, 130, 926–929 CrossRef.
- M. A. Hillmyer and R. H. Grubbs, Macromolecules, 1993, 26, 872–874 CrossRef CAS.
- M. A. Hillmyer and R. H. Grubbs, Macromolecules, 1995, 28, 8662–8667 CrossRef CAS.
- M. A. Hillmyer, S. T. Nguyen and R. H. Grubbs, Macromolecules, 1997, 30, 718–721 CrossRef CAS.
- C. W. Bielawski, D. Benitez, T. Morita and R. H. Grubbs, Macromolecules, 2001, 34, 8610–8618 CrossRef CAS.
- M. Yasir, P. Liu, I. K. Tennie and A. F. M. Kilbinger, Nat. Chem., 2019, 11, 488–494 CrossRef CAS PubMed.
- P. Liu, M. Yasir and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2019, 58, 15278–15282 CrossRef CAS PubMed.
- V. C. Gibson and T. Okada, Macromolecules, 2000, 33, 655–656 CrossRef CAS.
- J. B. Matson, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 3355–3362 CrossRef CAS PubMed.
- T. Zhang and W. R. Gutekunst, Polym. Chem., 2020, 11, 259–264 RSC.
- A. Mandal, I. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2022, 491–497 CrossRef CAS PubMed.
- H. Katayama, H. Urushima and F. Ozawa, Chem. Lett., 1999, 28, 369–370 CrossRef.
- H. Katayama, F. Yonezawa, M. Nagao and F. Ozawa, Macromolecules, 2002, 35, 1133–1136 CrossRef CAS.
- H. Katayama, Y. Fukuse, Y. Nobuto, K. Akamatsu and F. Ozawa, Macromolecules, 2003, 36, 7020–7026 CrossRef CAS.
- J. Louie and R. H. Grubbs, Organometallics, 2002, 21, 2153–2164 CrossRef CAS.
- P. Schwab, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1996, 118, 100–108 CrossRef CAS.
- E. K. Riga, D. Boschert, M. Vöhringer, V. T. Widyaya, M. Kurowska, W. Hartleb and K. Lienkamp, Macromol. Chem. Phys., 2017, 218, 1700273 CrossRef PubMed.
- A. E. Madkour, A. H. R. Koch, K. Lienkamp and G. N. Tew, Macromolecules, 2010, 43, 4557–4561 CrossRef CAS PubMed.
- K. Grudzień, M. Malinska and M. Barbasiewicz, Organometallics, 2012, 31, 3636–3646 CrossRef.
- V. Komanduri, D. R. Kumar, D. J. Tate, R. Marcial-Hernandez, B. J. Lidster and M. L. Turner, Polym. Chem., 2019, 10, 3497–3502 RSC.
- P. Lu, N. M. Alrashdi and A. J. Boydston, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2977–2982 CrossRef CAS.
- J. M. Ren, T. G. McKenzie, Q. Fu, E. H. H. Wong, J. Xu, Z. An, S. Shanmugam, T. P. Davis, C. Boyer and G. G. Qiao, Chem. Rev., 2016, 116, 6743–6836 CrossRef CAS PubMed.
- K. Khanna, S. Varshney and A. Kakkar, Polym. Chem., 2010, 1, 1171 RSC.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- R. S. Saunders, R. E. Cohen, S. J. Wong and R. R. Schrock, Macromolecules, 1992, 25, 2055–2057 CrossRef CAS.
- S. Gatard, S. Nlate, E. Cloutet, G. Bravic, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2003, 42, 452–456 CrossRef CAS PubMed.
- C. Zhou, C. Hou and J. Cheng, Polym. Chem., 2020, 11, 1735–1741 RSC.
- K. O. Kim, S. Shin, J. Kim and T.-L. Choi, Macromolecules, 2014, 47, 1351–1359 CrossRef CAS.
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS.
- N. A. Lynd, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef CAS.
- A. Mandal, I. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2021, 10, 1487–1492 CrossRef CAS PubMed.
- C. Cheng, E. Khoshdel and K. L. Wooley, Nano Lett., 2006, 6, 1741–1746 CrossRef CAS PubMed.
- J. B. Matson and R. H. Grubbs, Macromolecules, 2008, 41, 5626–5631 CrossRef CAS.
- C. W. Bielawski, T. Morita and R. H. Grubbs, Macromolecules, 2000, 33, 678–680 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- A. Rajakanthan, P. A. J. M. de Jongh, J. S. Town, P. Wilson and K. Kempe, Polym. Chem., 2019, 10, 5242–5250 RSC.
- S. Pal, A. Mandal, L. Hong, R. D. Ortuso, A. Petri-Fink, S. Salentinig and A. F. M. Kilbinger, Macromolecules, 2022, 55, 2854–2860 CrossRef CAS.
- T. C. Castle, L. R. Hutchings and E. Khosravi, Macromolecules, 2004, 37, 2035–2040 CrossRef CAS.
- S. Pal, I. Mandal and A. F. M. Kilbinger, ACS Macro Lett., 2022, 847–853 CrossRef CAS PubMed.
- A. Mandal, I. Mandal and A. F. M. Kilbinger, Macromolecules, 2022, 55, 7827–7833 CrossRef CAS.
Footnotes |
| † This work is dedicated to the memory of Professor Robert H. Grubbs. |
| ‡ Electronic supplementary information (ESI) available. Seehttps://doi.org/10.1039/d2sc04078f |
| This journal is © The Royal Society of Chemistry 2022 |