
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photo-oxidation driven proximity labeling strategy enables profiling of mitochondrial proteome dynamics in living cells†
He
Wang
 ab,
Zhiting
Wang
ab,
Hang
Gao
ab,
Jianhui
Liu
a,
Zichun
Qiao
ab,
Baofeng
Zhao
a,
Zhen
Liang
a,
Bo
Jiang
*a,
Lihua
Zhang
*a and
Yukui
Zhang
a
ab,
Zhiting
Wang
ab,
Hang
Gao
ab,
Jianhui
Liu
a,
Zichun
Qiao
ab,
Baofeng
Zhao
a,
Zhen
Liang
a,
Bo
Jiang
*a,
Lihua
Zhang
*a and
Yukui
Zhang
a
aCAS Key Laboratory of Separation Science for Analytical Chemistry, National Chromatographic R & A Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: jiangbo@dicp.ac.cn; lihuazhang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 28th September 2022
Abstract
Mapping the proteomic landscape of mitochondria with spatiotemporal precision plays a pivotal role in elucidating the delicate biological functions and complex relationship with other organelles in a variety of dynamic physiological processes which necessitates efficient and controllable chemical tools. We herein report a photo-oxidation driven proximity labeling strategy to profile the mitochondrial proteome by light dependence in living cells with high spatiotemporal resolution. Taking advantage of organelle-localizable organic photoactivated probes generating reactive species and nucleophilic substrates for proximal protein oxidation and trapping, mitochondrial proteins were selectively labeled by spatially limited reactions in their native environment. Integration of photo-oxidation driven proximity labeling and quantitative proteomics facilitated the plotting of the mitochondrial proteome in which up to 310 mitochondrial proteins were identified with a specificity of 64% in HeLa cells. Furthermore, mitochondrial proteome dynamics was deciphered in drug resistant Huh7 and LPS stimulated HMC3 cells which were hard-to-transfect. A number of differential proteins were quantified which were intimately linked to critical processes and provided insights into the related molecular mechanisms of drug resistance and neuroinflammation in the perspective of mitochondria. The photo-oxidation driven proximity labeling strategy offers solid technical support to a highly precise proteomic platform in time and finer space for more knowledge of subcellular biology.
Introduction
Eukaryotic cells are composed of diverse subcellular structures including organelles, vesicles and large protein assemblies, which perform their specific biological functions, relying on different protein compositions.1 Expounding spatiotemporal dynamic properties of the subcellular proteome is essential for a better understanding of various biological processes as protein localizations and expression levels change with the environment and their functions vary accordingly in physiological and pathological states.1,2 In addition, the precise regulation of the cell signaling network depends on protein alteration in defined space and time, such as assembly and degradation of protein complexes, translocation of transcription factors in the cytoplasm and nucleus and redistribution of proteins in response to cellular stress.3–5 Therefore, the delineation of subcellular proteome dynamics of organelles and other structures is indispensable for more knowledge of vital events in cells.Mitochondria, as the powerhouse of cells, were studied most in the proteome for their vital roles in many crucial processes, which impinged on health and disease, such as Ca2+ homeostasis, inflammation and cellular stress responses.6,7 The traditional strategy for mitochondrial proteome research was acquiring mitochondria by density gradient centrifugation,1,8 which showed the disadvantages of time-consumption, low purity and low coverage.9 Recently, the emergence of in situ protein chemical labeling technology provided a powerful tool for the realization of mitochondrial proteomics analysis with high specificity and high spatiotemporal resolution. Proximity labeling based on engineered enzymes (e.g., APEX and TurboID) to generate reactive intermediates for protein labeling parsed mitochondrial and even submitochondrial proteome,10–14 which was of great significance for discovering new components of mitochondria and determining subcellular localization of unknown proteins. Unfortunately, this required genetic manipulation to construct engineered enzymes and was not suitable for sensitive and complex tissue or cell samples that were difficult to transfect, including primary neurons, embryonic stem cells and macrophage cells.15 Chemical proteomics technology based on organelle-localizable reactive molecule (ORM) labeled proteins in specific subcellular compartments without gene transfection was developed for proteomic studies of various organelles such as nuclear, endoplasmic reticulum and mitochondria.16–18 However, the long reaction time and reaction species that are always “on” led to low spatiotemporal resolution and extra false positive labeling when passing by other subcellular structures. Accordingly, there was still an urgent demand to develop new chemical proteomic strategies for studying the mitochondrial proteome.
For high specificity and resolution, intelligently responsive chemical reactive species were applied to label proteins or other biomacromolecules in subcellular compartments.19–22 Controllable activation of reactive groups to form high reactive intermediates which generally were free radicals, excited singlet state species or highly electrophilic species reduced false positives and improved temporal resolution to a large extent via the short diffusion radius and fast reaction kinetics.23 A couple of photo-responsive (e.g., photoactivation24,25 and photocatalytic decaging26,27) and microenvironment responsive (e.g., localized metal ions,28,29 H2O2 (ref. 30 and 31) and protein aggregations32) probes were successively developed for subcellular proteome profiling. The recently reported strategy called bioorthogonal and photocatalytic decaging-enabled proximity labeling (CAT-Prox)26 was applied to the plotting of the mitochondrial proteome with improved spatiotemporal precision via an iridium catalyst allowing photocontrol, which showed the potential advantages of photo-responsive probes in the proteomic study of subcellular structures. Notably visible light photocatalysts to generate reactive species for proximal protein trapping, including photosensitive protein miniSOG,33 transition-metal iridium or ruthenium photocatalysts26,34 and organic photocatalysts,35–37 are burgeoning and promising due to their low biological disturbance and high spatiotemporal fidelity enabling the dynamic delineation of proteins in their native environment. Therefore, the photocatalytic strategy was expected to explore spatiotemporal dynamics in the mitochondrial proteome.
Herein we presented a photo-oxidation driven proximity labeling strategy for dissection of mitochondrial proteomics with high spatiotemporal resolution in a non-perturbed state exploiting a mitochondria-targeted probe with light dependence (Scheme 1). The photo-controllable probe could spontaneously accumulate in mitochondria of living cells and generate singlet oxygen locally under the irradiation of visible light. Spatially restricted reactions between propargylamine (nucleophile) and amino acid residues oxidized by abundant singlet oxygen (electrophilic active intermediates) allowed for protein tagging covalently in situ by an alkynyl handle, which enabled downstream modification by a biotin conjugate containing a cleavable linker (azo) for the enrichment and identification of mitochondrial proteins via copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC). Furthermore, by integrating a light controllable labeling probe and quantitative proteomics based on pseudo-isobaric dimethyl labeling,38 the mitochondrial proteome could be deciphered readily for a better understanding of mitochondrial function and its role in diverse physiological processes to elucidate molecular mechanisms of diseases. This strategy was capable of labeling and identifying mitochondrial proteins in different cells and profiling mitochondrial proteome dynamics in the process of drug resistance and neuroinflammation induced by lipopolysaccharide.
 | ||
| Scheme 1 Schematic illustration of a photo-oxidation driven proximity labeling strategy and workflow for mitochondrial proteome profiling. | ||
Results and discussion
For the mitochondria-targeted photoactivatable probe, dibromofluorescein (DBF), the eosin derivative with an appropriate 1O2 quantum yield (ΦΔ = 0.42) and hydrophobicity,39 was chosen as the photosensitive moiety to generate singlet oxygen via Dexter energy transfer under irradiation of green light and for indicating probe localization by fluorescence. With the triphenylphosphonium cation as the localizable moiety,40,41 we designed and synthesized mitochondria-targeted photoactivatable probe TPP-AcDBF (Scheme 1). In our experiment, DBF was acetylated to improve the ability of cell permeability and TPP-DBF was the active form in living cells which emerged via hydrolysis by intracellular esterase. TPP-DBF was characterized by measuring its spectral properties and the maximum absorption wavelength was 513 nm (Fig. S1A and B†). Singlet oxygen (1O2) was the prerequisite for mitochondrial protein activation and subsequent labeling. The quantification of 1O2 produced by TPP-DBF was evaluated with Si-DMA, a mitochondria localizable fluorescence probe for singlet oxygen detection that has drastically increased fluorescence intensity at 660 nm in the oxidation state42 (Fig. S1C†). As expected, the fluorescence intensity of Si-DMA increased along with time illuminated by a homemade green light irradiation device (Fig. 1A) whose wavelength peak was around 510 nm in the presence of TPP-DBF. Furthermore, we found a similar phenomenon in living HeLa cells where the fluorescence intensity of Si-DMA was much stronger after irradiation of green light for 10 min (Fig. 1B), indicating that the production of 1O2 was not perturbed by the complex intracellular matrix. Taken together, the efficient capability of singlet oxygen generation by TPP-DBF under vitro and physiological conditions was demonstrated.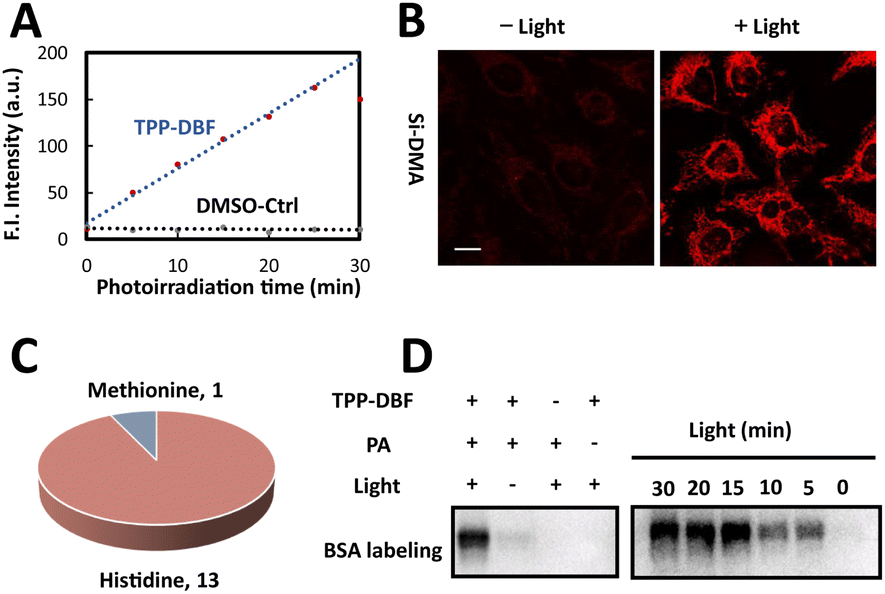 | ||
Fig. 1 Characterization and evaluation of the photoactivatable probe and labeling reagent PA. (A) Quantification of singlet oxygen production of TPP-DBF by Si-DMA in PBS/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 660 nm. (B) Confocal microscopy images of HeLa cells incubated with TPP-AcDBF and Si-DMA with or without irradiation of green light. Scale bar: 10 μm. (C) Labeled amino acid residues on BSA by TPP-DBF and PA. (D) Immunoblotting analysis of photo-oxidation driven labeled BSA by TPP-DBF and PA under different conditions. :1) at 660 nm. (B) Confocal microscopy images of HeLa cells incubated with TPP-AcDBF and Si-DMA with or without irradiation of green light. Scale bar: 10 μm. (C) Labeled amino acid residues on BSA by TPP-DBF and PA. (D) Immunoblotting analysis of photo-oxidation driven labeled BSA by TPP-DBF and PA under different conditions. | ||
After the proteins were activated, the next step was to label the highly reactive proteins. To trap oxidized amino acid residues, a set of labeling reagents with a high nucleophilic reactive moiety (amine) and different enrichment moieties (alkynyl, biotin and ferrocene) (Fig. S2A†) for protein tagging and capture were tested by modifications of peptides containing potential active amino acid residues (HMWY)36,43 with DBF generating 1O2 under irradiation of green light. An optimized labeling reagent was considered as one of the key factors to improve the labeling efficiency of mitochondrial proteins. As judged by MALDI-TOF/TOF-MS (Fig. S2C†), 1 (propargylamine, PA) showed the highest efficiency for peptides modified with histidine residues as the most prevalent modification site while peptides containing other amino acid residues (MWY) barely showed modification peaks. At the protein level, we examined the labeling ability of reagents 1-4 for BSA by LC-MS/MS under photooxidative conditions (Fig. S3A†). Except for oxidation modification, a relatively large number of PA modifications in histidine residues were mainly observed with a proportion of 10.4%, much more than those of other labeling regents (Fig. S3B†). The higher nucleophilicity of the aliphatic amine and better spatial accessibility of alkynyl enabled PA to label oxidative histidine residues efficiently. From a detailed analysis of labeling sites, it was found that 13 of all 17 histidine residues and 1 of all 5 methionine residues in BSA were labeled (Fig. 1C). The majority of histidine residues were modified by PA and the labeled residues were widely distributed on the surface of BSA (Fig. S4†) which further confirmed the high reactivity of PA. After the labeled proteins underwent a click reaction with azide-biotin, immunoblotting analysis was conducted to explore the labeling mechanism and efficiency. As shown in Fig. 1D, we found that BSA was labeled with a prerequisite that photoactivatable probe TPP-DBF, PA and irradiation all existed and the labeling intensity enhanced along with irradiation time and the concentration of TPP-DBF (Fig. S5A and B†), suggesting the feasibility of mitochondrial protein labeling. In addition, it was noted that BSA could be labeled with direct 1O2 in the absence of TPP-DBF and green light (Fig. S5C†) which indicated that the labeling reaction proceeded via1O2, enabling spatiotemporal control of protein labeling. Subsequently, the selectivity of the labeling reaction was determined by subcellular distribution of photoactivatable probe TPP-AcDBF and PA-modification.
The cell permeability and subcellular localization of photoactivatable probe TPP-AcDBF were evaluated by fluorescence co-localization analysis with confocal laser scanning microscopy (CLSM). The fluorescence of TPP-AcDBF merged well with Mito Tracker Deep Red and Pearson’s correlation coefficient was up to 0.88 (Fig. 2A), indicating its favorable specificity to target mitochondria spontaneously. Given that TPP-AcDBF is selectively localized in mitochondria, we next verified that PA addition to proteins occurred in mitochondria as well in a photoactivatable fashion with rhodamine-azide to visualize PA labeling. As a result, the cells showed strong green fluorescence in mitochondria in the presence of green light while there was negligible fluorescence of rhodamine without irradiation (Fig. 2B). As expected, this PA addition reaction was light dependent and had efficient mitochondrial specificity. After the click reaction with conjugated biotin, labeled proteins were quantified relatively by immunoblotting analysis, which showed that the intensity of labeled proteins increased compared with that of the control group (without green light) and drastically enhanced after enrichment in the case of illuminating by green light (Fig. S6†). It further demonstrated that mitochondrial protein specific labeling and capture in living cells was very feasible.
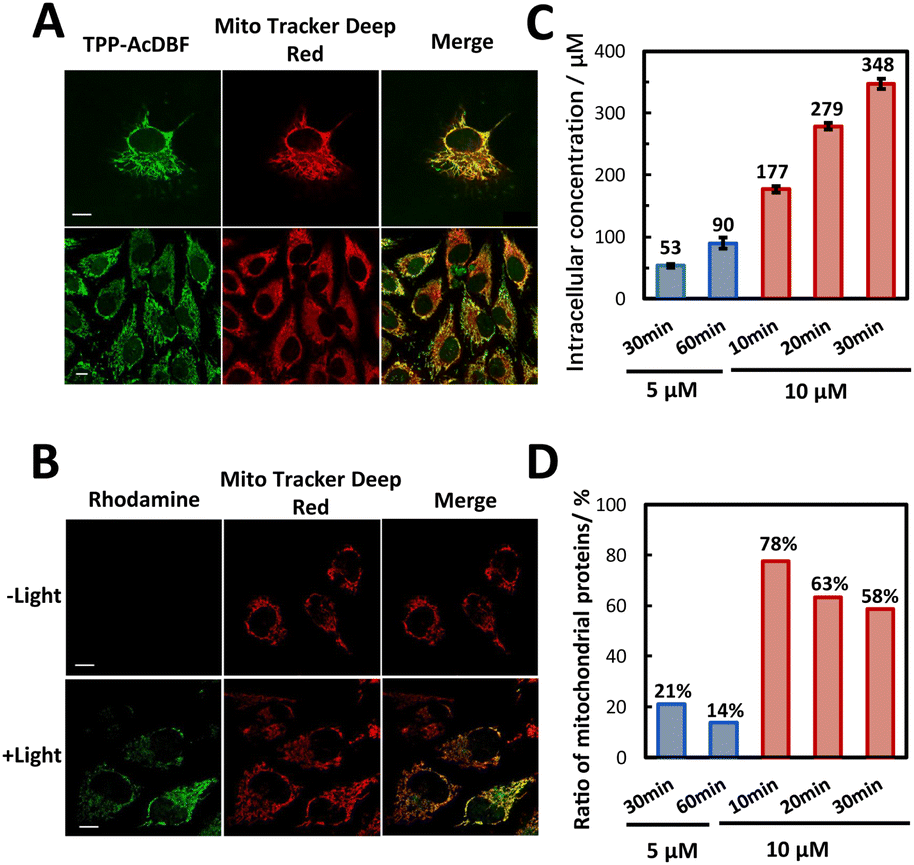 | ||
| Fig. 2 Mitochondria-targeting and protein labeling in vivo. (A) Confocal microscopy images of HeLa cells incubated with TPP-AcDBF (10 μM) and Mito Tracker Deep Red (100 nM). Scale bar: 10 μm. (B) Confocal microscopy images of fixed HeLa cells visualized by clicking with Azide-fluor 488 (10 μM) after incubating with TPP-AcDBF (10 μM) and PA (5 mM) in the presence or absence of green light. Scale bar: 10 μm. (C) Intracellular concentration of TPP-DBF in HeLa cells under various conditions, cells incubated with 5 or 10 μM TPP-AcDBF for different times ranging from 10 min to 1 h. (D) Ratio of mitochondrial proteins among all proteins identified by label-free MS quantification in photo-oxidation driven labeled HeLa cells under various incubation conditions. | ||
We next determined the final concentration of TPP-AcDBF inside cells after lysis and found that it was increasing along with incubation time and the initial concentration. The intracellular concentration in HeLa was 177 μM when incubated with 10 μM TPP-AcDBF for 10 min (Fig. 2C), which was the optimal incubation condition considering the final concentration and mitochondrial specificity. It was 18-fold higher than the initial concentration of TPP-AcDBF and would be much higher in mitochondria as the spontaneous aggregation of lipophilic cations is driven by the mitochondrial membrane potential. Additionally, the ratio of mitochondria-localizing proteins in all identified proteins was 78% (Fig. 2D) which was the highest evaluated by label-free MS quantitative analysis after the click reaction with the biotin-azide conjugate and enrichment with streptavidin agarose beads under this incubation condition. This strongly indicated the promising potential of the photo-oxidation driven proximity labeling strategy for subcellular protein profiling.
To evaluate the disturbance of photo-oxidation driven labeling on living cells, whole proteome analysis was performed under optimized conditions by label-free quantitation. The Pearson’s correlation coefficients between groups with or without probes and light were more than 0.98 (Fig. S7A†), which demonstrated that there was a subtle difference with the presence of labeling regents and green light. Proteins with a 2-fold (1.5-fold) difference in the experimental group against the blank group only accounted for a proportion of 0.56% (3.08%) of all quantified proteins (Fig. S7B and C†), indicating that the labeling strategy could hardly cause disturbance to the cell proteome and the biocompatibility was excellent.
Encouraged by successful attempts at protein labeling in vitro and in vivo, we capitalized on the photo-oxidation driven proximity labeling strategy to plot the mitochondrial proteome in HeLa cells with high-resolution MS. The PA-labeled proteins were biotinylated via a click reaction, enriched with streptavidin agarose beads and analyzed by LC-MS/MS. Reduction elution facilitated MS identification by removing biotin from proteins to reduce interference. As a result, we identified 488 labeled proteins and 310 proteins were localized to mitochondria explicitly with 1 location-unassigned protein, achieving a fairly good specificity of 64% for mitochondria (Fig. 3A and Data S1†). The MS data showed high reproducibility as well (Fig. S8 and Table S1†). Submitochondrial distribution illustrated that a large majority of identified mitochondrial proteins were localized in the matrix and inner membrane (Fig. 3B) in line with the matrix-targeting preference of lipophilic cations44 which might provide some insights into the submitochondrial location-unknown proteins. In the subsequent gene ontology (GO) enrichment analysis for identified proteins, it was found that these proteins were highly involved in many biological processes closely related to mitochondria such as translation and energy metabolism (Fig. 3C), indicating the fidelity of identification. Furthermore, we identified a number of protein subunits of ribosomes, respiratory chain complexes and inner membrane import complexes in the labeled proteome that exerted vital effects in mitochondrial biology (Fig. 3D). However, there were still 30–40% non-mitochondrial proteins in the labeled mitochondrial proteome, which might be the consequence of the random diffusion of the photoactivatable probe, the relatively low reaction efficiency of protein labeling by biotin, and the interference from endogenously biotinylated proteins within samples.45 Further efforts would be required to improve the specificity of subcellular proteome profiling.
 | ||
| Fig. 3 Identification of the mitochondrial proteome in living HeLa cells. (A) Mitochondrial specificity of proteome profiling by integrating the photo-oxidation driven proximity labeling strategy and quantitative mass spectrometry in HeLa cells with three independent biological replicates. (B) Submitochondrial localization of identified mitochondrial proteins in HeLa cells based on the MitoCarta 3.0 database and GO cell component terms. (C) Biological processes in GO enrichment analysis for identified proteins in HeLa cells. (D) Protein complexes enriched in the mitochondrial proteome and protein–protein interactions (gray lines) which were annotated by the STRING database. (E) Overlap of identified mitochondrial proteins from HeLa cells by our method, ORMs (organelle-localizable reactive molecules) and CAT-Prox (bioorthogonal and photocatalytic decaging-enabled proximity labeling strategy). (F) Abundance of mitochondrial proteins identified by our method or the other two methods. The abundance data were referenced to a previous dataset.46 | ||
Compared with APEX10 or TurboID,13 our strategy performed a little worse in coverage and specificity for mitochondrial proteins (Table S2†), which might be caused by the lower reaction kinetics of the abiotic catalytic process than engineered enzymes in reactive species generation. On the other hand, in contrast to MRMs17 and CAT-Prox,26 the two representative chemical proteomic methods for mitochondria without genetic operation, this strategy plotted the most comprehensive mitochondrial proteome with comparable specificity (Table S2†). Due to the complex cellular environment, diverse targeting molecules would aggregate into different micro-regions within the mitochondria, thus resulting in various types of protein labeling. Accordingly, there was a substantial portion of distinct mitochondrial proteins identified solely by the photo-oxidation driven proximity labeling strategy and this was complementary with different methods to a certain extent (Fig. 3E). In addition, the mitochondrial proteins identified by our method had lower abundance generally46 (Fig. 3F) owing to the efficient 1O2 production of TPP-AcDBF and the high reactivity and excellent spatial accessibility caused by the small size of PA, which emphasized the special superiority of this strategy.
To further confirm the feasibility of quantitative analysis of this method, we determined the mitochondrial quantitative landscape of sorafenib-resistant hepatocellular carcinoma by labeling proteins in sorafenib-resistant and sensitive Huh7 cells (Huh7-R and Huh7 cells) (Fig. S9A†). A variety of mechanisms are responsible for sorafenib-resistance, including dysregulation of apoptosis/cell cycle, epithelial–mesenchymal transition and hypoxic environment, strongly linked with mitochondrial function.47,48 As conducted above, 89 mitochondria-localizable proteins were quantified among 141 identified proteins in sorafenib-resistant Huh7 cells (Data S1†), suggesting the excellent reproducibility and selectivity of photo-oxidation driven proximity labeling. GO enrichment analysis of identified proteins indicated that synthesis and degradation of bio-macromolecules, mitochondrial RNA regulation and mitochondrial biogenesis might have potential implications for sorafenib-resistance (Fig. S9B†). Of note, there were a number of differential proteins in quantified proteins compared with those in sensitive Huh7 cells (Fig. S9C†), among which some proteins were of substantial significance, such as GSTK1, CPS1, MGST3 and HK2. These are strongly linked to cell metabolism,49,50 regulation of drug resistance, protection against oxidative stress51,52 and the HIF-1 signaling pathway related to formation of a hypoxia microenvironment,53 that eventually resulted in sorafenib-resistance in hepatocellular carcinoma.
The identification results of HeLa and drug-resistant Huh7 cells verified the generality and reliability of our strategy, and we speculate that it may have a unique advantage in genetically difficult-to-transfect cell lines. As resident macrophages in the central nervous system (CNS), microglia exert a critical effect on innate immune responses for neuroprotection along with monitoring the microenvironment and maintaining homeostasis of the CNS.54 It is generally acknowledged that the progression of almost all neurological disorders is implicated in neuroinflammation caused by persistent activation of microglia55–57 in which mitochondria play an active role.58 To comprehend neuroinflammation from the perspective of mitochondria, we utilized human microglia cell line HMC3 which is hard for genetic operation to implement the photo-oxidation driven proximity labeling strategy in different stages of lipopolysaccharide (LPS) stimulation with normal HMC3 cells as the control group (Fig. 4A).
 | ||
| Fig. 4 Revealing mitochondrial proteome dynamics in LPS-stimulated microglia cells. (A) Workflow of dynamic mitochondrial proteome profiling in HMC3 cells stimulated by LPS. (B) Immunoblotting analysis of NLRP3 in HMC3 cells under stimulation of LPS (1 μg mL−1). (C) Ratio distribution of identified proteins by comparing stimulation groups (stimulated by LPS for 24 h and 48 h) with the control group (stimulated by LPS for 0 h). (D) Heat map of identified mitochondrial proteins expressed using converse trends in the first and next 24 h. (E) Volcano plot of quantified proteins in the group stimulated for 48 h compared with those in the group stimulated for 24 h. | ||
As deduced from cytotoxicity tests, there was a negligible influence on cell viability when HMC3 cells were incubated with 1μg mL−1 LPS for 24 h and 48 h (Fig. S10A†). The increased expression of NOD-like receptor thermal protein domain associated protein 3 (NLRP3) in HMC3 under LPS stimulation enabled the neuroinflammation context to be more reliable (Fig. 4B). With quantitative mass spectrometry analysis, 744 proteins were identified and quantified with 383 mitochondrial proteins annotated (Data S1†). These proteins were highly enriched in biological processes related to cellular metabolism and material transport (Fig. S10B†) for high energy and material demands in the development of inflammation.59 It was noted that the most identified mitochondrial proteins were down-regulated after stimulation with LPS while there was a recovery when stimulating for a longer time (Fig. 4C). We reasoned that it might be due to the decrease in oxidative phosphorylation and the marked increase in glycolysis in the preliminary pro-inflammatory stage.60,61 And there was possibly a transformation of proinflammatory microglia cells into anti-inflammatory phenotype62,63 with prolongation of stimulation time enabling cellular metabolic reprograming, which was indicated by immunoblotting analysis as well that the expression level of NLRP3 increased at first while decreased in the next stimulation stage. Focusing on the mitochondrial proteins expressed using converse trends along with stimulation time (Fig. 4D), they were intimately correlated with events related to energy metabolism and stress responses facilitating the emergence of transformation. Noteworthily, some differential proteins such as SNAP29, MTOR and LONP1 (Fig. 4E and Table S3†) participated in autophagy/mitophagy64–67 which were closely involved in the regulation of neuroinflammation. PCK2, CHCHD2 (renamed MNRR1) and GPT2 were up-regulated in long time stimulation which impacted energy metabolic shift, activation of macrophages68,69 and tricarboxylic acid cycle70 through glutamine metabolism thus affecting the formation of an inflammatory microenvironment. PON2, a lactonase displaying anti-inflammatory actions71,72 was down-regulated, which suggested the decline of oxidative stress in microglia cells after a long time of stimulation which further supported the anti-inflammatory transformation. These results were supported by research findings that autophagy and abnormalities in energy metabolism and material equilibrium of mitochondria correlated greatly with neuroinflammation and contributed to the development and progression of neurodegenerative disease.73–76 Besides, expressions of several proteins (FPGS, ACSF2, ACOT9, PRORP, LRPPRC, and MTHFD2) associated with folate and one-carbon metabolism and transcription were significantly changed but research about their functions in inflammation and immune response was still patchy. Focusing on these representative proteins and the unclear roles they play in neuroinflammation is advantageous for the mechanism research of neurodegenerative diseases and may give us some interesting information that we ignored before. Together, the delineation of mitochondrial proteome dynamics in the context of neuroinflammation provides some meaningful views into the link between the immune response mediated by mitochondria and neurological disorders.
Conclusions
In conclusion, we developed a photo-oxidation driven proximity labeling strategy to profile mitochondrial proteome dynamics in living cells with the compelling advantages of high spatiotemporal precision, avoidance of genetic operation and low false positives because of visible light-dependence. The photoactivated probe displayed excellent mitochondrial specificity and ability of generating singlet oxygen in situ allowing the oxidative residues that were confined to mitochondria to be covalently tagged by the nucleophilic substrate. This MS-based strategy was successfully applied to reveal mitochondrial proteome dynamics in different cells, including microglia cells which were hard for transfection, and provided some molecular insights into the biological processes with regard to drug resistance and neuroinflammation. Additionally, the modular structure of the mitochondria-targeting photoactivated probe allows alteration of localizable and reactive moieties to realize more efficient subcellular proteomic mapping of other organelles and even finer sub-organelle or microregions with special physicochemical properties in cells or tissues. This strategy also promises to facilitate the plotting of the protein interactome and low-abundance post-translational modifications at the subcellular level by integrating with upstream crosslink or downstream enrichment approaches. Although the modification sites of proteins in living cell environments demanded further investigation, more information about structural features of labeled proteins can be expected by developing probes that have higher reaction efficiency as well as more specific and stable labeling products. Furthermore, higher subcellular specificity is achievable with efforts in developing more efficient labeling reagents for one-step reactions and improved enrichment systems (e.g., host–guest binding pair45). Given the mounting concern about cellular processes for medical research, the strategy has great application potential in elucidating the vital functions of subcellular structures and their spatiotemporal interactions in a healthy or disease state.Data availability
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD036630.Author contributions
Bo Jiang, Lihua Zhang and Yukui Zhang conceived and designed the project. He Wang synthesized the probe, carried out all the characterization studies and proteomic experiments and wrote the manuscript. Hang Gao, Zhiting Wang, Jianhui Liu and Zichun Qiao got involved in the quantitative proteomics. All authors participated in data analysis, discussion of results and manuscript revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation (21725506, 32088101, and 22074140), National Key R&D Program of China (2020YFE0202200) and Talent innovation support program of Dalian (2019CT07).Notes and references
- E. Lundberg and G. H. H. Borner, Nat. Rev. Mol. Cell Biol., 2019, 20, 285–302 CrossRef CAS PubMed.
- J. A. Christopher, C. Stadler, C. E. Martin, M. Morgenstern, Y. Pan, C. N. Betsinger, D. G. Rattray, D. Mahdessian, A.-C. Gingras, B. Warscheid, J. Lehtiö, I. M. Cristea, L. J. Foster, A. Emili and K. S. Lilley, Nat. Rev. Methods Primers, 2021, 1, 32 CrossRef CAS PubMed.
- J. D. Scott and T. Pawson, Science, 2009, 326, 1220–1224 CrossRef CAS PubMed.
- J. Zhang, H. Feng, J. Zhao, E. R. Feldman, S. Y. Chen, W. Yuan, C. Huang, O. Akbari, S. A. Tibbetts and P. Feng, Cell Rep., 2016, 16, 405–418 CrossRef CAS PubMed.
- H. An, A. Ordureau, M. Korner, J. A. Paulo and J. W. Harper, Nature, 2020, 583, 303–309 CrossRef CAS PubMed.
- J. B. Spinelli and M. C. Haigis, Nat. Cell Biol., 2018, 20, 745–754 CrossRef CAS PubMed.
- J. Nunnari and A. Suomalainen, Cell, 2012, 148, 1145–1159 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, B. Li and Q. Y. He, Expert Rev. Proteomics, 2017, 14, 891–903 CrossRef CAS PubMed.
- H. Zhu, T. Tamura and I. Hamachi, Curr. Opin. Chem. Biol., 2019, 48, 1–7 CrossRef CAS PubMed.
- H. W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328–1331 CrossRef CAS PubMed.
- V. Hung, P. Zou, H. W. Rhee, N. D. Udeshi, V. Cracan, T. Svinkina, S. A. Carr, V. K. Mootha and A. Y. Ting, Mol. Cell, 2014, 55, 332–341 CrossRef CAS PubMed.
- V. Hung, S. S. Lam, N. D. Udeshi, T. Svinkina, G. Guzman, V. K. Mootha, S. A. Carr and A. Y. Ting, Elife, 2017, 6, e24463 CrossRef PubMed.
- T. C. Branon, J. A. Bosch, A. D. Sanchez, N. D. Udeshi, T. Svinkina, S. A. Carr, J. L. Feldman, N. Perrimon and A. Y. Ting, Nat. Biotechnol., 2018, 36, 880–887 CrossRef CAS PubMed.
- H. Li, A. M. Frankenfield, R. Houston, S. Sekine and L. Hao, J. Am. Soc. Mass Spectrom., 2021, 32, 2358–2365 CrossRef CAS PubMed.
- K. A. Hajj and K. A. Whitehead, Nat. Rev. Mater., 2017, 2, 17056 CrossRef CAS.
- Y. Yasueda, T. Tamura and I. Hamachi, Chem. Lett., 2016, 45, 265–267 CrossRef CAS.
- Y. Yasueda, T. Tamura, A. Fujisawa, K. Kuwata, S. Tsukiji, S. Kiyonaka and I. Hamachi, J. Am. Chem. Soc., 2016, 138, 7592–7602 CrossRef CAS PubMed.
- A. Fujisawa, T. Tamura, Y. Yasueda, K. Kuwata and I. Hamachi, J. Am. Chem. Soc., 2018, 140, 17060–17070 CrossRef CAS PubMed.
- K. Sakurai, Asian J. Org. Chem., 2015, 4, 116–126 CrossRef CAS.
- B. L. Stocker and M. S. Timmer, Chembiochem, 2013, 14, 1164–1184 CrossRef CAS PubMed.
- Y. Li, M. B. Aggarwal, K. Nguyen, K. Ke and R. C. Spitale, ACS Chem. Biol., 2017, 12, 2709–2714 CrossRef CAS PubMed.
- T. Ding, L. Zhu, Y. Fang, Y. Liu, W. Tang and P. Zou, Angew. Chem., Int. Ed. Engl., 2020, 59, 22933–22937 CrossRef CAS PubMed.
- E. Skovsen, J. W. Snyder, J. D. Lambert and P. R. Ogilby, J. Phys. Chem. B, 2005, 109, 8570–8573 CrossRef CAS PubMed.
- D. He, X. Xie, F. Yang, H. Zhang, H. Su, Y. Ge, H. Song and P. R. Chen, Angew. Chem., Int. Ed. Engl., 2017, 56, 14521–14525 CrossRef CAS PubMed.
- L. Li, J. Liang, H. Luo, K. M. Tam, E. C. M. Tse and Y. Li, Chem. Commun., 2019, 55, 12340–12343 RSC.
- Z. Huang, Z. Liu, X. Xie, R. Zeng, Z. Chen, L. Kong, X. Fan and P. R. Chen, J. Am. Chem. Soc., 2021, 143, 18714–18720 CrossRef CAS PubMed.
- J. Wang, Y. Liu, Y. Liu, S. Zheng, X. Wang, J. Zhao, F. Yang, G. Zhang, C. Wang and P. R. Chen, Nature, 2019, 569, 509–513 CrossRef CAS PubMed.
- T. Miki, M. Awa, Y. Nishikawa, S. Kiyonaka, M. Wakabayashi, Y. Ishihama and I. Hamachi, Nat. Methods, 2016, 13, 931–937 CrossRef CAS PubMed.
- S. Lee, C. Y. Chung, P. Liu, L. Craciun, Y. Nishikawa, K. J. Bruemmer, I. Hamachi, K. Saijo, E. W. Miller and C. J. Chang, J. Am. Chem. Soc., 2020, 142, 14993–15003 CrossRef CAS PubMed.
- H. Zhu, T. Tamura, A. Fujisawa, Y. Nishikawa, R. Cheng, M. Takato and I. Hamachi, J. Am. Chem. Soc., 2020, 142, 15711–15721 CrossRef CAS PubMed.
- H. Iwashita, E. Castillo, M. S. Messina, R. A. Swanson and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018513118 CrossRef CAS PubMed.
- D. Shen, W. Jin, Y. Bai, Y. Huang, H. Lyu, L. Zeng, M. Wang, Y. Tang, W. Wan, X. Dong, Z. Gao, H. L. Piao, X. Liu and Y. Liu, Angew. Chem., Int. Ed. Engl., 2021, 60, 16067–16076 CrossRef CAS PubMed.
- T. L. To, K. F. Medzihradszky, A. L. Burlingame, W. F. DeGrado, H. Jo and X. Shu, Bioorg. Med. Chem. Lett., 2016, 26, 3359–3363 CrossRef CAS PubMed.
- K. Nakane, S. Sato, T. Niwa, M. Tsushima, S. Tomoshige, H. Taguchi, M. Ishikawa and H. Nakamura, J. Am. Chem. Soc., 2021, 143, 7726–7731 CrossRef CAS PubMed.
- M. Muller, F. Grabnitz, N. Barandun, Y. Shen, F. Wendt, S. N. Steiner, Y. Severin, S. U. Vetterli, M. Mondal, J. R. Prudent, R. Hofmann, M. van Oostrum, R. C. Sarott, A. I. Nesvizhskii, E. M. Carreira, J. W. Bode, B. Snijder, J. A. Robinson, M. J. Loessner, A. Oxenius and B. Wollscheid, Nat. Commun., 2021, 12, 7036 CrossRef CAS PubMed.
- T. Tamura, M. Takato, K. Shiono and I. Hamachi, Chem. Lett., 2020, 49, 145–148 CrossRef CAS.
- H. Wang, Y. Zhang, K. Zeng, J. Qiang, Y. Cao, Y. Li, Y. Fang, Y. Zhang and Y. Chen, JACS Au, 2021, 1, 1066–1075 CrossRef CAS PubMed.
- J. Liu, Y. Zhou, X. Hou, C. Liu, B. Zhao, Y. Shan, Z. Sui, Z. Liang, L. Zhang and Y. Zhang, Anal. Chem., 2022, 94, 7637–7646 CrossRef CAS PubMed.
- J. T. Ngo, S. R. Adams, T. J. Deerinck, D. Boassa, F. Rodriguez-Rivera, S. F. Palida, C. R. Bertozzi, M. H. Ellisman and R. Y. Tsien, Nat. Chem. Biol., 2016, 12, 459–465 CrossRef CAS PubMed.
- W. Xu, Z. Zeng, J. H. Jiang, Y. T. Chang and L. Yuan, Angew. Chem., Int. Ed. Engl., 2016, 55, 13658–13699 CrossRef CAS PubMed.
- H. Huang, C. Dong, M. Chang, L. Ding, L. Chen, W. Feng and Y. Chen, Exploration, 2021, 1, 50–60 CrossRef.
- S. Kim, T. Tachikawa, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 11707–11715 CrossRef CAS PubMed.
- P. Di Mascio, G. R. Martinez, S. Miyamoto, G. E. Ronsein, M. H. G. Medeiros and J. Cadet, Chem. Rev., 2019, 119, 2043–2086 CrossRef CAS PubMed.
- R. A. Smith, R. C. Hartley and M. P. Murphy, Antioxid. Redox Signaling, 2011, 15, 3021–3038 CrossRef CAS PubMed.
- D. W. Lee, K. M. Park, M. Banerjee, S. H. Ha, T. Lee, K. Suh, S. Paul, H. Jung, J. Kim, N. Selvapalam, S. H. Ryu and K. Kim, Nat. Chem., 2011, 3, 154–159 CrossRef CAS PubMed.
- M. Beck, A. Schmidt, J. Malmstroem, M. Claassen, A. Ori, A. Szymborska, F. Herzog, O. Rinner, J. Ellenberg and R. Aebersold, Mol. Syst. Biol., 2011, 7, 549 CrossRef PubMed.
- L. Niu, L. Liu, S. Yang, J. Ren, P. B. S. Lai and G. G. Chen, Biochim. Biophys. Acta, Rev. Cancer, 2017, 1868, 564–570 CrossRef CAS PubMed.
- M. I. Hernandez-Alvarez and A. Zorzano, Cancers, 2021, 13, 2571 CrossRef CAS PubMed.
- F. Morel and C. Aninat, Drug Metab. Rev., 2011, 43, 281–291 CrossRef CAS PubMed.
- The Cancer Genome Atlas Research Network, Cell, 2017, 169, 1327–1341 CrossRef PubMed.
- J. Zhang, C. Xu, Y. Gao, Y. Wang, Z. Ding, Y. Zhang, W. Shen, Y. Zheng and Y. Wan, Front. Oncol., 2020, 10, 215 CrossRef PubMed.
- L. G. Higgins and J. D. Hayes, Drug Metab. Rev., 2011, 43, 92–137 CrossRef CAS PubMed.
- C. Mendez-Blanco, F. Fondevila, A. Garcia-Palomo, J. Gonzalez-Gallego and J. L. Mauriz, Exp. Mol. Med., 2018, 50, 1–9 CrossRef CAS PubMed.
- M. Colonna and O. Butovsky, Annu. Rev. Immunol., 2017, 35, 441–468 CrossRef CAS PubMed.
- B. Vezzani, M. Carinci, S. Patergnani, M. P. Pasquin, A. Guarino, N. Aziz, P. Pinton, M. Simonato and C. Giorgi, Biomolecules, 2020, 10, 1437 CrossRef CAS PubMed.
- M. T. Heneka, M. P. Kummer and E. Latz, Nat. Rev. Immunol., 2014, 14, 463–477 CrossRef CAS PubMed.
- H. Keren-Shaul, A. Spinrad, A. Weiner, O. Matcovitch-Natan, R. Dvir-Szternfeld, T. K. Ulland, E. David, K. Baruch, D. Lara-Astaiso, B. Toth, S. Itzkovitz, M. Colonna, M. Schwartz and I. Amit, Cell, 2017, 169, 1276–1290 CrossRef CAS PubMed.
- V. Bader and K. F. Winklhofer, Semin. Cell Dev. Biol., 2020, 99, 163–171 CrossRef CAS PubMed.
- E. L. Mills, B. Kelly and L. A. J. O'Neill, Nat. Immunol., 2017, 18, 488–498 CrossRef CAS PubMed.
- L. Peruzzotti-Jametti and S. Pluchino, Trends Mol. Med., 2018, 24, 838–855 CrossRef CAS PubMed.
- S. Galvan-Pena and L. A. O'Neill, Front. Immunol., 2014, 5, 420 Search PubMed.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS PubMed.
- T. A. Wynn and K. M. Vannella, Immunity, 2016, 44, 450–462 CrossRef CAS PubMed.
- R. Liu, X. Zhi and Q. Zhong, Autophagy, 2015, 11, 847–849 CrossRef CAS PubMed.
- Q. Tang, P. Gao, T. Arzberger, M. Hollerhage, J. Herms, G. Hoglinger and T. Koeglsperger, Cell Death Dis., 2021, 12, 854 CrossRef CAS PubMed.
- X. Ye, M. Zhu, X. Che, H. Wang, X. J. Liang, C. Wu, X. Xue and J. Yang, J. Neuroinflammation, 2020, 17, 18 CrossRef CAS PubMed.
- L. Gibellini, A. De Gaetano, M. Mandrioli, E. Van Tongeren, C. A. Bortolotti, A. Cossarizza and M. Pinti, Int. Rev. Cell Mol. Biol., 2020, 354, 1–61 CAS.
- H. Dong, Y. Feng, Y. Yang, Y. Hu, Y. Jia, S. Yang, N. Zhao and R. Zhao, Front. Cell Dev. Biol., 2021, 9, 726931 CrossRef PubMed.
- L. I. Grossman, N. Purandare, R. Arshad, S. Gladyck, M. Somayajulu, M. Huttemann and S. Aras, Oxid. Med. Cell Longev., 2017, 2017, 6739236 Search PubMed.
- M. Kim, J. Gwak, S. Hwang, S. Yang and S. M. Jeong, Oncogene, 2019, 38, 4729–4738 CrossRef CAS PubMed.
- G. Giordano, L. Tait, C. E. Furlong, T. B. Cole, T. J. Kavanagh and L. G. Costa, Free Radical Biol. Med., 2013, 58, 98–108 CrossRef CAS PubMed.
- L. G. Costa, R. de Laat, K. Dao, C. Pellacani, T. B. Cole and C. E. Furlong, Neurotoxicology, 2014, 43, 3–9 CrossRef CAS PubMed.
- P. Su, J. Zhang, D. Wang, F. Zhao, Z. Cao, M. Aschner and W. Luo, Neuroscience, 2016, 319, 155–167 CrossRef CAS PubMed.
- G. Lou, K. Palikaras, S. Lautrup, M. Scheibye-Knudsen, N. Tavernarakis and E. F. Fang, Trends Mol. Med., 2020, 26, 8–20 CrossRef CAS PubMed.
- M. Harland, S. Torres, J. Liu and X. Wang, J. Neurosci., 2020, 40, 1756–1765 CrossRef CAS PubMed.
- L. G. de Oliveira, Y. S. Angelo, A. H. Iglesias and J. P. S. Peron, Front. Immunol., 2021, 12, 624919 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc04087e |
| This journal is © The Royal Society of Chemistry 2022 |