
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne step synthesis of unsymmetrical 1,3-disubstituted BCP ketones via nickel/photoredox-catalyzed [1.1.1]propellane multicomponent dicarbofunctionalization†
Weichen
Huang
 a,
Sebastian
Keess
b and
Gary A.
Molander
*a
a,
Sebastian
Keess
b and
Gary A.
Molander
*a
aRoy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia 19104-6323, Pennsylvania, USA. E-mail: gmolandr@sas.upenn.edu
bMedicinal Chemistry Department, Neuroscience Discovery Research, AbbVie Deutschland GmbH & Co. KG, Ludwigshafen 67061, Germany
First published on 3rd October 2022
Abstract
Bicyclo[1.1.1]pentanes (BCPs), utilized as sp3-rich bioisosteres for tert-butyl- and aryl groups as well as internal alkynes, have gained considerable momentum in drug development programs. Although many elegant methods have been developed to access BCP amines and BCP aryls efficiently, the methods used to construct BCP ketones directly are relatively underdeveloped. In particular, the preparation of unsymmetrical 1,3-disubstituted-BCP ketones remains challenging and still requires multiple chemical steps. Herein, a single-step, multi-component approach to versatile disubstituted BCP ketones via nickel/photoredox catalysis is reported. Importantly, installing a boron group at the carbon position adjacent to the BCP structure bypasses the limitation to tertiary BF3K coupling partners, thus expanding the scope of this paradigm. Further transformation of disubstituted-BCP ketones into a variety of other BCP derivatives demonstrates the synthetic value of this developed method.
Three-dimensional (3D) molecular scaffolds have received considerable attention in drug molecular design to improve physicochemical properties of drug candidates.1 Among the promising 3D scaffolds in this area are the bicyclo[1.1.1]pentanes (BCPs), which serve as bioisosteres of aromatic rings as well as tert-butyl- and alkyne groups in medicinal chemistry.2 In Stepan's pioneering work,2a the replacement of the fluorinated aryl ring of a gamma secretase inhibitor with a BCP moiety resulted in improved permeability and kinetic solubility. Since this landmark work, the number of patents published with BCP-containing drugs has skyrocketed. Despite considerable interest from the medicinal chemistry community, the incorporation of BCPs into specific structural classes found in bioactive molecules remains an unsolved challenge.
BCP ketones could be considered as bioisosteres of aryl ketones, which widely exist in FDA-approved drugs (Fig. 1A).3 They can also be used as vehicles for the synthesis of other important BCP derivatives, including BCP amides and BCP esters through efficient transformations. Nevertheless, the methods that are used to construct BCP ketones efficiently are relatively underdeveloped, especially compared with well-developed approaches to access amino BCPs and aryl BCPs (Fig. 1B).4 Specifically, the Wiberg,5a Walsh,5b and Pan5c groups have reported methods for acylation of [1.1.1]propellane with aldehydes to form monosubstituted-BCP ketones. In contrast, the preparation of unsymmetrically 1,3-disubstituted-BCP ketones remains challenging and still requires multiple chemical steps. For example, Wills and coworkers reported a method for the synthesis of BCP ketones by reacting [1.1.1]propellane and Grignard reagents, followed by addition to an aldehyde and oxidation with MnO2 (Fig. 2A).6a This method requires the use of metal reagents and multiple synthesis steps, which are incompatible with the construction of complex targets containing sensitive functional groups. The Knochel group developed a similar two-step strategy to construct 1,3-disubstituted BCP ketones by opening the [1.1.1]propellane with allylzinc halides, followed by addition to acyl chlorides (Fig. 2A).6b However, this method is only suitable for some special organozinc reagents, which limits the diversity of the BCP ketones. Chemists at SpiroChem also reported a two-step method for construction of 1,3-disubstituted BCP ketones through a process involving radical addition to [1.1.1]propellane, followed by engagement with different arylmetal reagents (Fig. 2A).6c In this case, the other substituent on the BCP ring is limited to an ester functional group. Furthermore, there are some individual examples showing that disubstituted BCP ketones can be obtained from the corresponding BCP redox active ester. Specifically, the Ohmiya group developed the N-heterocyclic carbene-catalyzed acylation of BCP redox active ester, but the yield was only 20% (Fig. 2A).6d The Yuan group also conducted the cross-coupling of BCP redox active esters with pyridyl esters to access BCP ketones (Fig. 2A).6e Considering the five-step synthesis of BCP ketones from [1.1.1]propellane, these methods cannot meet the requirements of rapid synthesis of a library of products in the medicinal chemistry setting. Clearly, the drawbacks of stepwise synthetic approaches to 1,3-disubstituted BCP ketones hamper the broad application of bicyclo[1.1.1]pentanes. Thus, more efficient methods for the preparation of disubstituted BCP ketones are urgently needed.
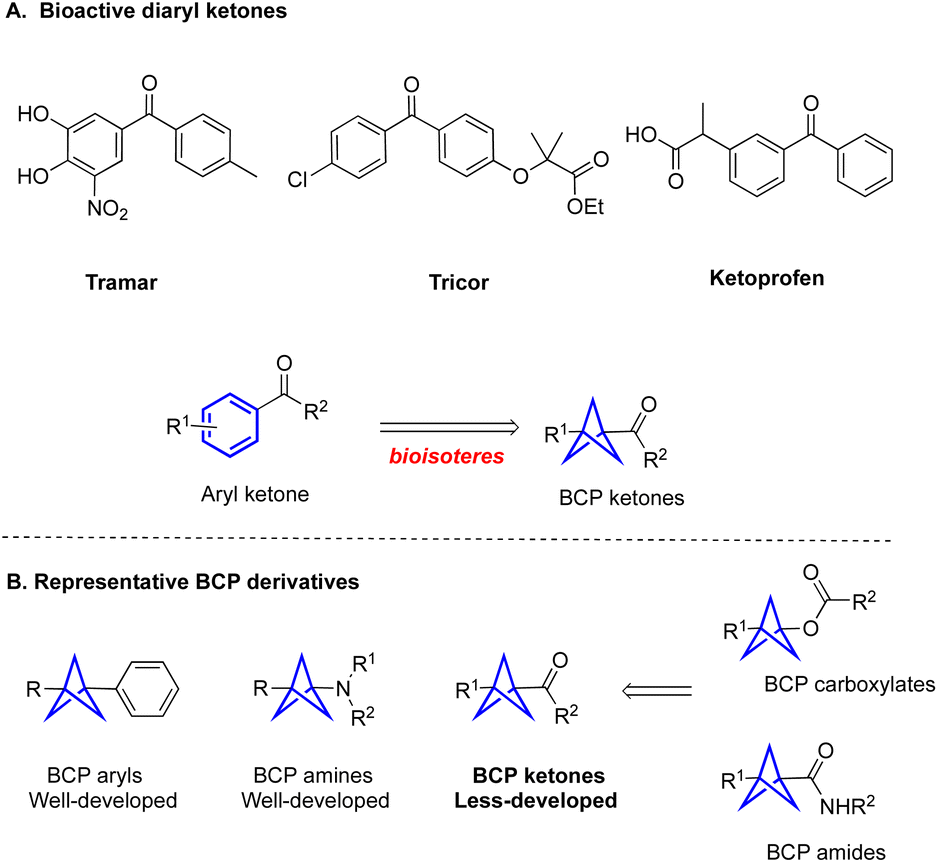 | ||
| Fig. 1 (A) Examples of bioactive diaryl ketones. (B) Representative BCP derivatives. | ||
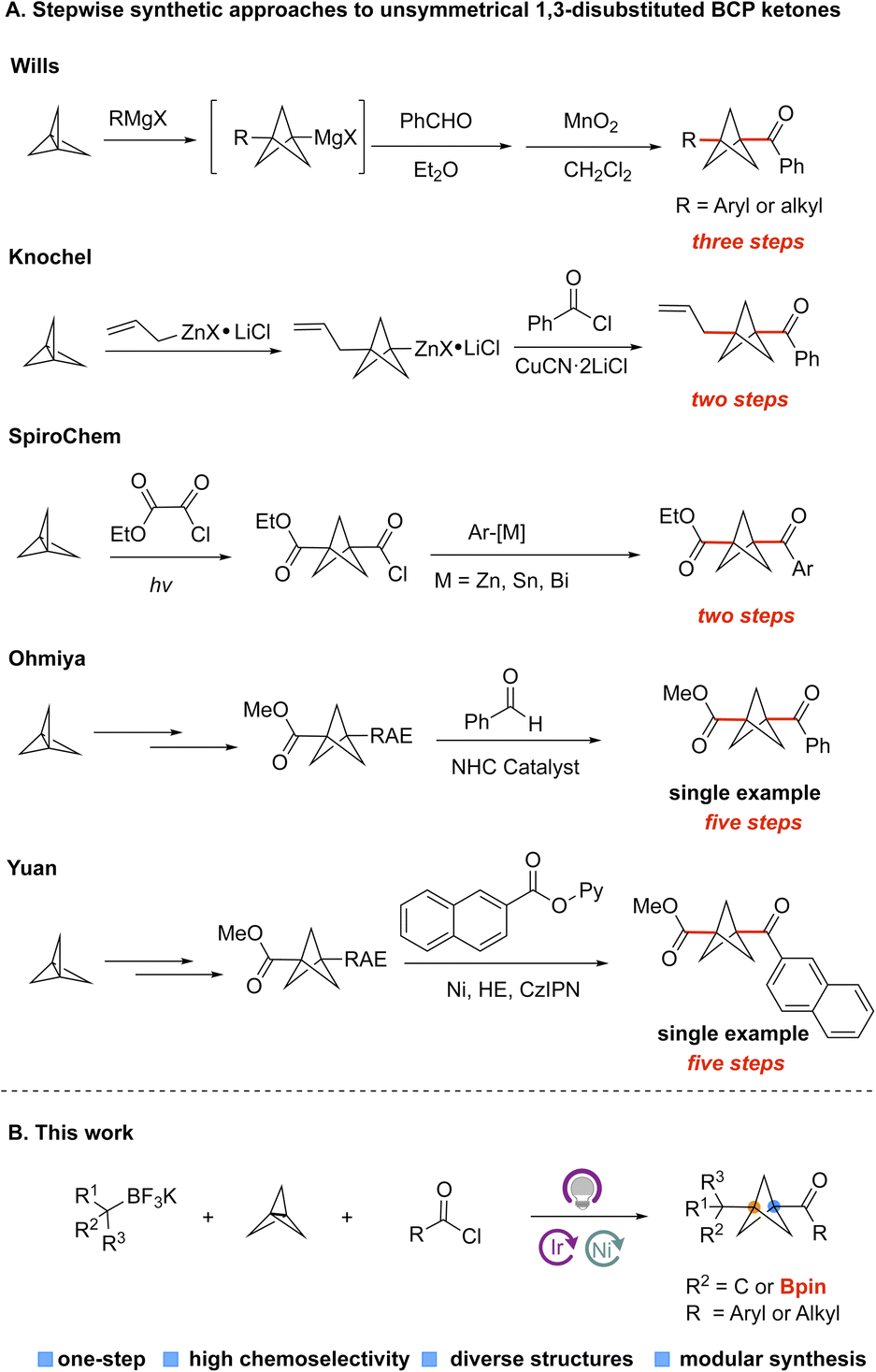 | ||
| Fig. 2 (A) Previous strategies to access unsymmetrically 1,3 disubstituted BCP ketones. (B) Research reported herein. HE = Hantzsch ester; RAE = Redox active ester [N-(acyloxy)phthalimide]; NHC = N-heterocyclic carbene; CzIPN = 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene. | ||
Multicomponent reactions (MCRs) that allow one-step access to complex and diverse disubstituted BCP products are synthetically advantageous to current stepwise approaches to BCP derivatives. However, achieving such a transformation is still challenging because of competing two-component coupling or propellane oligomerization. Uchiyama,7a MacMillan,7b and our group7c,d have successfully developed multi-component approaches to versatile BCP derivatives based on the differentiated reactivity of BCP radicals and substrate alkyl radicals. In our previous report,7d we successfully took advantage of the slow capture of tertiary radicals by Ni species as a key mechanistic aspect to achieve a one-step, multicomponent reaction for the synthesis of BCP-aryl derivatives. Meanwhile, our group has successfully developed an efficient photoredox/Ni dual catalysis paradigm for transition metal-catalyzed cross-couplings of alkylboron- or alkylsilicon reagents with various electrophiles, including aryl halides, acyl chlorides, alkenyl halides, and isocyanates based on a single-electron transfer (SET) transmetalation pathway.8 Inspired by these results, we questioned whether acyl chlorides or other electrophiles could also serve as partners in the three-component radical coupling of [1.1.1]propellane to access a diverse array of BCP derivatives of high importance in the pharmaceutical industry. Herein we report a one-step, three-component radical coupling of [1.1.1]propellane to afford diversely functionalized bicycles using various electrophiles.
To determine the chemoselectivity of the proposed MCR pathway, the reactivity of tertiary alkyl and BCP radicals in the nickel/photoredox-catalyzed cross-couplings with acyl chlorides was first examined (Fig. 3). The results indicated that BCP bridgehead radicals engage the nickel catalyst to enter the cross-coupling catalytic cycle, generating the product BCP ketone, while acyclic tertiary radicals do not take part in this catalytic cycle. Encouraged by this promising reactivity pattern, we explored the possibility of achieving a multi-component reaction forging two C–C bonds in a single operation using [1.1.1]propellane.
 | ||
| Fig. 3 Control experiments. | ||
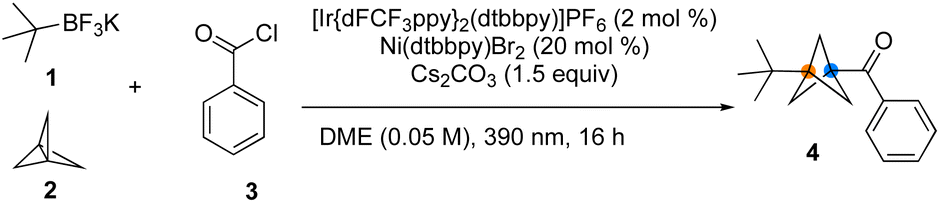
Initial investigations utilized t-BuBF3K, [1.1.1]propellane, and benzoyl chloride as a model reaction to optimize the reaction conditions (Table 1). Reaction screening indicated that the reaction was best performed with [Ir(dFCF3ppy)2dtbbpy]PF6 (2.0 mol%), Ni(dtbbpy)Br2 (20 mol%), and Cs2CO3 (1.5 equiv.) in DME (0.05 M) irradiated by a 390 nm LED light at room temperature for 16 h (Table 1, entry 1). The selected results of these studies led to the conditions provided in Table 1. As expected, control studies confirmed that this MCR was indeed dual catalytic in nature (Table 1, entries 8−10) and that all the components of the reaction were necessary to ensure successful difunctionalization of [1.1.1]propellane. Notably, initial trials with tertiary alkyl carboxylates as radical precursors proved unsuccessful in this MCR process.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | NMR yield (%) |
| a Optimization of reaction conditions: 1 (0.15 mmol), 2 (0.3 mmol), 3 (0.10 mmol) under purple Kessil irradiation (λ max = 390 nm) for 16 h at rt; NMR yield was calculated using 1,3,5-trimethoxybenzene as an internal standard (IS) from the crude mixture. | ||
| 1 | None | 63 |
| 2 | No base | 32 |
| 3 | 0.01 M | 52 |
| 4 | 0.025 M | 55 |
| 5 | 427 mm | 25 |
| 6 | 2 mol% [Ir] cat. 10 mol% [Ni] | 49 |
| 7 | 2 mol% [Ir] cat. 20 mol% [Ni] | 58 |
| 8 | No [Ni] catalyst | 0 |
| 9 | No [Ir] catalyst | 0 |
| 10 | No light | 0 |
| 11 | t-BuCOOCs, instead of 1 | 0 |
With suitable conditions in hand, the generality of this metallaphotoredox protocol with respect to a broad range of aliphatic- and aromatic acyl chlorides was investigated. As summarized in Fig. 4, both electron-rich and electron-poor aromatic acyl chlorides were coupled under the developed reaction conditions with 28−64% yields (4–15). For example, aromatic acyl chlorides containing common functional groups such as ether (5), fluoro (6), chloro (7), trifluoromethoxy (8), cyano (10) and trifluoromethylthio (12) proved to be suitable. Heteroaromatic acyl chlorides (14, 15) also react smoothly to afford the desired product in acceptable yield. Furthermore, the success of the reaction with ethyl succinyl chloride (19), which was not compatible utilizing previous methods employing metal reagents, further demonstrates the functional group compatibility of this protocol. Notably, alkyl bromide or -chloride handles (20, 21) have been incorporated, thus enabling further modification by substitution. Finally, other electrophiles including isocyanates and alkenyl halides (24, 25) have been embedded within the substrates, although the efficiency is not ideal in these cases.
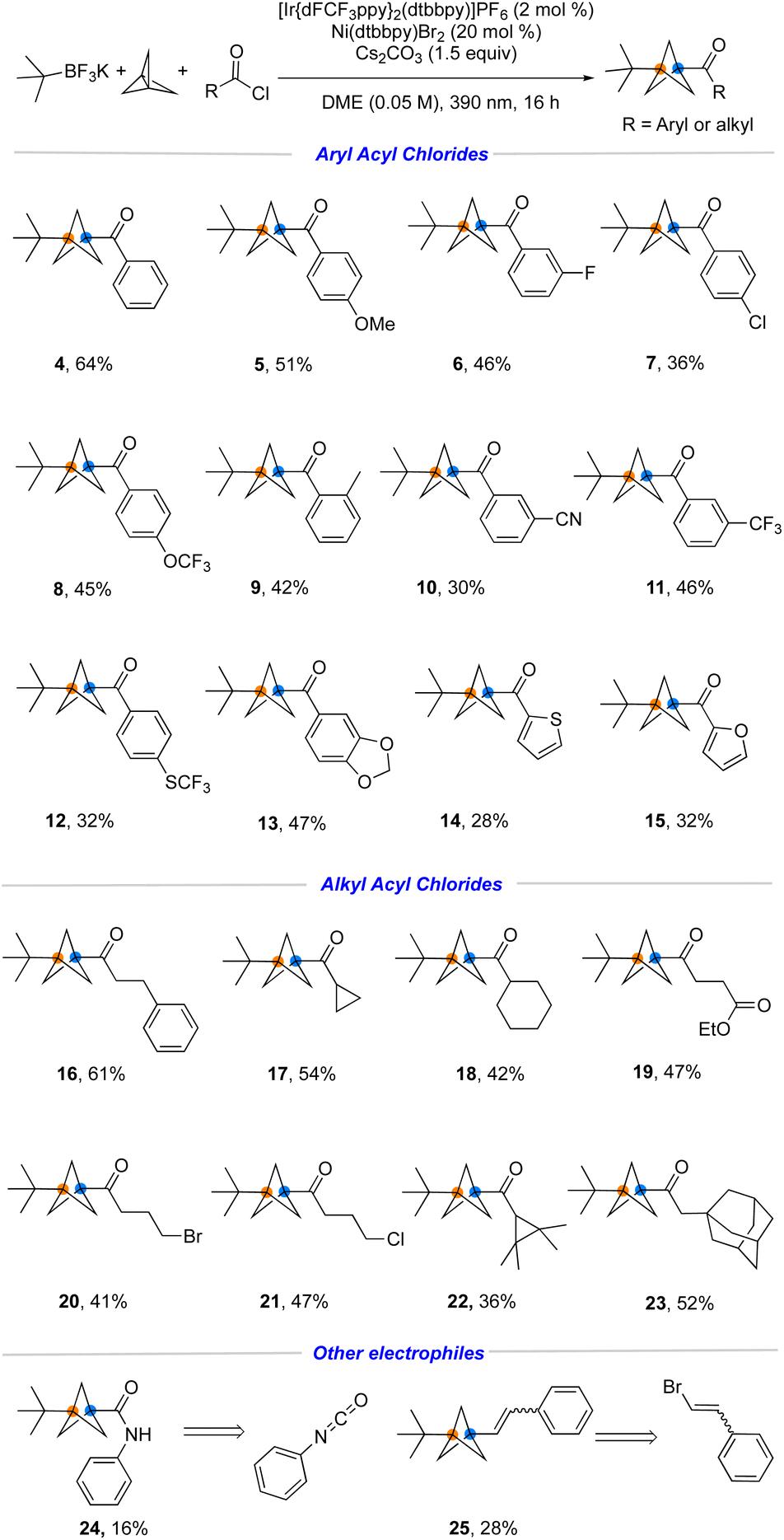 | ||
| Fig. 4 Scope of aliphatic and aromatic acyl chloride substrates. Reaction conditions: acyl chlorides (0.30 mmol, 1.0 equiv.), [1.1.1]propellane (0.90 mmol, 3.0 equiv.), alkyltrifluoroborates (0.45 mmol, 1.5 equiv.), [Ir(dFCF3ppy)2dtbbpy]PF6 (2 mol%, 0.006 mmol), Ni(dtbbpy)Br2 (20 mol%, 0.06 mmol), Cs2CO3 (1.5 equiv., 0.45 mmol), DME (0.05 M), irradiating with purple Kessil irradiation (λ max = 390 nm) for 16 h at rt. | ||
To explore the generality of this transformation further, a variety of structurally diverse tertiary- and secondary alkyltrifluoroborates were investigated using the developed conditions. Thanks to the development of powerful synthetic methods, tertiary boronate esters9 are quite readily available from diverse feedstocks including carboxylic acids, alkenes, alkyl halides, and ketones.10 As demonstrated in Fig. 5, ester-, nitrile-, ketone-, alkene-, and even hydroxyl-containing trifluoroborates were incorporated into the established protocol (26–32). These sensitive functional groups would be difficult to integrate within previously reported synthetic methods, especially those using metal reagents as the coupling partner. Additionally, alkyltrifluoroborates possessing various ring sizes reacted smoothly to afford the coupled products (33–39). Interestingly, the secondary radical derived from a benzyltrifluoromethyl-substituted alkyltrifluoroborate was engaged in this MCR process (40), with no evidence for formation of the two-component product. Aliphatic acyl chlorides were also tested and found to be compatible with the reaction conditions, affording the corresponding products 41–43. Finally, we applied this method to the late-stage modification of drug-like molecules. Several alkyltrifluoroborate-containing natural products and drug scaffolds were incorporated under the standard conditions to afford the desired products in moderate to good yields (44–48), demonstrating the applicability of the developed method in complex molecular settings.
 | ||
| Fig. 5 Scope of aliphatic- and aromatic acyl chloride substrates. Reaction conditions: acyl chlorides (0.30 mmol, 1.0 equiv.), [1.1.1]propellane (0.90 mmol, 3.0 equiv.), alkyltrifluoroborates (0.45 mmol, 1.5 equiv.), [Ir(dFCF3ppy)2dtbbpy]PF6 (2 mol%, 0.006 mmol), Ni(dtbbpy)Br2 (20 mol%, 0.06 mmol), Cs2CO3 (1.5 equiv., 0.45 mmol), DME (0.05 M), irradiating with purple Kessil irradiation (λ max = 390 nm) for 16 h at rt. | ||
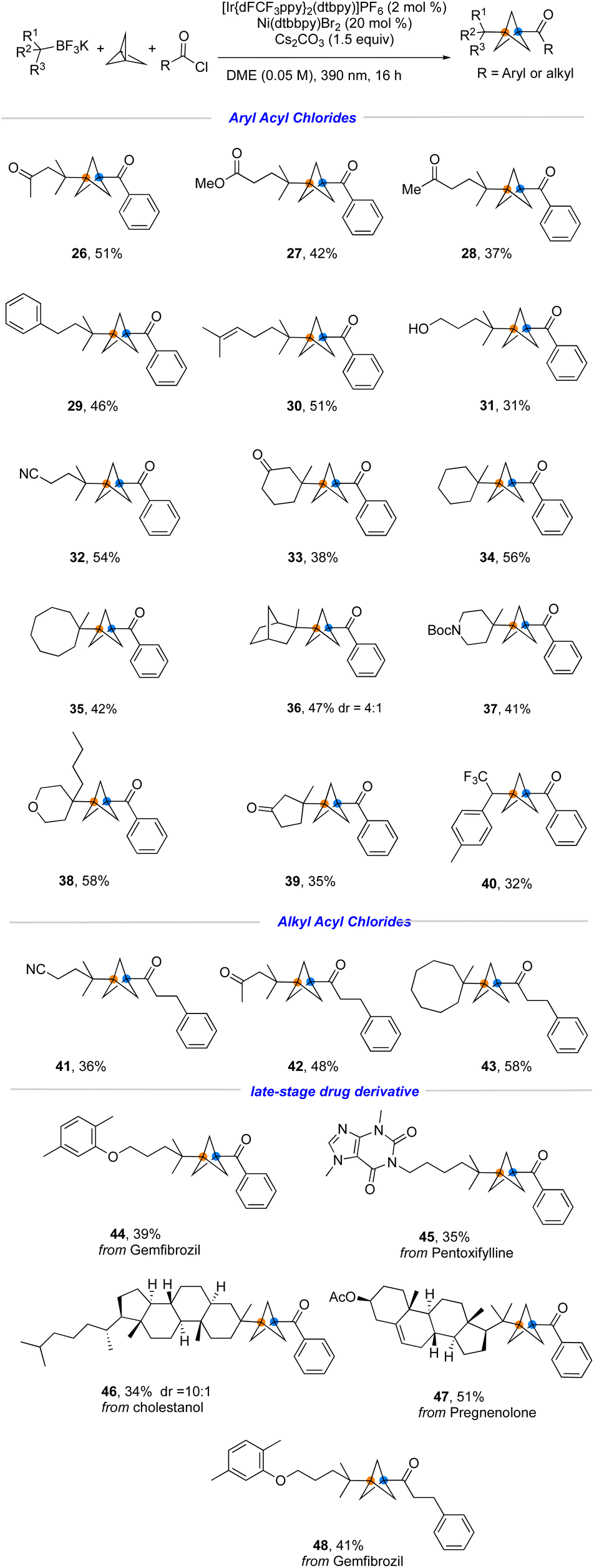
Although the established one-step, three-component radical coupling enabled by nickel/photoredox dual catalysis provides an efficient method for rapid construction of disubstituted BCP ketones, this protocol was only applicable to tertiary radicals or a specific secondary radical that limits its generality. To resolve this issue, we examined the feasibility of incorporating a substituent on the carbon adjacent to the BF3K group that would serve as a versatile surrogate group. As an example, if a Bpin group was installed into this position, it could be proto-deborylated or even further manipulated in downstream transformations, greatly expanding the scope of the overall process. The Masarwa group reported a method for the desymmetrization of gem-diborylalkanes,11 allowing ready access to the requisite trifluoroborates. Gratifyingly, when the desymmetrized 1,1-dibora substrate was subjected to the developed reaction conditions with an aromatic- and aliphatic acyl chloride, the desired products 49 and 50 were formed in good yield (Fig. 6A).
 | ||
| Fig. 6 (A) the Synthesis of β-Bpin-substituted BCP ketones. (B) Further transformations. | ||
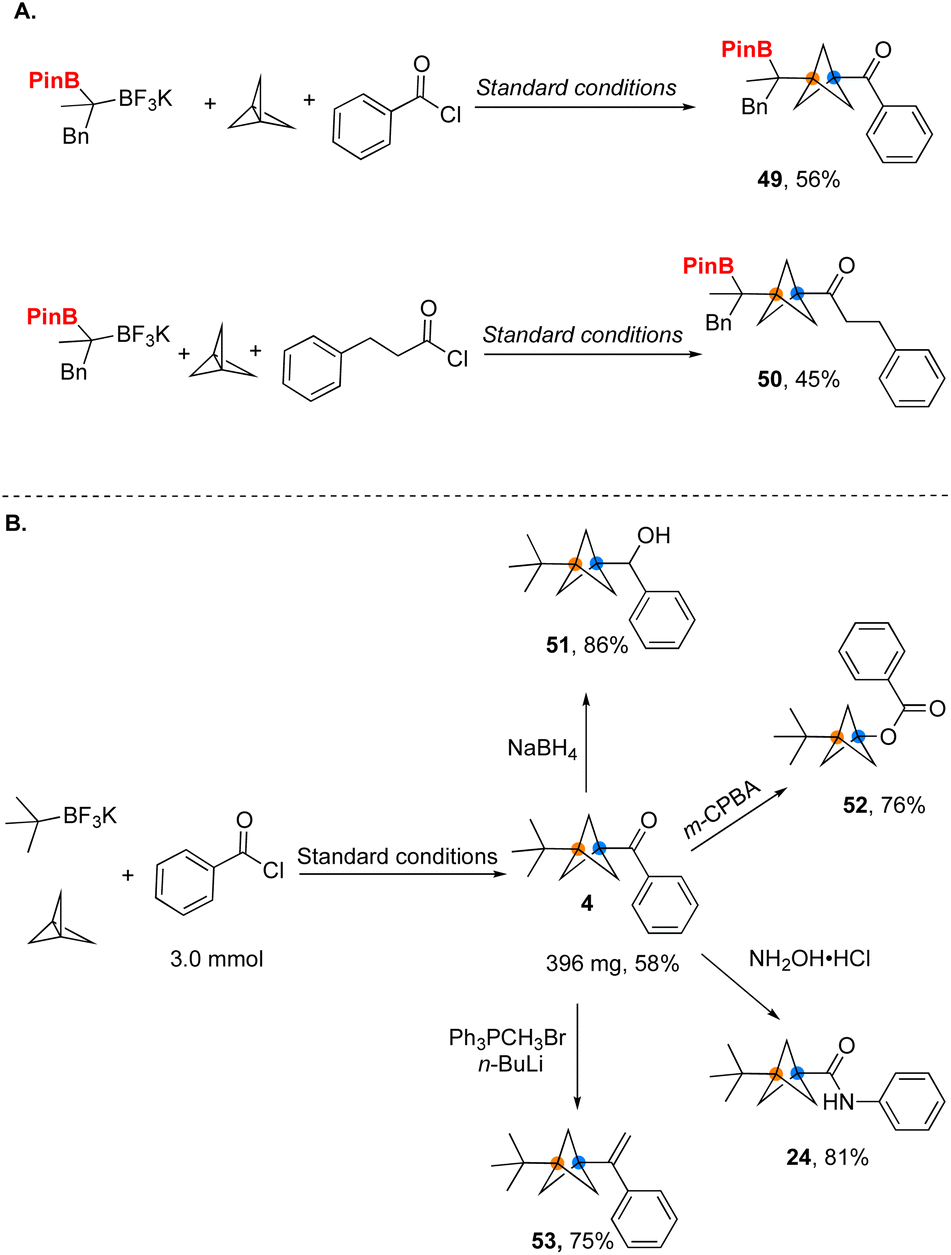
Ketones have long been used as important intermediates to provide access to other functional groups. We have utilized the efficient access to BCP ketones provided by the method developed herein to demonstrate their conversion into a variety of diverse BCP substructures, including carboxylates and amides through classical functional group interconversions (Fig. 6). Considering the lack of efficient methods for synthesis of such building blocks, the current protocol takes on added significance. Reduction of ketones with NaBH4 produces the corresponding secondary alcohol 51 with a good yield. By using a Baeyer–Villiger oxidation, the corresponding BCP carboxylate 52 was formed. Alternatively, the ketone was further transformed into BCP amide 24via a Beckmann rearrangement. Finally, a BCP ketone was used to generate the corresponding alkene in 75% yield through a Wittig olefination.
To gain insights into the reaction mechanism, we conducted a series of control experiments. First, competition experiments demonstrated that a tertiary radical participates in the three-component reaction exclusively, while a secondary radical was only involved in the classical cross-coupling reaction (Fig. 7A). TEMPO trapping experiments showed that the reaction was completely suppressed in the presence of this reagent, and only TEMPO adducts 56 derived from the radical precursors were observed (Fig. 7B). The reaction of the alkyltrifluoroborate generated from verbenone under the standard conditions afforded ring-opened product 58 (Fig. 7C).12 Therefore, the radical nature of the MCR process was confirmed. Based on these results and previous reports,10 a plausible reaction mechanism for this dual nickel/photoredox catalyzed three-component cross-coupling is depicted in Fig. 7D. Initially, under light irradiation, the photocatalyst is excited to provide *Ir(III). The alkyltrifluoroborates reductively quench the excited photocatalyst *Ir(III) to generate tertiary alkyl radical V. Because the metal–carbon bond between the nickel center and tertiary alkyl group is quite fragile, the acyclic tertiary radical favorably dissociates from the Ni(III) center to form free alkyl tertiary radicals.13 Tertiary alkyl radical V undergoes irreversible radical addition to [1.1.1]propellane, leading to BCP radical VI, which is then trapped by Ni(0), forming an alkyl Ni(I) species VIII. Subsequently, VIII undergoes rapid oxidative addition with acyl chlorides. Alternatively, as shown in blue, BCP radical VI can also be captured by Ni(II) oxidative addition complex IX. Both pathways lead to Ni(III) complex X, which subsequently undergoes rapid and productive C–C bond formation to yield the BCP ketone products.
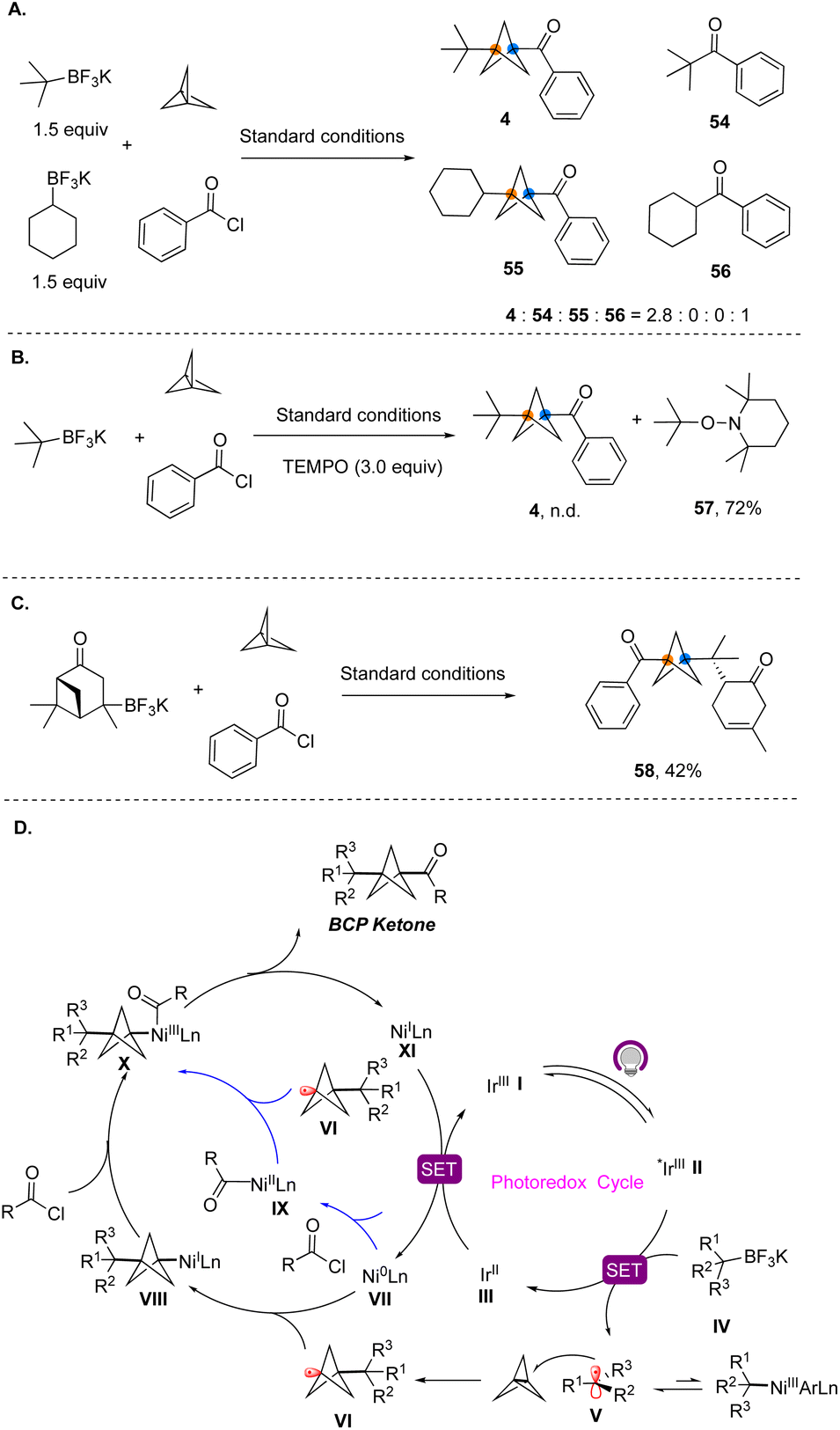 | ||
| Fig. 7 Mechanistic study. (A) Secondary versus tertiary radical competition (the ratio was determined by GC-MS analysis). (B) Radical-trapping experiment. (C) Radical ring-opening reaction. (D) Proposed mechanism. | ||
In conclusion, the multi-component radical cross-coupling reaction involving [1.1.1]propellane reported herein enables rapid access to a diverse array of disubstituted BCP ketones and offers an expedient alternative to traditional routes for the synthesis of BCP ketones via pre-functionalization of [1.1.1]propellane. The method exhibits several advantages over previously reported routes, including excellent chemoselectivity, mild reaction conditions, and good functional group tolerance. Importantly, the usefulness of this method is further boosted by installing boronate esters (Bpin) at the carbon adjacent to the BCP substructure, which in principle could be protodeborylated or even further manipulated in downstream transformations. Overall, the reaction described herein enables access to unprecedented BCP structures of interest to the organic chemistry synthetic community, especially in the drug discovery sector.
Data availability
The ESI† includes all experimental details, including optimization of the synthetic method, synthesis and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products reported are included as well.Author contributions
Weichen Huang conceived the chemistry and carried out the experiments under the guidance and mentorship of Professor Molander and Dr Keess. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Dr Charles W. Ross, III (University of Pennsylvania) for obtaining HRMS data. We acknowledge Kessil Lighting for lights used in this study. The authors are grateful for the financial support from NIH General Medical Sciences (R35 GM 131680 to G. M.). An SIOC fellowship to W. H. from the Shanghai Institute of Organic Chemistry (CAS) is also gratefully acknowledged. Financial support for this research was also provided in part by AbbVie. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplements awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology, supported the purchase of the NMRs used in this study.References
- (a) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471 RSC; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS; (c) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC; (d) F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- (a) A. F. Stepan, C. Subrama-nyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414–3442 CrossRef CAS PubMed; (b) M. V. Westphal, B. T. Wolfstadter, J.-M. Plancher, J. Gatfield and E. M. Carreira, ChemMedChem, 2015, 10, 461–469 CrossRef CAS PubMed; (c) N. D. Measom, K. D. Down, D. J. Hirst, C. Jamieson, E. S. Manas, V. K. Patel and D. O. Somers, ACS Med. Chem. Lett., 2017, 8, 43–48 CrossRef.
- (a) K. K. Koh, S. H. Han, M. J. Quon, J. Y. Ahn and E. K. Shin, Diabetes Care, 2005, 28, 1419–1424 CrossRef CAS PubMed; (b) M. Kurth, C. Adler, M. S. Hilaire, C. Singer, C. Waters, P. LeWitt, D. Chernik, E. Dorflinger and K. Yoo, Neurology, 1997, 48, 81–87 CrossRef CAS; (c) M. W. Walter, Nat. Prod. Rep., 2002, 19, 278–291 RSC.
- For selected reviews of BCP synthesis, see: (a) J. Kanazawa and M. Uchiyama, Synlett, 2019, 30, 1–11 CrossRef CAS; (b) X. Ma and L. N. Pham, Asian J. Org. Chem., 2020, 9, 8–22 CrossRef CAS; (c) F.-S. He, S. Xie, Y. Yao and J. Wu, Chin. Chem. Lett., 2020, 31, 3065–3072 CrossRef CAS; (d) J. M. Anderson, N. D. Measom, J. A. Murphy and D. L. Poole, Angew. Chem., Int. Ed., 2021, 60, 24754–24769 CrossRef CAS PubMed.
- Methods reported to access monosubstituted-BCP ketones: (a) K. B. Wiberg and S. T. Waddell, J. Am. Chem. Soc., 1990, 112, 2194–2216 CrossRef CAS; (b) N. Trongsiriwat, Y. Pu, Y. Nieves-Quinones, R. A. Shelp, M. C. Kozlowski and P. J. Walsh, Angew. Chem., Int. Ed., 2019, 58, 13416–13420 CrossRef CAS PubMed; (c) Q. Li, L. Li, Q. L. Xu and F. Pan, Org. Lett., 2022, 24, 4292–4297 CrossRef CAS.
- Methods reported to access unsymmetrically 1,3-disubsitituted BCP Ketones (a) V. K. Vyas, G. j. Clarkson and M. Wills, Org. Lett., 2021, 23, 3179–3183 CrossRef CAS; (b) K. Schwärzer, H. Zipse, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 20235–20241 CrossRef PubMed; (c) Y. Suzuki, D. Jimenez-teja, C. Salome and T. Fessard, WO2017157932 A1, Sep 21, 2017; (d) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS; (e) X. Xi, Y. Luo, W. Li, M. Xu, H. Zhao, Y. Chen, S. Zheng, X. Qi and W. Yuan, Angew. Chem., Int. Ed., 2022, 61, e202114731 CAS.
- (a) J. Kanazawa, K. Maeda and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 17791–17794 CrossRef CAS PubMed; (b) X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220–226 CrossRef CAS PubMed; (c) W. Dong, E. Yen-Pon, L. Li, A. Bhattacharjee, A. Jolit and G. A. Molander, Nat. Chem., 2022, 14, 1068–1077 CrossRef CAS; (d) W. Huang, S. Keess and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 12961–12969 CrossRef CAS.
- (a) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed; (b) J. Amani, E. Sodagar and G. A. Molander, Org. Lett., 2016, 18, 732–735 CrossRef CAS PubMed; (c) J. Amani and G. A. Molander, J. Org. Chem., 2017, 82, 1856–1863 CrossRef CAS PubMed; (d) N. R. Patel, C. B. Kelly, M. Jouffroy and G. A. Molander, Org. Lett., 2016, 18, 764–767 CrossRef CAS; (e) S. Zheng, D. N. Primer and G. A. Molander, ACS Catal., 2017, 7, 7957–7961 CrossRef CAS PubMed; (f) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS.
- (a) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CrossRef CAS; (b) S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed; (c) G. A. Molander, J. Org. Chem., 2015, 80, 7837–7848 CrossRef CAS PubMed; (d) G. A. Molander and L. Jean-Gerard, Org. React., 2012, 79, 1–316 Search PubMed.
- Selected methods reported to access tertiary boronate esters: (a) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (b) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef PubMed; (c) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed; (d) G. A. Molander and S. A. McKee, Org. Lett., 2011, 13, 4684–4687 CrossRef CAS; (e) Y. Yang, J. Tsien, A. Ben David, J. M. E. Hughes, R. R. Merchant and T. Qin, J. Am. Chem. Soc., 2021, 143, 471–480 CrossRef CAS.
- N. Kumar, R. R. Reddy and A. Masarwa, Chem. - Eur. J., 2019, 25, 8008–8012 CrossRef CAS PubMed.
- A. Fernandez-Mateos, P. H. Teijon and R. R. Gonzalez, Tetrahedron, 2011, 67, 9529–9534 CrossRef CAS.
- (a) M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS; (b) L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, J. Am. Chem. Soc., 2020, 142, 20390–20399 CrossRef CAS; (c) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069–20078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05100a |
| This journal is © The Royal Society of Chemistry 2022 |