
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed alkoxycarbonylation of unactivated alkyl chlorides†
Peng
Wang
,
Yaxin
Wang
,
Helfried
Neumann
 * and
Matthias
Beller
*
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse e. V., Albert-Einstein-Straße 29a, Rostock 18059, Germany. E-mail: helfried.neumann@catalysis.de; matthias.beller@catalysis.de
First published on 8th November 2022
Abstract
A general rhodium-catalyzed selective carbonylative coupling of unactivated alkyl chlorides with aliphatic alcohols or phenols to the corresponding esters is presented for the first time. Crucial for this transformation is the addition of sodium iodide, which provides in situ more active alkyl iodides. In the presence of a Rh(I)-DPPP catalyst system diverse esters (81 examples) including industrially relevant acetates from chloro- and dichloromethane can be prepared in a straightforward manner in up to 95% isolated yield. The used ligand not only affects the selectivity of the carbonylation reaction but also controls the selectivity of the preceding halide exchange step.
Introduction
Palladium-catalyzed carbonylative coupling reactions, especially of (hetero)aryl halides have become a powerful tool for the preparation of all kinds of (hetero)aryl carboxylic acid derivatives including esters, amides, ketones and so on.1 Compared to these methods the related carbonylation of alkyl halides is still in its infancy despite the industrial importance of such reactions, e.g. acetic acid production via methanol carbonylation in the presence of iodide (Monsanto and Cativa processes).2 In fact, most work in this area has been performed in industry from the mid-1960s on, when Monsanto introduced cis-[Rh(CO)2I2]− as an active catalyst for methanol carbonylation. In contrast, in academia only few carbonylations of alkyl iodides (Scheme 1a) and bromides (Scheme 1b) were realized and alkyl chlorides as substrates remained a challenge for such transformations until to date.3,4 Early carbonylative coupling reactions of alkyl iodides required either high pressure of CO (50 atm) at elevated temperatures or intense irradiation was needed.5 As an example of such photocatalytic transformations, Ryu and co-workers reported a Pd–carbene catalyzed and photo-induced atom transfer carbonylation of alkyl iodides using ionic liquids as reaction media.6 More recently, in 2019, Gong and co-workers reported copper-catalyzed and indium-mediated methoxycarbonylation of unactivated alkyl iodides with CO at ambient pressure. This procedure is suitable for carbonylation of primary, secondary, and tertiary alkyl iodides as substrates, thereby also allowing the formation of esters with quaternary carbon centers.7 In 2017, the group of Mankad reported a bimetallic Cu/Mn catalyzed carbonylative Suzuki–Miyaura coupling with alkyl iodides.8 The catalytic system showed an excellent tolerance for both aryl boronic ester nucleophiles and primary/secondary alkyl iodide electrophiles. Soon after, the same group reported Cu-catalysed hydroxymethylation of alkyl iodides with CO to produce one-carbon-extended alcohols,9 as well as carbonylative silylations of alkyl iodides to give alkyl-substituted acylsilanes in high yield.10 | ||
| Scheme 1 Selected metal-catalyzed alkoxycarbonylations of alkyl halides with alcohols and phenols. | ||
Notably, several functional groups are tolerated under the mild conditions for these two reactions. Already in 1984, the carbonylative coupling of alkyl bromides (Scheme 1b) and organoborates was disclosed by Alper and co-workers utilizing palladium and rhodium catalysts.11 More recently, this transformation could be improved according to Alexanian and co-workers by employing palladium/carbene complexes with strongly electron-donating NHC ligands under mild conditions (2 atm CO, 50 °C). Mechanistic investigations support the proposed organometallic–radical pathway.12 In 2020, Wu and co-workers reported another interesting rhodium-catalyzed carbonylative coupling of alkyl bromides and iodides with phenols at low CO pressure.13 Their catalytic system (RhCl3/DPPP) exhibited high functional group tolerance and the corresponding aryl esters were obtained in good to excellent yields. In the same year, they further described a copper/iron co-catalyzed alkoxycarbonylation of unactivated primary, secondary, and tertiary alkyl bromides to give the corresponding aliphatic esters in good yields.14
Inspired by the latter works, we herein report the first rhodium-catalyzed alkoxycarbonylation of primary alkyl chlorides with different O-nucleophiles in the presence of sodium iodide to produce diverse esters at low CO pressure with high conversion and selectivity (Scheme 1c). Apart from controlling the metal-catalyzed carbonylation step the used DPPP ligand facilitates specifically the preceding halide exchange reaction.
Results and discussion
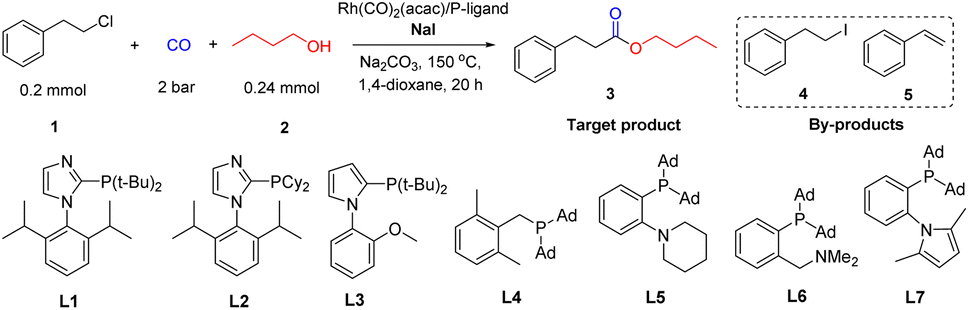
The rhodium-catalyzed alkoxycarbonylation of 2-chloroethylbenzene with n-butanol was investigated as a non-activated model system, while carbonylations of more activated substrates such benzyl and allyl chlorides are established.15a,b At the start of this work, we studied the activity of different phosphine ligands in the presence of Rh(acac)(CO)2. This commercially available complex is a typical precursor for Rh-catalyzed carbonylations.16 The initial experiments were carried out at comparably low CO pressure (2 bar) to avoid formation of halide-containing carbonyl clusters, which are known to be less active for the insertion into C–X (X = Cl) bonds.17a–c To promote this difficult reaction step and to avoid HX formation resulting in corrosion problems, NaI and Na2CO3 were added to the mixture. As shown in Table 1, in the absence of phosphine ligand only formation of 1-iodo-2-phenylethane (4) and styrene (5) are observed as by-products (entry 1). In contrast, using PPh3 as ligand led to the desired carbonylative product 3 in 68% yield; however still significant amounts of by-products were detected (entry 2). Different kinds of sterically hindered and electron-rich alkyl phosphines as well as aryl/alkyl phosphines gave worse results (entries 3–11). Similarly, privileged bidentate ligands such as Xantphos, DPPE, and DPPF, which have been found to allow carbonylation of aryl chlorides in other systems,17d–i led to mainly by-products (entries 12, 13 and 15). In contrast, DPPB and DPPP performed better and the latter ligand provided the desired product in 86% isolated yield (entries 14 and 16). Notably, the presence of base and iodide are both crucial for the success of the carbonylative coupling reaction (entries 17–18).13–18 The selectivity towards 3 decreased when reducing the amount of catalyst precursor and NaI (entries 19–20). The influence of different reaction parameters including variations of the metal precursor, additives, base, and solvents is shown in Table S1 (ESI†).|
|
|||||
|---|---|---|---|---|---|
| Entry | P-ligand | Convb. (%) | Selb. 3 (%) | Selb. 4 (%) | Selb. 5 (%) |
| a 2-Chloroethylbenzene (0.2 mmol), n-butanol (0.24 mmol; 1.2 eq.), 5 mol% Rh(acac)(CO)2, S/Rh mol ratio = 20, 15 mol% P-ligand, [Rh]/P-ligand molar ratio = 1/6, Na2CO3 (0.24 mmol; 1.2 eq.), NaI (0.20 mmol; 1.0 eq.), CO 2 bar, N2 20 bar, 150 °C, solvent 0.5 mL, 20 h. b Determined by GC and GC-MS, n-dodecane (C12) as internal standard. c Isolated yield is 86%. d Without Na2CO3. e Without NaI. f 1 mol% Rh(acac)(CO)2, S/Rh mol ratio = 100, 3 mol% P-ligand. g NaI (0.04 mmol; 0.2 eq.). h 100 °C. | |||||
| 1 | — | 64 | 0 | 95 | 5 |
| 2 | PPh3 | 99 | 68 | 12 | 20 |
| 3 | P(t-Bu)3 | 99 | 11 | 71 | 18 |
| 4 | BuPAd2 | 99 | 16 | 73 | 11 |
| 5 | L1 | 40 | 0 | 92 | 8 |
| 6 | L2 | 84 | Trace | 92 | 7 |
| 7 | L3 | 99 | 10 | 37 | 53 |
| 8 | L4 | 82 | 2 | 92 | 6 |
| 9 | L5 | 79 | 0 | 92 | 8 |
| 10 | L6 | 99 | 6 | 82 | 12 |
| 11 | L7 | 61 | Trace | 95 | 5 |
| 12 | Xantphos | 99 | 26 | 12 | 62 |
| 13 | DPPE | 99 | 10 | 72 | 18 |
| 14 | DPPB | 99 | 73 | 19 | 8 |
| 15 | DPPF | 99 | 6 | 80 | 14 |
| 16c | DPPP | 99 | 91 | 0 | 9 |
| 17d | DPPP | 90 | 13 | 80 | 7 |
| 18e | DPPP | 9 | 0 | 0 | 100 |
| 19f | DPPP | 99 | 34 | 57 | 9 |
| 20g | DPPP | 18 | 17 | 62 | 21 |
| 21h | DPPP | 16 | 0 | 74 | 26 |
Mechanistic investigations and control experiments
According to our mechanistic proposal, initially the model substrate 1-chloro-2-phenylethane (1) will be transformed to 1-iodo-2-phenylethane (4) (Scheme 2A). In general, the conversion of alkyl chlorides to alkyl iodides proceeds only under special conditions, e.g., using microwave or the traditional Finkelstein reaction.19,20 Indeed, without catalyst and additives present only 24% of 4 is observed (entry 1 in Scheme 2A). When Rh(acac)(CO)2 or Na2CO3 were added into the reaction, similar conversion of the substrate is detected (28–30% of 4). Under the standard conditions in the presence of catalyst and additives 4 is obtained in only 9%. Here, styrene (5) is detected in 86% yield because of base mediated β-elimination. Thus, without Na2CO3 the selectivity towards 4 is greatly improved (78%). | ||
| Scheme 2 Mechanistic studies. | ||
However, much to our surprise, even using the ligand DPPP alone the chlorine/iodide exchange reaction proceeded in excellent yield and selectivity (entry 6 in Scheme 2A). Thus after 2 h reaction time, in the presence of the phosphine ligand significant conversion is observed and the proposed reactive intermediate 4 is obtained in high selectivity (entries 7–12 in Scheme 2A). Apparently, the phosphine ligand not only stabilizes the catalytically active metal center for the carbonylation step, but also promotes the chlorine–iodide exchange reaction. Indeed, various phosphines promote the formation of 4 under reaction conditions similar to catalytic experiments. Notably, depending on the phosphine ligand the selectivity towards 4 varied and DPPP gave the best results for the halide exchange reaction (entries 13–15 vs. 6 in Scheme 2A).
In previous studies Wu and coworkers described the negative effect of the free radical scavenger TEMPO on the carbonylation of alkyl bromides/iodides (Scheme 2B).13 We observed a similar behavior (TEMPO, 1.0–3.0 equiv.), which is explained by simple nucleophilic substitution reaction of TEMPO with 4.
To proof that the reaction does not occur through well-known alkoxycarbonylation of styrene as reaction intermediate the corresponding reaction was performed under the standard conditions. As shown in Scheme 2C no conversion took place, which basically excludes styrene as an active intermediate. Finally, the reaction of 1-iodo-2-phenylethane gave the desired product in 93% yield, which confirmed this compound as a key intermediate (Scheme 2D).
To gain more insight into the mechanism and to identify the rate-determining step of the reaction, a kinetic profile of the model reaction was carried out under the standard conditions. As shown in Fig. 1, in the first 2 h, 30% of (2-chloroethyl)benzene is converted; however, only small amounts of the target product 3 (6%) were detected. At this time the major product is 4. As the reaction continued, the conversion of substrate and the selectivity of the product increased gradually, while 4 decreased steadily. After 20 h, complete conversion is achieved and the selectivity of the ester 3 increased to 91%. During the whole process minor amounts (10%) of styrene (5) are formed via classic β-elimination of 1 or 4. In this reaction step, the base is consumed, and the elimination process becomes slower. Based on all these results and previous reports,13–16 the following reaction mechanism is proposed: as shown in Scheme 3, the reaction starts by nucleophilic substitution of the alkyl chloride with NaI to form the corresponding alkyl iodide.
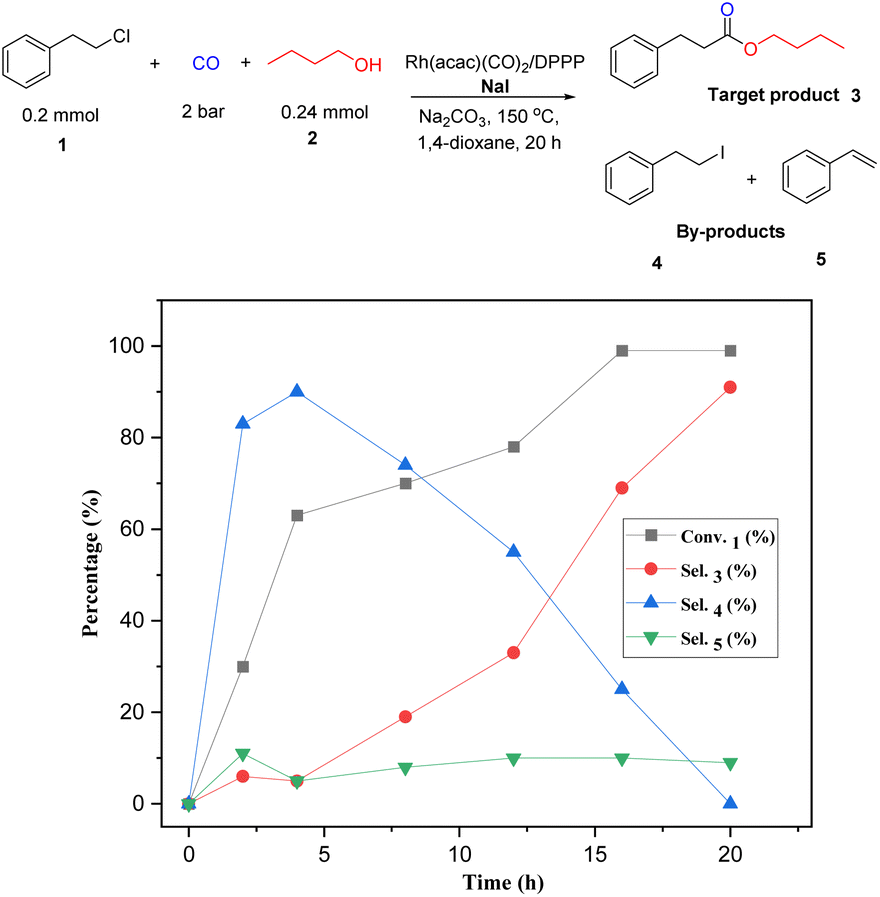 | ||
| Fig. 1 Kinetic profile of the rhodium-catalyzed alkoxycarbonylation of (2-chloroethyl)benzene with CO and n-butanol (determined by GC, n-dodecane as internal standard). | ||
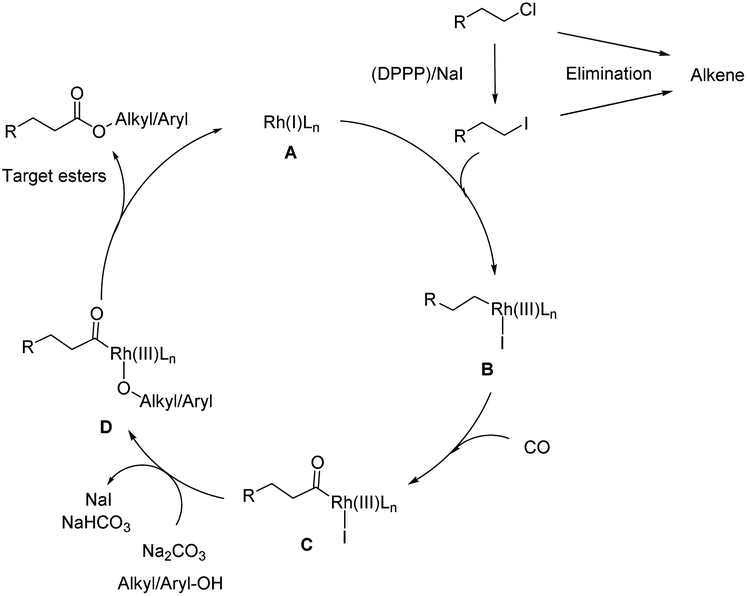 | ||
| Scheme 3 Plausible mechanism for the alkoxycarbonylation of unactivated alkyl chlorides with alkyl/aryl-OH. | ||
Then, oxidative addition of the alkyl iodide with the active Rh(I) complex gives the alkyl Rh(III) intermediate B followed by CO coordination and insertion to give the alkyl Rh complex C. Finally, nucleophilic attack by different alcohols or phenols forms the target esters along with the active Rh(I) species left for the next catalytic cycle with the assistance of base Na2CO3. Notably, olefins, which can be easily formed by β-elimination are not active intermediates but represent a dead end in this process (Scheme S1†).
Substrate scope and synthetic applications
Under the optimal conditions (0.2 mmol alkyl-Cl, 0.24 mmol alcohol, 1.2 eq., 5.0 mol% Rh(acac)(CO)2, 15 mol% DPPP, 0.20 mmol NaI, 1.0 eq., 0.24 mmol Na2CO3, 1.2 eq., CO 2 bar, N2 20 bar, 150 °C, 0.5 mL 1,4-dioxane, 20 h) different alcohols and alkyl chlorides were tested to showcase the substrate scope of this novel methodology. At first, different primary and secondary aliphatic alcohols were selected. As shown in Scheme 4, 2-chloroethylbenzene is conveniently transformed to the corresponding esters in moderate to very good yields (6–23). In general, aliphatic alcohols reacted well with 1 to give the target esters 6–21 in high yield. Apparently, the presence of electron-withdrawing (e.g. fluoro) or electron-donating (e.g. alkoxyl) substituents didn't have a strong influence on the yield of the products (56–83%, 15–21). | ||
| Scheme 4 Alkoxycarbonylation of unactivated alkyl chlorides with aliphatic alcohols using Rh(acac)(CO)2/DPPP/NaI catalytic system. a Alkyl chlorides (0.2 mmol), alkyl-OH (0.24 mmol), Rh(acac)(CO)2 (0.01 mmol), DPPP (0.03 mmol), NaI (0.2 mmol), Na2CO3 (0.24 mmol), CO 2 bar, N2 20 bar, 150 °C, 1,4-dioxane 0.5 mL, 20 h; b determined by GC and GC-MS (n-dodecane was used as internal standard), isolated yield was calculated. c Chloroethane as substrate, 2.0 mol L−1 in THF. | ||
Compared to primary aliphatic alcohols, secondary ones can be also transformed into the corresponding esters 22–23, albeit in lower yield. Interestingly, unsaturated alcohols 24–27 reacted selectively to the corresponding esters (66–83% yield) without affecting the double bond. This distinct this methodology from many other carbonylations as Rh complexes are known to be excellent catalysts for carbonylations of olefins.16,21 Notably, even highly reactive allyl alcohol as coupling partner showed very good chemoselectivity and the catalyst specifically activates the C–Cl bond (70% yield of 27). Apart from the model substrate other linear alkyl chlorides provided the respective esters 28–32 in high yields (80–90%; for other substrates see Scheme 6).
Apart from aliphatic alcohols, phenols represent interesting oxygen nucleophiles.22–24 Despite the lower nucleophilicity the carbonylation reaction of (2-chloroethyl)benzene proceeded smoothly under identical conditions (for variations of different metal precursors and additives see Table S2†). As shown in Scheme 5, several alkyl phenol esters were obtained in good to very good yields. Exemplarily, phenyl 3-phenylpropanoate (33) was isolated in 87% yield. Similarly, utilizing 1-, or 2-naphthol (34, 35) as well as alkyl chlorides containing biphenyl groups (36–38) the desired products were obtained in 75–82% yield. Interestingly, using 2-hydroxycarbazole as nucleophilic coupling reagent selective ester formation took place without affecting the N–H group and 39 was achieved in 85%. Furthermore, phenols containing electron-withdrawing (fluoro, cyano, acyl) or electron-donating (methyl, tert-butyl, alkoxyl) groups (40–53) were converted to the target products in high yields (71–82%). At the same time, different linear alkyl chlorides (54-57) provided the corresponding alkyl aryl esters in good to high yields (79–91%).
 | ||
| Scheme 5 Alkoxycarbonylation of unactivated alkyl chlorides with phenols using Rh(acac)(CO)2/DPPP/NaI catalytic system. a Alkyl chlorides (0.2 mmol), aryl-OH (0.24 mmol), Rh(acac)(CO)2 (0.01 mmol), DPPP (0.03 mmol), NaI (0.2 mmol), Na2CO3 (0.24 mmol), CO 2 bar, N2 20 bar, 150 °C, 1,4-dioxane 0.5 mL, time 20 h; b determined by GC and GC-MS (n-dodecane was used as internal standard), isolated yield was calculated. c Chloroethane as substrate, 2.0 mol L−1 in THF. | ||
The selective carbonylation of alkyl chlorides offers the possibility for interesting applications both for the modification of bioactive molecules as well as the synthesis of bulk chemicals. As an example, chlorination or oxychlorination of methane to give chloromethane is well precedented as a viable route for functionalization of this notoriously difficult substrate.24 As shown in Scheme 6a, the carbonylation of CH3Cl allows for the synthesis of acetic acid esters, which are commercially very important. Overall, this constitutes a formal two-step process for valorization of methane in which the halide can be recycled.25 Gratifyingly, the carbonylation of chloromethane proceeded smoothly and we obtained different acetic acid esters (58–62) in good to very good yields from 69–92%. Next, the carbonylation of dichloromethane (CH2Cl2) with different aliphatic alcohols was investigated. Surprisingly, under the standard set of conditions only the monocarbonylation products were observed, and the second C–Cl bond is hydrogenated. This result is explained by the fast hydrogenation of the intermediate chloroacetate. It is well known that Rh catalysts show excellent activity in many hydrogenation reactions. After the first carbonylation step, the second C–Cl bond becomes activated for this hydrogenation reaction. Thus, in the presence of our catalytic system acetic acid esters (63–69) are obtained in yields from 40–80% without further optimization.
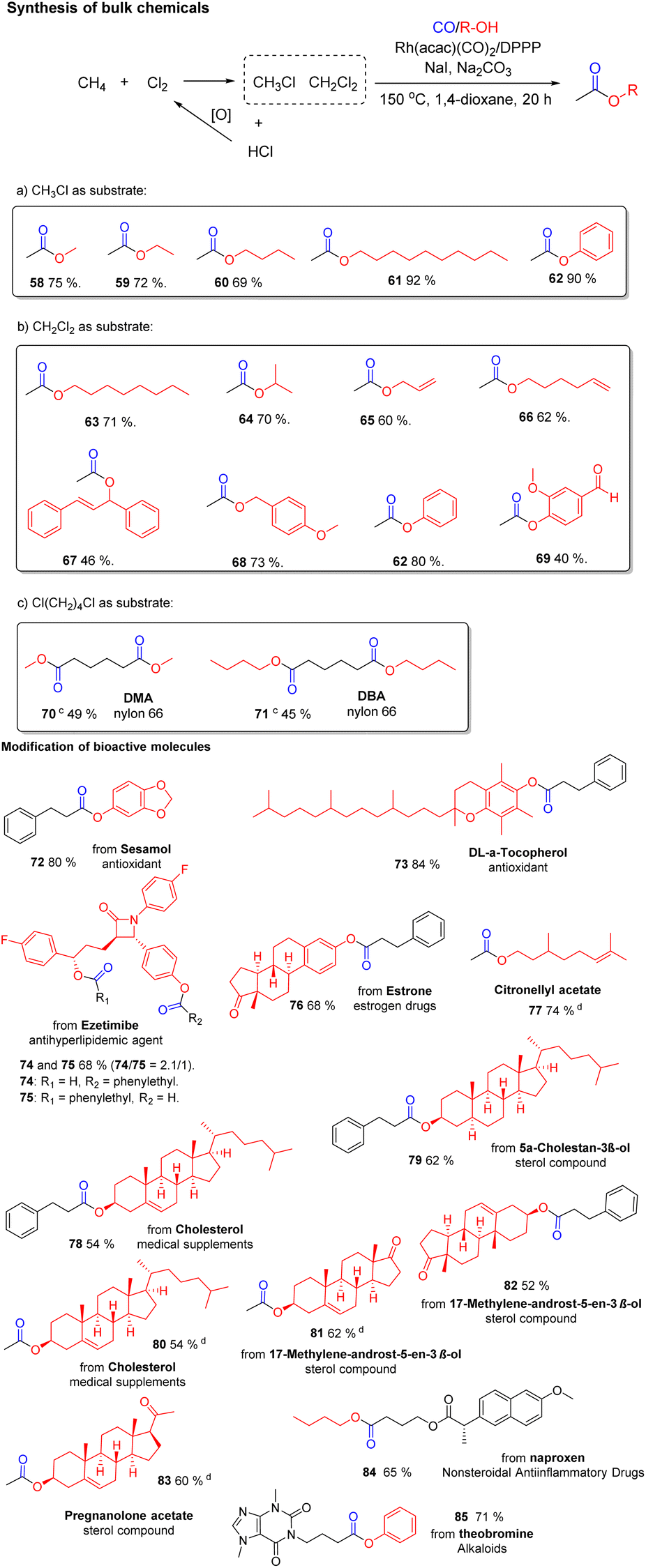 | ||
| Scheme 6 Selected applications: Synthesis of bulk chemicals and modification of bioactive molecules. a Alkyl chlorides (0.2 mmol), aryl-OH or alkyl-OH (0.24 mmol), Rh(acac)(CO)2 (0.01 mmol), DPPP (0.03 mmol), NaI (0.2 mmol), Na2CO3 (0.24 mmol), CO 2 bar, N2 20 bar, 150 °C, 1,4-dioxane 0.5 mL, 20 h; b isolated yield was calculated. c Twice the amount of catalyst. d Chloromethane as substrate. | ||
This unexpected finding is interesting as chlorination of methane provides mixtures of chlorinated products, which then do not need to be further purified. Indeed, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH3Cl and CH2Cl2 can be directly carbonylated to provide selectively decyl acetate (61) in 80% yield. To the best of our knowledge, this is the first example of such selective carbonylation of mixtures of alkyl chlorides. In addition, preliminary tests on the alkoxycarbonylation of chloroform (CHCl3) with octanol were performed. Under standard conditions we obtained 63 in 43% and octyl chloroacetate in 45% yield. Even highly inert tetrachloromethane showed some reactivity under these conditions and provided decyl chloroacetate according to GC-MS. Other important industrial esters like dimethyl adipate (70) and dibutyl adipate (71) were also synthesized following our protocol and provided the corresponding esters in 45–49% yield.
:1 mixture of CH3Cl and CH2Cl2 can be directly carbonylated to provide selectively decyl acetate (61) in 80% yield. To the best of our knowledge, this is the first example of such selective carbonylation of mixtures of alkyl chlorides. In addition, preliminary tests on the alkoxycarbonylation of chloroform (CHCl3) with octanol were performed. Under standard conditions we obtained 63 in 43% and octyl chloroacetate in 45% yield. Even highly inert tetrachloromethane showed some reactivity under these conditions and provided decyl chloroacetate according to GC-MS. Other important industrial esters like dimethyl adipate (70) and dibutyl adipate (71) were also synthesized following our protocol and provided the corresponding esters in 45–49% yield.
Apart from the synthesis of current products of the chemical industry, this methodology can be applied for functionalization of bioactive molecules to the respective esters (72–85) in 52–84% yield. As shown in Scheme 6, antioxidants Sesamol and Vitamin E were applied to the reaction and 72 and 73 were obtained in 80–84% yield. Other pharmaceuticals such as Ezetimibe and Estrone also delivered the corresponding carbonylation products 74–76. In case of Ezetimibe carbonylation of both the aryl-OH and alkyl-OH are observed (74/75 = 2.1/1). Citronellyl acetate (77) was synthesized in 74% yield. Natural products with secondary alcohol groups are tolerated by our catalytic system and gave 78–83 in yields from 54–62%. In addition, naproxen, one of the most important non-steroidal anti-inflammatory drugs, is smoothly carbonylated to give 84 in 65% yield. Finally, theobromine as an example of alkaloids was applied to the reaction and 85 was obtained in 71% yield.
Experimental
In a typical experiment (2-chloroethyl)benzene (0.2 mmol), Rh(acac)(CO)2 (0.01 mmol), DPPP (0.03 mmol), NaI (0.20 mmol), n-butanol (0.2 mmol) and Na2CO3 (0.24 mmol) were added into 1,4-dioxane (0.5 mL) sequentially. The obtained mixture in a 50 mL stainless steel autoclave was purged with CO (2 bar) for three times and pressured to 2 bar, then N2 was flushed into the reactor (20 bar). To prevent the solvent from being ejected from the reaction system, 20 bar N2 is needed. The reaction mixture was stirred at the appointed temperature for 20 h. Upon completion, the autoclave was cooled down to room temperature and depressurized carefully. The reaction solution was analyzed by GC and GC-MS to determine the conversions and the chemical selectivity (n-dodecane (C12) as internal standard), then the separation yield was obtained by column chromatography and 1H NMR and 13C NMR to identify the structure of the products.Conclusions
In conclusion, the first low-pressure rhodium-catalyzed alkoxycarbonylation of unactivated alkyl chlorides has been developed. Under the optimized standard conditions a variety of different alcohols and phenols can be used in this carbonylative transformation and various esters were prepared with high chemical/regioselectivity. Carbonylation of mixtures of industrially relevant chloromethane allows for a selective synthesis of acetates, which paths the way for a two-step methane to acetic acid derivatives. From a mechanistic point of view, the addition of NaI and the use of a Rh-DPPP catalyst are crucial for the success of the carbonylation process. Apart from stabilization of the metal center the specific ligand also promotes the chlorine–iodide exchange reaction as shown by detailed control experiments.Author contributions
Peng Wang developed this chemistry, synthesized the most compounds and has written a draft. Yaxin Wang gave support in chemical syntheses with regard to referee report. Helfried Neumann has rewrote and corrected the draft. Matthias Beller has written the final version of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the BMBF and the State of Mecklenburg-Vorpommern for financial and general support. We gratefully acknowledge the National Natural Science Foundation of China (No. 21901250). We thank the analytical team of the Leibniz-Institut für Katalyse e. V., especially Sandra Leiminger for their excellent service.Notes and references
- (a) Y. B. David, M. Portnoy and D. Milstein, J. Am. Chem. Soc., 1989, 111, 8742 CrossRef; (b) W. Mägerlein, A. F. Indolese and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 2856 CrossRef; (c) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed; (d) D. A. Watson, X. X. Fan and S. L. Buchwald, J. Org. Chem., 2008, 73, 7096 CrossRef CAS PubMed; (e) X. F. Wu, RSC Adv., 2016, 6, 83831 RSC; (f) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979 CrossRef CAS PubMed; (g) P. L. L. Tremblay, A. Fabrikant and B. A. Arndtsen, ACS Catal., 2018, 8, 5350 CrossRef; (h) Z. P. Yin, J. X. Xu and X. F. Wu, ACS Catal., 2020, 10, 6510 CrossRef CAS; (i) J. B. Peng, H. Q. Geng and X. F. Wu, Chem, 2019, 5, 526 CrossRef CAS; (j) P. Wang, J. Yang, K. K. Sun, H. Neumann and M. Beller, Org. Chem. Front., 2022, 9, 2491–2497 RSC.
- (a) G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293 CrossRef CAS; (b) J. R. Zoeller, V. H. Agreda, S. L. Cook, N. L. Lafferty, S. W. Polichnowski and D. M. Pond, Catal. Today, 1992, 13, 73 CrossRef CAS; (c) A. W. Budiman, J. S. Nam, J. H. Park, R. I. Mukti, T. S. Chang, J. W. Bae and M. J. Choi, Catal. Surv. Asia, 2016, 20, 173 CrossRef CAS.
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563 CrossRef CAS PubMed.
- J. B. Peng, H. Q. Geng and X. F. Wu, Chem, 2019, 5, 526 CAS.
- (a) H. Urata, H. Maekawa, S. Takahashi and T. Fuchikami, J. Org. Chem., 1991, 56, 4320 CrossRef CAS; (b) T. Fukuyama, S. Nishitani, T. Inouye, K. Morimoto and I. Ryu, Org. Lett., 2006, 8, 1383 CrossRef CAS PubMed; (c) A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Chem.–Eur. J., 2012, 18, 9415 CrossRef CAS PubMed; (d) T. Kondo, Y. Tsuji and Y. Watanabe, Tetrahedron Lett., 1988, 29, 3833 CrossRef CAS; (e) T. Kondo, Y. Sone, Y. Tsuji and Y. Watanabe, J. Organomet. Chem., 1994, 473, 163 CrossRef CAS; (f) T. Ishiyama, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1991, 32, 6923 CrossRef; (g) Y. P. Jia, Y. N. Cui, J. M. Yin, G. Y. Zhou, S. M. Li, D. B. Gao and X. S. Wang, Chin. Chem. Lett., 2010, 21, 1033 CrossRef CAS; (h) H. W. Wang, Y. P. Jia, D. B. Gao, J. M. Yin, G. Y. Zhou and S. M. Li, Chin. Chem. Lett., 2007, 18, 795 CrossRef CAS.
- T. Fukuyama, T. Inouye and I. Ryu, J. Organomet. Chem., 2007, 692, 685 CrossRef CAS.
- Y. C. Chen, L. Su and H. G. Gong, Org. Lett., 2019, 21, 4689 CrossRef CAS PubMed.
- D. R. Pye, L. J. Cheng and N. P. Mankad, Chem. Sci., 2017, 8, 4750 RSC.
- S. L. Zhao and N. P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 5867 CrossRef CAS PubMed.
- L. J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2020, 142, 80 CrossRef CAS PubMed.
- K. E. Hashem, J. B. Woell and H. Alper, Tetrahedron Lett., 1984, 25, 4879 CrossRef CAS.
- B. T. Sargent and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 7520 CrossRef CAS PubMed.
- H. J. Ai, H. Wang, C. L. Li and X. F. Wu, ACS Catal., 2020, 10, 5147 CrossRef CAS.
- Y. H. Li and X. F. Wu, Commun. Chem., 2018, 1, 39 CrossRef.
- (a) P. Wang, Z. S. Cao, Y. X. Wang, H. Neumann and M. Beller, Eur. J. Org. Chem., 2022, 30, e202200663 Search PubMed; (b) M. Foà, F. Francalanci, A. Gardano, G. Cainelli and A. Umani-Ronchi, J. Organomet. Chem., 1983, 248, 225 CrossRef.
- (a) X. Y. Ren, Z. Y. Zheng, L. Zhang, Z. Wang, C. G. Xia and K. L. Ding, Angew. Chem., Int. Ed., 2017, 56, 310 CrossRef CAS PubMed; (b) X. Wang and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 19080 CrossRef CAS PubMed; (c) V. Agabekov, W. Seiche and B. Breit, Chem. Sci., 2013, 4, 2418 RSC.
- (a) M. T. Rahman, T. Fukuyama, N. Kamata, M. Satoa and I. Ryu, Chem. Commun., 2006, 21, 2236 RSC; (b) F. Q. Zhao, H. J. Ai and X. F. Wu, Chin. J. Chem., 2021, 39, 927 CrossRef CAS; (c) L. J. Cheng and N. P. Mankad, Acc. Chem. Res., 2021, 54, 2261 CrossRef CAS PubMed; (d) W. Fang, H. Zhu, Q. Deng, S. Liu, X. Liu, Y. Shen and T. Tu, Synthesis, 2014, 46, 1689 CrossRef; (e) X. F. Wu, H. Neumann and M. Beller, Chem.–Eur. J., 2010, 16, 9750 CrossRef CAS PubMed; (f) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979 CrossRef CAS PubMed; (g) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed; (h) P. L. L. Tremblay, A. Fabrikant and B. A. Arndtsen, ACS Catal., 2018, 8, 5350 CrossRef; (i) Y. B. David, M. Portnoy and D. Milstein, J. Am. Chem. Soc., 1989, 111, 8742 CrossRef.
- J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed.
- A. Jasper, D. Schepmann, K. Lehmkuhl, J. M. Vela, H. Buschmann, J. Holenz and B. Wünsch, Eur. J. Med. Chem., 2012, 53, 327 CrossRef CAS PubMed.
- (a) G. H. Coleman and C. R. Hauser, J. Am. Chem. Soc., 1928, 50, 1193 CrossRef CAS; (b) H. Finkelstein, Ber. Dtsch. Chem. Ges., 1910, 43, 1528 CrossRef CAS; (c) B. H. Lipshutz, M. R. Wood and R. Tirado, Org. Synth., 1999, 76, 252 CrossRef CAS; (d) A. Streitwieser, Chem. Rev., 1956, 56, 571 CrossRef CAS.
- (a) R. Lang, J. L. Wu, L. J. Shi, C. G. Xia and F. W. Li, Chem. Commun., 2011, 47, 12553 RSC; (b) J. B. Peng, F. P. Wu and X. F. Wu, Chem. Rev., 2019, 119, 2090 CrossRef CAS PubMed; (c) S. W. Yuan, H. Han, Y. L. Li, X. L. Wu, X. G. Bao, Z. Y. Gu and J. B. Xia, Angew. Chem., Int. Ed., 2019, 58, 8887 CrossRef CAS PubMed.
- P. Pongrácza, A. A. Seni, L. T. Mikac and L. Kollára, Mol. Catal., 2017, 438, 15 CrossRef.
- C. Zhu, J. Liu, M. B. Li and J. E. Bäckvall, Chem. Soc. Rev., 2020, 49, 341 RSC.
- C. Dumont, F. Belva, R. M. Gauvin and M. Sauthier, ChemSusChem, 2018, 11, 3917 CrossRef CAS PubMed.
- (a) Y. F. Fan, D. Ma and X. H. Bao, Catal. Lett., 2009, 130, 286 CrossRef CAS; (b) D. Bajec, M. Grom, D. L. Jurkovic, A. Kostyniuk, M. Huš, M. Grilc, B. Likozar and A. Pohar, Processes, 2020, 8, 443 CrossRef CAS; (c) K. X. Wang, H. F. Xu, W. S. Li, C. T. Au and X. P. Zhou, Appl. Catal., A, 2006, 304, 168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04103k |
| This journal is © The Royal Society of Chemistry 2022 |