
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical synthesis of 1,2,4-triazoles via addition reaction of triplet intermediates to diazoalkanes and azomethine ylide intermediates†
Bao-Gui
Cai‡
a,
Ye-Peng
Bao‡
a,
Chao
Pei
b,
Qian
Li
a,
Lei
Li
a,
Rene M.
Koenigs
 *b and
Jun
Xuan
*ac
*b and
Jun
Xuan
*ac
aAnhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, College of Chemistry & Chemical Engineering, Anhui University, Hefei, Anhui 230601, China. E-mail: xuanjun@ahu.edu.cn
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
cKey Laboratory of Structure and Functional Regulation of Hybrid Materials, (Anhui University), Ministry of Education, Hefei, Anhui 230601, China
First published on 24th October 2022
Abstract
The reactivity of diazoalkanes most commonly proceeds through the formation of carbene intermediates or dipolar cycloaddition reactions. The reaction of diazoalkanes with intermediates with unpaired electrons, however, is much less elaborated. Herein, we report on the photochemical reaction of acceptor-only diazoalkanes with azodicarboxylates. Photoexcitation of the latter results in the formation of a triplet species, which undergoes an addition reaction with diazoalkanes and formation of an azomethine ylide followed by dipolar cycloaddition reaction with organic nitriles to give a 1,2,4-triazole. The application of this transformation was elaborated in a broad and general substrate scope (48 examples), including scale-up via flow chemistry and downstream transformations. Experimental and computational studies were performed to elucidate the reaction mechanism and to rationalize the reaction outcome.
Introduction
Diazoalkanes are versatile reagents in organic synthesis to introduce a carbene fragment onto organic building blocks.1 Their reaction most commonly involves the generation of a free (3) or metal-bound (2) carbene intermediate that engages in a rich portfolio of organic reactions, such as cyclopropanation, rearrangement reactions or C–H functionalization reactions (Scheme 1a).1–3 More recently, photocatalysis emerged as a new stratagem to expand this classic reactivity towards radical reactivity via a proton coupled electron transfer process4 On the contrary, the reactivity of diazoalkanes with reagents possessing unpaired electrons, such as radical or triplet intermediates, is vastly overlooked and only singular reports discuss the reaction of a diazoalkane with such species.5 The most prominent example of such radical addition reactions to diazoalkanes can be found in the polymerization reaction of diazoalkanes, where a metal-bound radical intermediate adds to another molecule of diazoalkanes.6 A second approach discusses the UV-mediated reaction of diazomethane with haloalkanes in substitution reactions as reported by Urry and Bilow in 1964 (5, Scheme 1c).7 Such radical addition approaches have recently gained interest using iodine-based reagents to allow addition reactions of radical species in a more systematic fashion, yet with the necessity of high reaction temperatures in combination with suitable reagents, catalysts (or promotors), and leaving groups to access the radical intermediate.8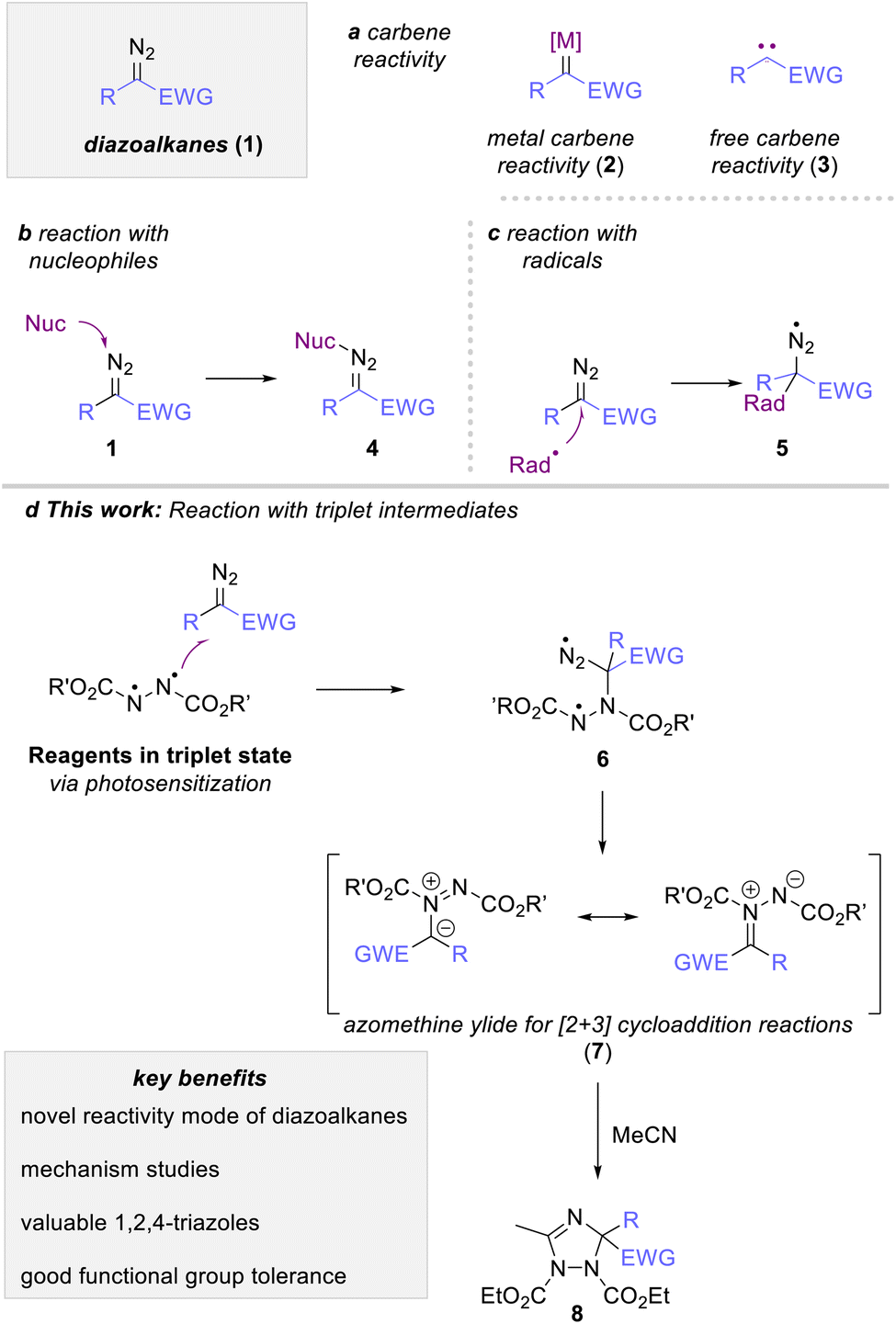 | ||
| Scheme 1 Photochemical carbene transformation reactions of diazoalkanes and our reaction design. | ||
Owing to the close relationship of radicals and triplet intermediates, we envisioned that the triplet spin state of a suitable reagent could be used to leverage a radical-type addition reaction to diazoalkanes. This strategy would in turn facilitate approaches towards this reaction pathway and at the same time improve on the overall reaction sustainability as this approach would require a rather simple photoexcitation of suitable reagents from ground to triplet spin state (Scheme 1d).
Against this background and our ongoing research interest in low-energy visible light-promoted organic synthesis,9 we envisioned that photoexcitation of intensely colored azo dicarboxylates could be a practical a handle to access a triplet intermediate, which in turn should engage in an addition reaction with ethyl diazoacetate. Following extrusion of nitrogen gas, such reaction would result in the formation of an azomethine ylide, which itself can undergo dipolar cycloaddition reactions to form valuable azole heterocycles. Specifically, the reaction with nitriles should proceed through [3 + 2]-addition to construct 1,2,4-triazole derivatives, an important heterocycle with rich bioactivities (Scheme 1d).10
Results and discussion
Reaction design and reaction mechanism
A requirement to achieve such transformation lies within the use of a suitable organic building block that can undergo facile photoinduced formation of a triplet intermediate. In this context, we considered azo dicarboxylates, which show a strong absorption in the visible light region with an absorption maximum at 400 nm, which expands into the visible light region up to more than 500 nm.At the same time, we analyzed a set of commonly used acceptor-only diazoalkanes, including ethyl diazoacetate 10a, 2-diazo-1,1,1-trifluoroethane 10b, and diazoacetonitrile 10c (Scheme 2a). 10c presents an almost identical absorbance spectra in the visible region compared with the well-studied donor–acceptor diazoalkanes, e.g. ethyl 2-diazo-2-phenylacetate 12a (Scheme 2a, dark green line and red line). The other two acceptor-only diazoalkanes 10a and 10b also have absorption in the visible region (Scheme 2a, light green line and pink line).
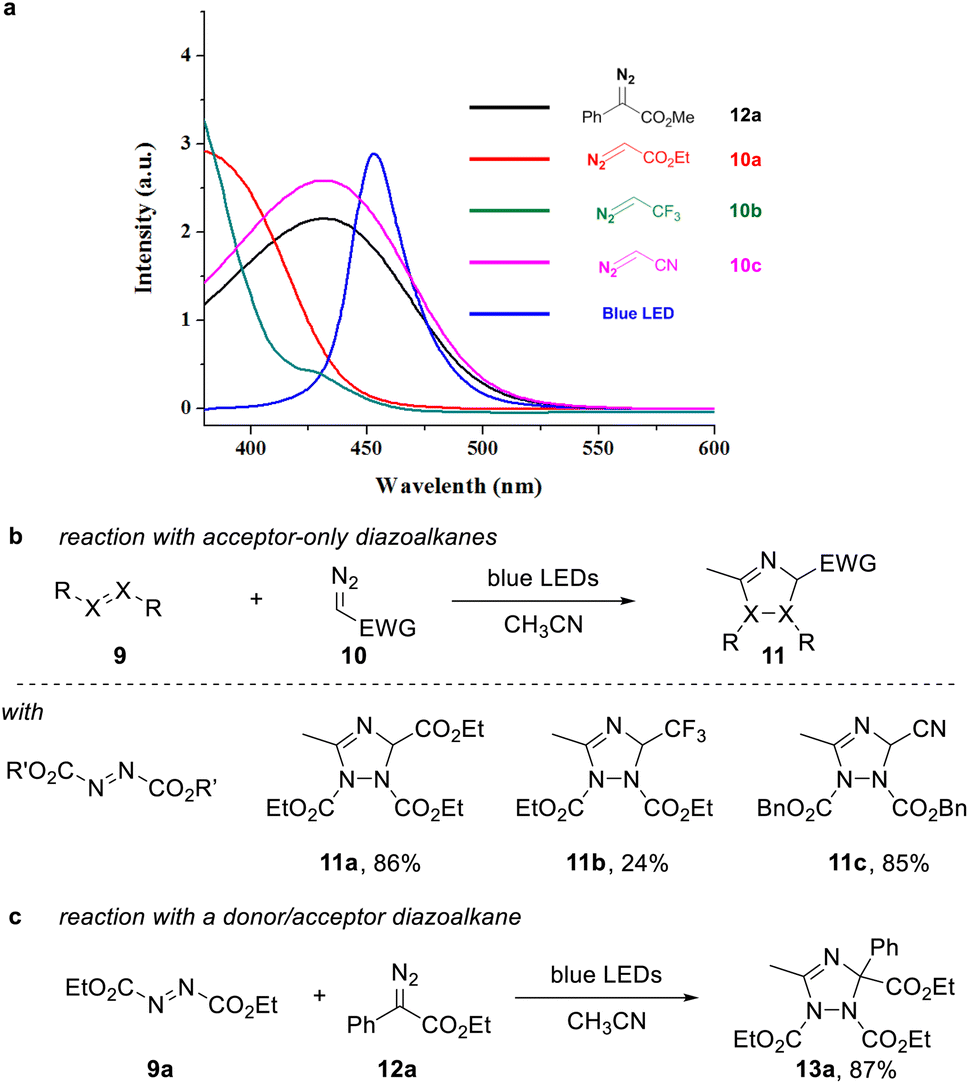 | ||
| Scheme 2 (a) UV-vis absorbance spectra of commonly used three acceptor-only diazoalkanes in CH3CN (c = 0.2 M). (b) Three component cascade reaction of acceptor diazoalkanes, CH3CN and DEAD. (c) Reaction of donor/acceptor diazoalkane 12a, CH3CN and DEAD. | ||
For proof of concept, we next examined the reaction of acceptor-only diazoalkanes 10a–c with azodicarboxylates 9 in acetonitrile, which serves as both solvent and dipolarophile. In agreement to our initial hypothesis, we were able to obtain the desired 1,2,4-triazole products 11a–c in good isolated yield after 12 h reaction time (Scheme 2b, for details of optimization, see the ESI†). Control experiments indicated that no desired 1,2,4-triazole product was observed when the reaction was conducted in dark. Notably, also the reaction of ethyl phenyldiazoacetate proved very efficient to give the corresponding triazole 13a in high yield (Scheme 2c).
The outcome of these reactions triggered our interest in understanding the reaction mechanism as the formation of the 1,2,4-triazole can be explained by either photoexcitation of azodicarboxylate 9 as outlined in the introduction, or by photolysis reaction of the diazoalkane and formation of a nitrile ylide followed by dipolar cycloaddition of the nitrile ylide (Scheme 3a).
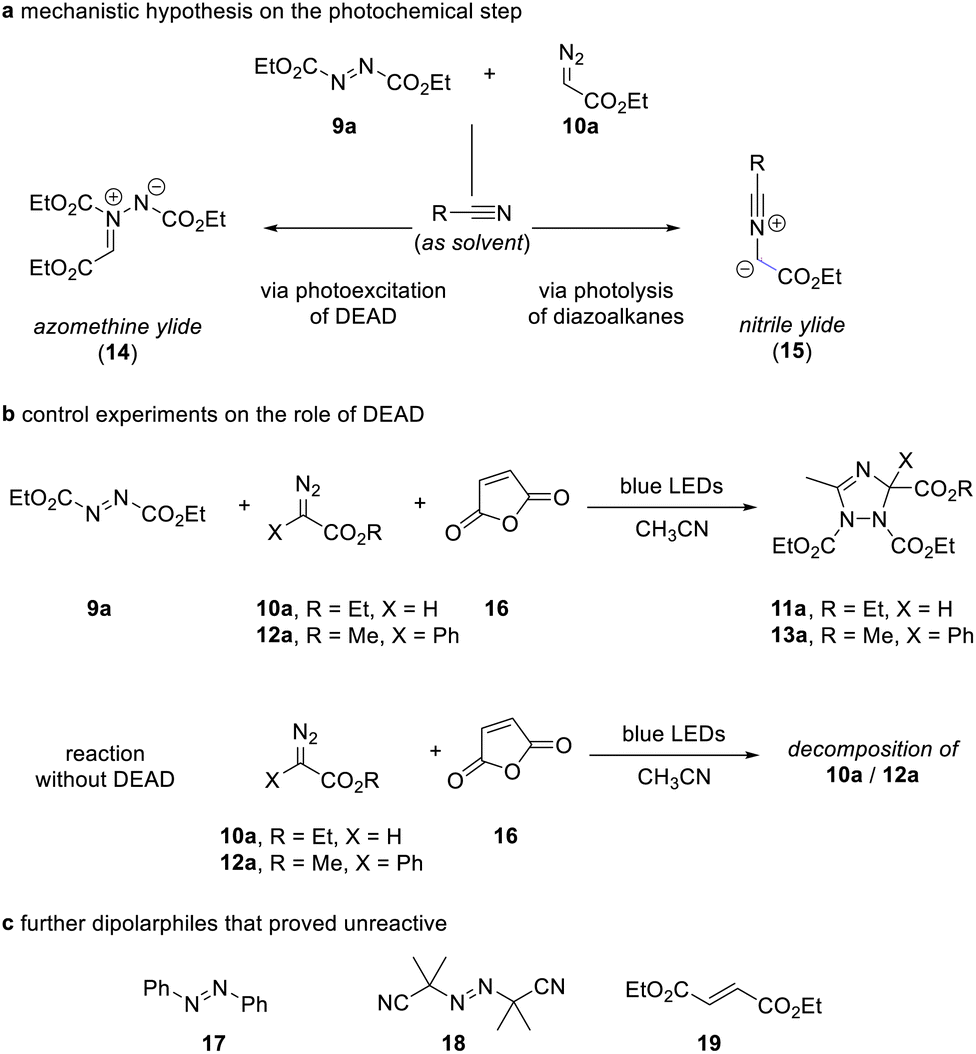 | ||
| Scheme 3 Control experiments. | ||
For deconvolution of reaction pathways, we first conducted the reaction in acetonitrile solvent in the presence of maleic anhydride. When the reaction is carried out in the presence of additional maleic anhydride, only the formation of the 1,2,4-triazole 11a/13a was observed and no reaction with maleic anhydride occurred. We then performed the same reaction in the absence of azodicarboxylate 9a. Here, no reaction occurred, which provides evidence on the participation of azodicarboxylate 9a in the course of the reaction and now suggests that a nitrile ylide is not formed as a reaction intermediate (Scheme 3b). Similar, the reaction with other olefins, such as azobenzene, 2,2′-azobis(2-methylpropionitrile), and diethyl maleate proved unsuccessful and show the relevance of photoexcitation of azodicarboxylate 9a in this transformation (Scheme 3c).
We finally conducted experiments in the presence of TEMPO as a radical scavenger and were able to observe a notable reduction of the yield of triazole 11a upon addition of stoichiometric amounts of TEMPO (for details, see the Experimental ESI†), which hints at the participation of species with unpaired electrons.
Further analysis then involved computational studies. First, we analyzed the photoexcitation of DEAD and EDA and formation of DEAD-T and EDA-T using (TD)-DFT methods. These show that visible light photoexcitation is more favorable for DEAD under visible light conditions; the energy required for the π–π* transition was calculated to be 58.6 kcal mol−1 (λmax = 488 nm) for DEAD, while a much higher energy is required for photoexcitation of EDA (70.4 kcal mol−1; λmax = 410 nm) (for details, see the ESI†).
With this data, we next embarked on computational studies of the reaction mechanism (Scheme 4). After photoexcitation of DEAD, the triplet state DEAD-T can undergo a radical-type addition reaction with acetonitrile or with EDA. The addition to acetonitrile, however, requires a high activation free energy of 28.5 kcal mol−1 (TS1) and is thus kinetically and thermodynamically unfavorable. In contrast, a similar radical addition onto EDA was identified much more facile and proceeds with a low energy barrier of 13.6 kcal mol−1 (TS2) and leads to diradical INT2 in an exergonic reaction. Further calculations indicate the formation of INT3-T through denitrogenation of INT2 (TS3, ΔG‡ = 0.8 kcal mol−1) as a nearly barrierless process. Subsequently, intersystem crossing of INT3-T affords the more stable singlet species INT3-S – or azomethine ylide. This process was identified as a near barrierless step as shown by minimum energy crossing point (MECP) calculations. Final [3 + 2] cycloaddition of azomethine ylide INT3-S with acetonitrile will generate the 1,2,4-triazole.
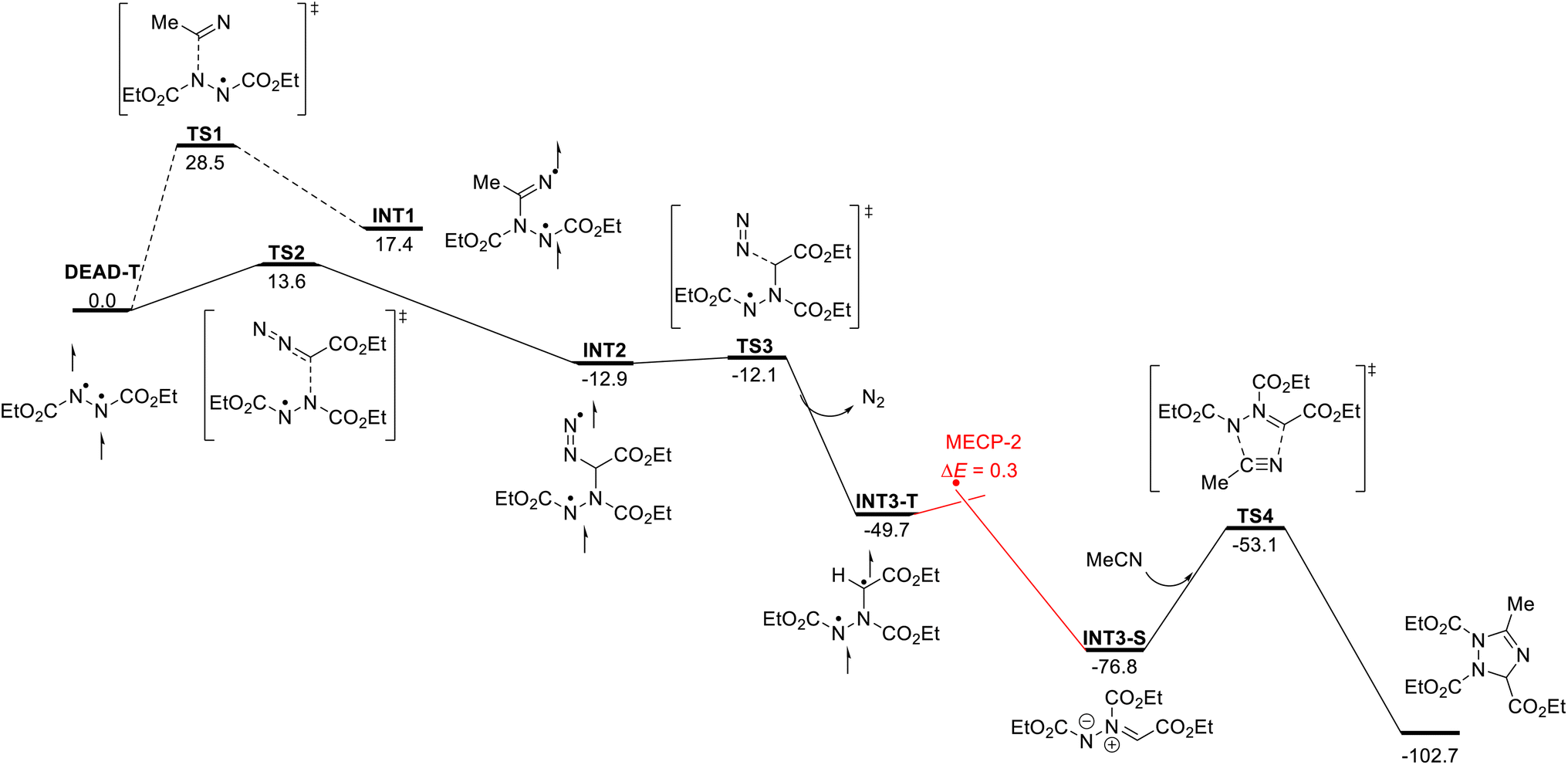 | ||
| Scheme 4 Reaction mechanism. | ||
Application
In further studies we focused on the application of this concept in organic synthesis and examined the generality and limitations of this light-promoted 1,2,4-triazole synthesis protocol. We first investigated the reactivity of acceptor-only diazoalkanes by using different alkyl 2-diazoacetates. It was found that diazoacetates containing different alkyl groups smoothly underwent azomethine ylide formation to afford the corresponding 1,2,4-triazoles in generally good yields (Scheme 5a). In further investigations, we found that nitrile solvents bearing different alkyl groups (11k and l), cycloalkyl groups (11m–p), alkyl groups with sensitive carbon–carbon double bonds (11q and r) and aryl group (11s) showed good compatibility to produce the corresponding 1,2,4-triazoles products in good yields (Scheme 5b). It is well known that incorporation of deuterium atoms into drug molecules are one the most important approaches for their activity modification.11 To our delight, the deuterated 1,2,4-triazole 11t could be obtained in 61% yield with CD3CN as carbene trapping reagent. We also studied the effect of substituents on azodicarboxylates to the reaction efficiency (Scheme 5c). Replacement of DEAD to more sterically demanding diisopropyl azodicarboxylate (DIAD) and di-tert-butyl azodicarboxylate (DTBAD) caused slightly decreased reaction yields (11u and v). The more electrophilic DBAD remained highly reactive toward cycloaddition to give 1,2,4-triazole 11w in 61% yield. Importantly, different acceptor groups, such as a nitrile, a ketone, or a CF3 group on the diazoalkane component were examined and found compatible to the reaction conditions (see also Scheme 2b).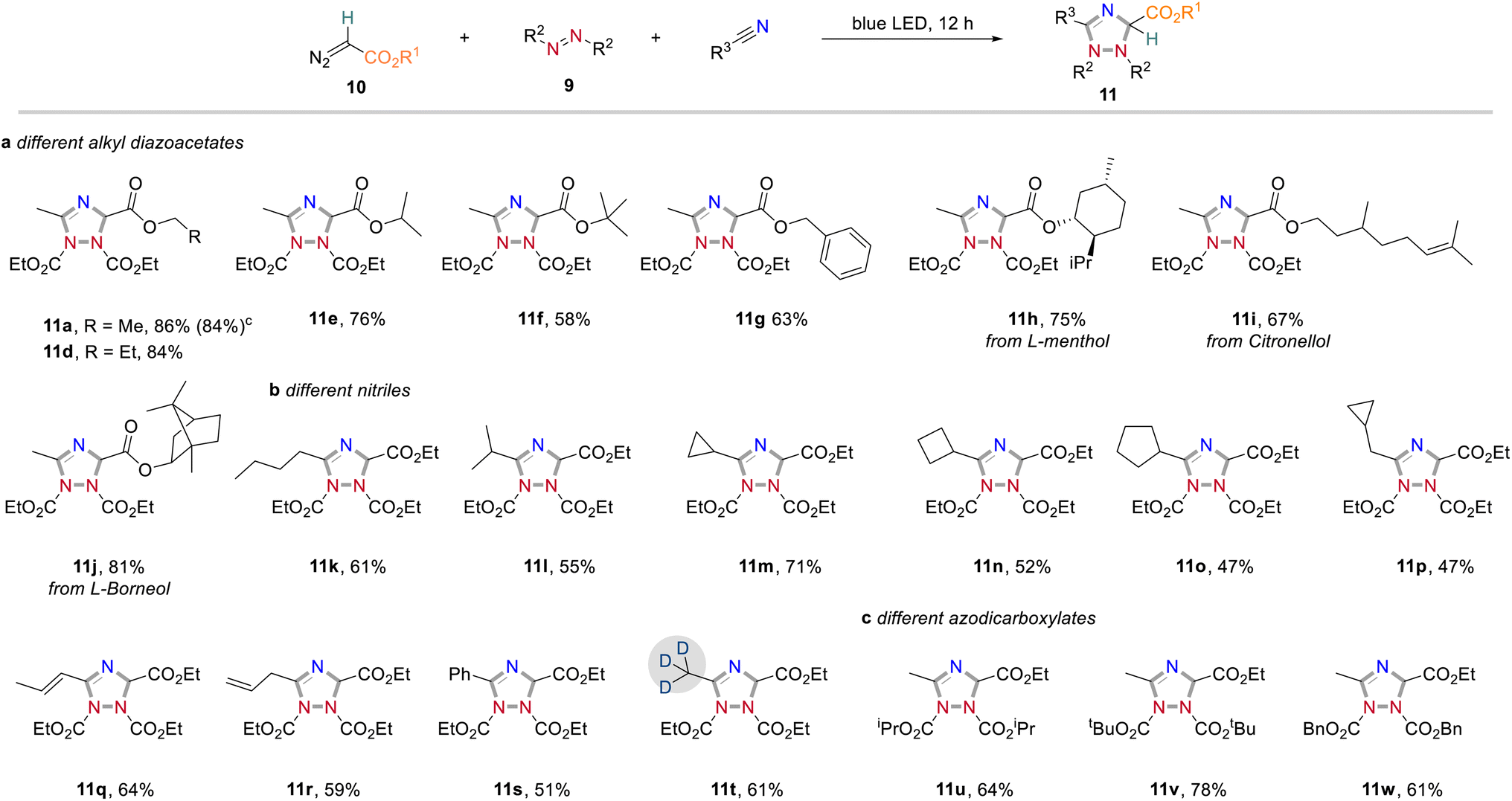 | ||
| Scheme 5 Reactivity exploration of acceptor-only diazoalkanes. Reaction conditions: a Reaction performed with diazoalkanes (0.4 mmol), azodicarboxylates (0.1 mmol) in dry nitrile solvent (1.0 mL) at rt under irradiation with 24 W blue LEDs for 12 h. b Isolated yield. c Performed at 1.0 mmol scale. | ||
Apart from acceptor-only diazoalkanes, the reactivity of donor–acceptor diazoalkanes was then studied (Scheme 6, for an overview of unsuccessful examples, see page S3 in the Experimental ESI†). Generally, a wide range of 1,2,4-triazole derivatives were accessed in good yields when using aryldiazoesters as carbene precursors (Scheme 6a). For instance, the three-component reaction of methyl 2-diazo-2-phenylacetate, CH3CN and DEAD gave the cycloaddition product 13a in 87% yield. Similarly, different electron-withdrawing groups (–F, –Cl, –Br, –CF3) at the para or ortho positions on the phenyl ring of aryldiazoacetate were all well-tolerated and the corresponding 1,2,4-triazoles were obtained in good yields (13c–f, 89–95% yields). In contrast, the electron-rich substituents on the phenyl ring (13b, g and h), or a naphthyl substituent, led to a slightly decreased the reaction yield (13j).
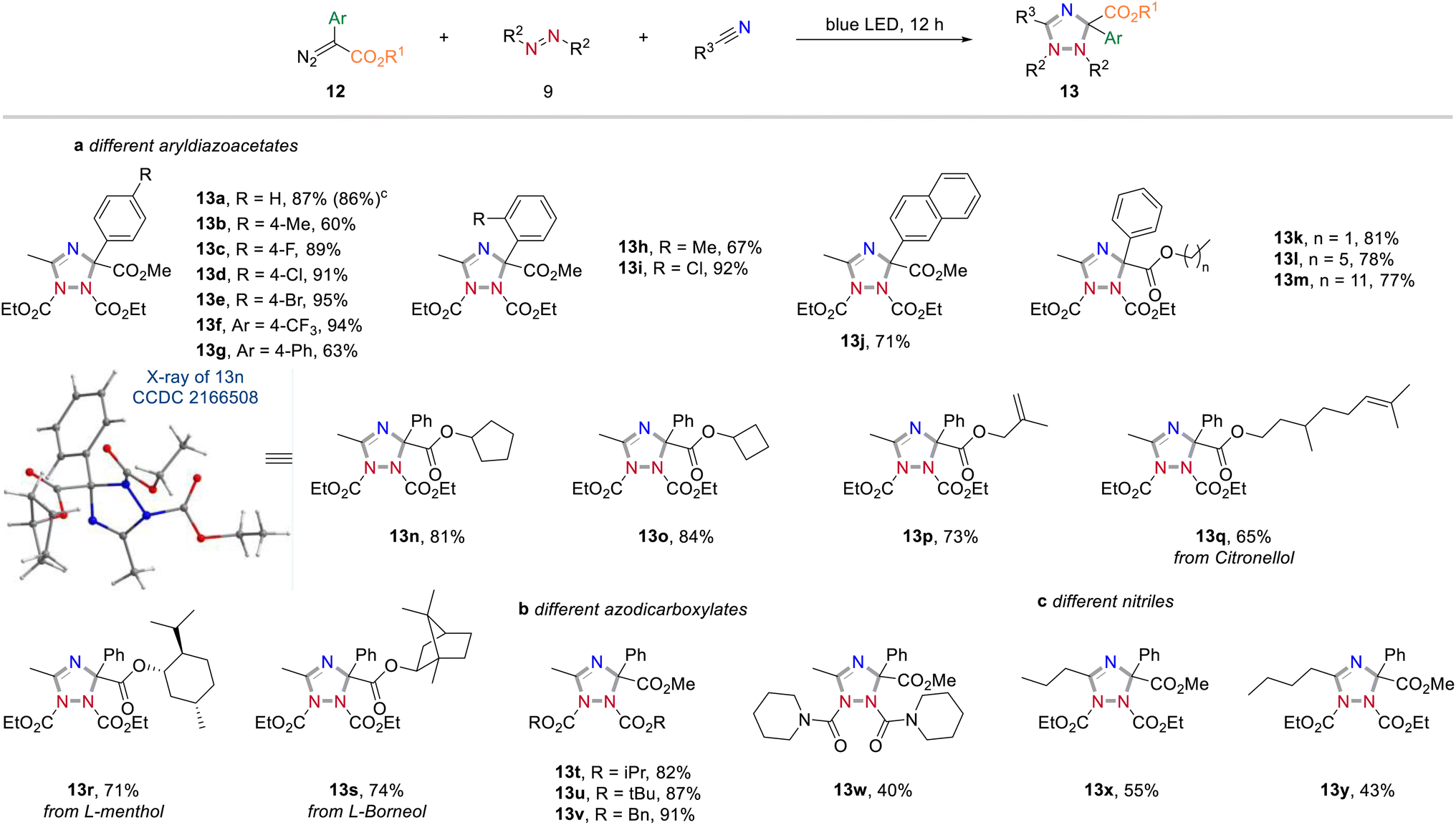 | ||
| Scheme 6 Reactivity exploration of donor/acceptor diazoalkanes. Reaction conditions: aReaction performed with diazoalkanes (0.4 mmol), azodicarboxylates (0.1 mmol) in dry nitrile solvent (1.0 mL) at rt under irradiation with 24 W blue LEDs for 12 h. bIsolated yield. cPerformed at 1.0 mmol scale. | ||
We then examined the scope of different ester groups of donor/acceptor diazoalkanes and found that a wide range of linear alkyl groups (13k–m), cyclic alkyl groups (13n and o), and linear alkyl groups bearing sensitive C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (13p and q) were tolerated in the present reaction leading to the corresponding heterocycles without an obvious effect on the reaction yield. The structure of 13n was confirmed by X-ray diffraction (CCDC 2166508). More significantly, the introduction of Citronellol, L-menthol, and L-(−)-borneol fragments into final 1,2,4-triazoles further proves the synthetic value of the current method (13q–s). Changing the protecting group of the azodicarboxylates (13t–w) or prolonging the carbon chain of nitrile solvents (13x and y) all proved to be successful. Notably, also 1,1'-(azodicarbonyl)-dipiperidine was found compatible under the optimal reaction conditions, albeit with relatively low yield (13w, 40%).
C bond (13p and q) were tolerated in the present reaction leading to the corresponding heterocycles without an obvious effect on the reaction yield. The structure of 13n was confirmed by X-ray diffraction (CCDC 2166508). More significantly, the introduction of Citronellol, L-menthol, and L-(−)-borneol fragments into final 1,2,4-triazoles further proves the synthetic value of the current method (13q–s). Changing the protecting group of the azodicarboxylates (13t–w) or prolonging the carbon chain of nitrile solvents (13x and y) all proved to be successful. Notably, also 1,1'-(azodicarbonyl)-dipiperidine was found compatible under the optimal reaction conditions, albeit with relatively low yield (13w, 40%).
To elaborate the synthetic utility of this visible light-promoted three-component annulation process, a series follow-up chemistry was conducted (Scheme 7). The reaction could be easily scaled-up under continuous-flow photochemical conditions to give 11a and 13a in 81% and 85% yield, respectively (for detailed information of the flow chemistry apparatus, see the Experimental ESI†). A subsequent decarboxylative aromatization under batch conditions of the formed 1,2,4-triazoles under basic reaction conditions afforded 20 and 21 in excellent yields. These triazoles 20 and 21 are important synthetic intermediates with applications in drug discovery. For instance, 20 can be used to the synthesis of taselisib (GDC-0032), an effective small molecule PI3K β-isoform sparing inhibitor for the treatment of various cancers.12a Dipeptidyl peptidase IV inhibitors can also be synthesized by simple transformation of 21 in several steps.12b
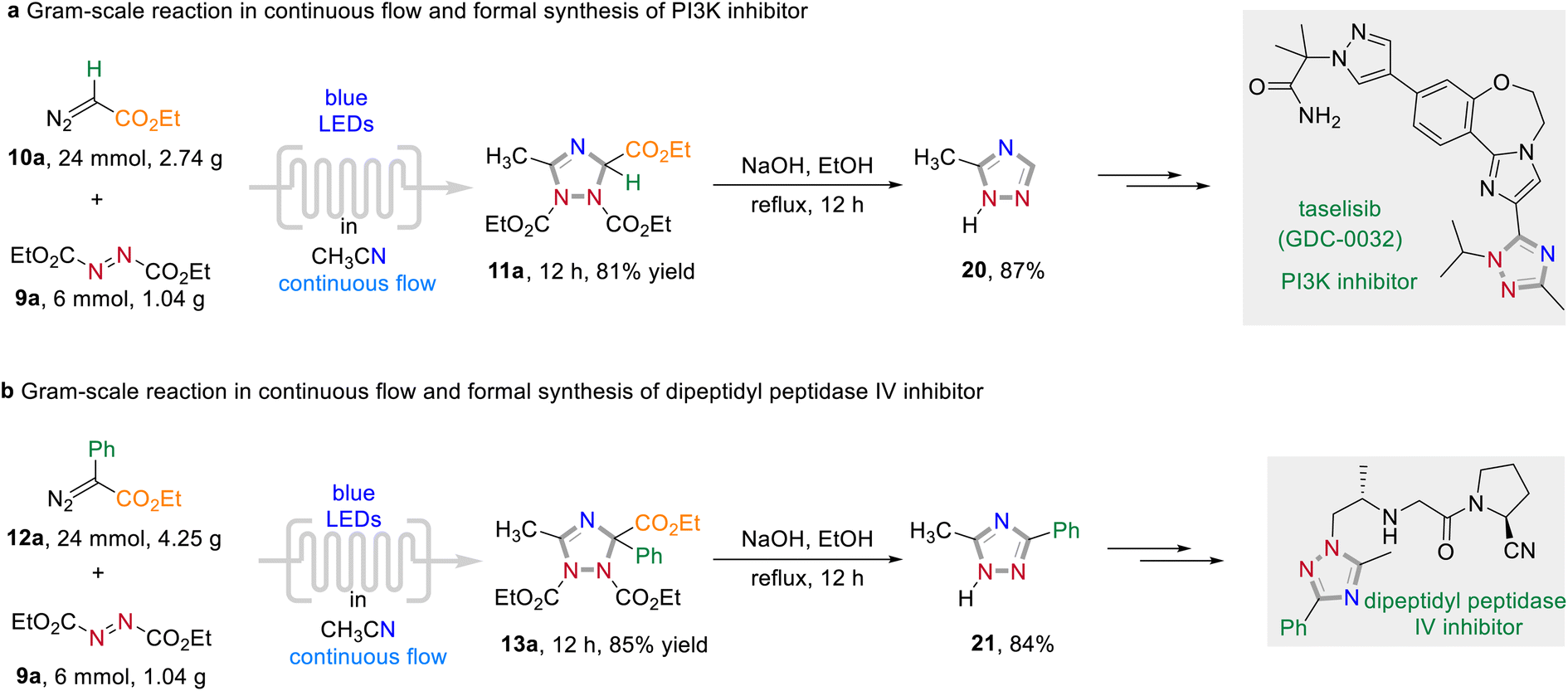 | ||
| Scheme 7 Follow-up chemistry. | ||
Conclusions
In summary, we herein report on a photochemical multi-component reaction that allows the synthesis of important 1,2,4-triazoles under mild reaction conditions. Photochemical excitation of azodicarboxylates was identified as a key trigger to initiate this reaction via formation of DEAD in triplet spin state. This triplet intermediate reacts with diazoalkanes to an azomethine ylide, which is a potent 1,2-dipole for subsequent cycloaddition reactions with nitrile to give 1,2,4-triazoles. We explored the application of this transformation in a broad substrate scope. Computational and experimental studies were performed to support the mechanistic hypothesis and suggest that an addition reaction of a triplet species to the diazoalkane reaction partner is key in this transformation.Conflicts of interest
There are no conflicts to declare.Data availability
Experimental data have been provided in the ESI.†Author contributions
The manuscript was written through contributions of all authors. B. G. C., Y. P. B. and Q. L. carried out experimental work. B. G. C. and L. L. analyzed the data. C. P. carried out all computational studies. J. X. and R. M. K. conceived the ideas and designed the project. All authors have given approval to the final version of the manuscript.Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 21971001, 21702001) for financial support. CP gratefully acknowledges the China Scholarship Council for financial support.Notes and references
- (a) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (b) M. P. Doyle, R. Duffy, M. Ratnikov and Z. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (c) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (d) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506 RSC; (e) C. Damiano, P. Sonzini and E. Gallo, Chem. Soc. Rev., 2020, 49, 4867 RSC.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) S.-F. Zhu and Q.-L. Zhou, Natl. Sci. Rev., 2014, 1, 580 CrossRef CAS; (c) Y. Zhang and J. Wang, Coord. Chem. Rev., 2010, 254, 941 CrossRef CAS; (d) S. Jana, Y. Guo and R. M. Koenigs, Chem. – Eur. J., 2021, 27, 1270 CrossRef CAS PubMed; (e) H. U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151 CrossRef CAS PubMed.
- (a) Ł. W. Ciszewski, K. Rybicka-Jasinska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432 RSC; (b) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833 RSC; (c) B.-G. Cai and J. Xuan, Chin. J. Org. Chem., 2021, 41, 4565 CrossRef; (d) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895 CrossRef CAS; (e) C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788 RSC.
- (a) Ł. W. Ciszewski, J. Durka and D. Gryko, Org. Lett., 2019, 21, 7028 CrossRef PubMed; (b) Y.-L. Su, G.-X. Liu, J.-W. Liu, L. Tram, H. Qiu and M. P. Doyle, J. Am. Chem. Soc., 2020, 142, 13846 CrossRef CAS PubMed; (c) N. Ma, L. Guo, D. Qi, F. Gao, C. Yang and W. Xia, Org. Lett., 2021, 23, 6278 CrossRef CAS PubMed; (d) K. Rybicka-Jasińska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, J. Am. Chem. Soc., 2016, 138, 15451 CrossRef PubMed; (e) X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636 CrossRef CAS PubMed; (f) F. Li, S. Zhu and R. M. Koenigs, Chem. Commun., 2022, 58, 7526 RSC; (g) Z. Zhang, N. Kvasovs, A. Dubrovina and V. Gevorgyan, Angew. Chem., Int. Ed., 2022, 61, e202110924 CAS; (h) F. Li, C. Pei and R. M. Koenigs, Angew. Chem., Int. Ed., 2022, 61, e202111892 CAS.
- (a) N. Wang, W. Hao, T. Zhang, G. Li, Y. Wu, S. Tu and B. Jiang, Chem. Commun., 2016, 52, 5144 RSC; (b) H.-S. Dang and B. P. Roberts, J. Chem. Soc., Perkin Trans. 1, 1996, 769 RSC.
- J. Luo and K. J. Shea, Acc. Chem. Res., 2010, 43, 1420 CrossRef CAS PubMed.
- W. H. Urry and N. Bilow, J. Am. Chem. Soc., 1964, 86, 1815 CrossRef CAS.
- (a) S. Kim and J. R. Cho, Bull. Korean Chem. Soc., 1993, 14, 664 CAS; (b) G. Verardo, P. Geatti and A. Gambi, J. Phys. Org. Chem., 2009, 22, 24 CrossRef CAS; (c) Y. Wang, L. Ma, M. Ma, H. Zheng, Y. Shao and X. Wan, Org. Lett., 2016, 18, 5082 CrossRef CAS PubMed.
- (a) B.-G. Cai, J. Xuan and W.-J. Xiao, Sci. Bull., 2019, 64, 337 CrossRef CAS; (b) J. Xuan, X.-K. He and W.-J. Xiao, Chem. Soc. Rev., 2020, 49, 2546 RSC; (c) S.-X. Wang, L. Tang, B.-G. Cai, Z. Yin, Y. Li, L. Xiong, X. Kang, J. Xuan, Y. Pei and M.-Z. Zhu, J. Am. Chem. Soc., 2022, 144, 3787 CrossRef CAS PubMed; (d) Z.-L. Chen, J.-R. Chen and J. Xuan, Chin. Chem. Lett., 2022, 33, 2763 CrossRef CAS.
- (a) X. Cao, F. Li, M. Hu, W. Lu, G.-A. Yu and S.-H. Liu, J. Agric. Food Chem., 2008, 56, 11367 CrossRef CAS PubMed; (b) A. Moulin, M. Bibian, A.-L. Blayo, S. E. Habnouni, J. Martinez and J.-A. Fehrentz, Chem. Rev., 2010, 110, 1809 CrossRef CAS PubMed; (c) T. Remarchuk, R. Angelaud, D. Askin, A. Kumar, A. S. Thompson, H. Cheng, J. F. Reichwein, Y. Chen and F. St-Jean, Org. Process Res. Dev., 2019, 23, 775 CrossRef CAS; (d) G. Basarab, P. Doig, C. J. Eyermann, V. Galullo, G. Kern, A. Kimzey, A. Kutschke, M. Morningstar, V. Schuck, K. Vishwanathan, F. Zhou, M. Gowravaram and S. Hauck, J. Med. Chem., 2020, 63, 11882 CrossRef CAS PubMed; (e) Y. Sicak, Med. Chem. Res., 2021, 30, 1557 CrossRef CAS.
- (a) A. Katsnelson, Nat. Med., 2013, 19, 656 CrossRef CAS PubMed; (b) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219 CrossRef CAS PubMed; (c) X. Wang, W.-Y. Tong, B. Huang, S. Cao, Y. Li, J. Jiao, H. Huang, Q. Yi, S. Qu and X. Wang, J. Am. Chem. Soc., 2022, 144, 4952 CrossRef CAS PubMed.
- (a) T. Remarchuk, R. Angelaud, D. Askin, A. Kumar, A. S. Thompson, H. Cheng, J. F. Reichwein, Y. Chen and F. St-Jean, Org. Process Res. Dev., 2019, 23, 775 CrossRef CAS; (b) B. Markus, H. Daniel, K. Holger, L. B. Michael, S. Ramakanth and W. H. Assignee, Preparation of N-Aminoacetyl-Substituted Pyrrolidines as dipeptidyl Peptidase IV Inhibitors. PCT Int. Appl., 2003 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2166508. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc04720a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |