
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBuffering the local pH via single-atomic Mn–N auxiliary sites to boost CO2 electroreduction†
Yan
Yang
abe,
Tang
Tang
 a,
Zhen-Hua
Lyu
ae,
Li-Rong
Zheng
c,
Qing-Hua
Zhang
d,
Jiaju
Fu
*a and
Jin-Song
Hu
*aef
a,
Zhen-Hua
Lyu
ae,
Li-Rong
Zheng
c,
Qing-Hua
Zhang
d,
Jiaju
Fu
*a and
Jin-Song
Hu
*aef
aBeijing National Laboratory for Molecular Sciences (BNLMS), CAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: fujiaju@iccas.ac.cn; hujs@iccas.ac.cn
bZhejiang Tiandi Environmental Protection Technology Co., Ltd, Hangzhou 310003, China
cInstitute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
dInstitute of Physics, Chinese Academy of Sciences, Beijing 100190, China
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
fDalian National Laboratory for Clean Energy, Dalian 116023, China
First published on 2nd November 2022
Abstract
Electrocatalytic CO2 reduction driven by renewable energy has become a promising approach to rebalance the carbon cycle. Atomically dispersed transition metals anchored on N-doped carbon supports (M-N-C) have been considered as the most attractive catalysts to catalyze CO2 to CO. However, the sluggish kinetics of M-N-C limits the large-scale application of this type of catalyst. Here, it is found that the introduction of single atomic Mn–N auxiliary sites could effectively buffer the locally generated OH− on the catalytic interface of the single-atomic Ni–N–C sites, thus accelerating proton-coupled electron transfer (PCET) steps to enhance the CO2 electroreduction to CO. The constructed diatomic Ni/Mn–N–C catalysts show a CO faradaic efficiency of 96.6% and partial CO current density of 13.3 mA cm−2 at −0.76 V vs. RHE, outperforming that of monometallic single-atomic Ni–N–C or Mn–N–C counterparts. The results suggest that constructing synergistic catalytic sites to regulate the surface local microenvironment might be an attractive strategy for boosting CO2 electroreduction to value-added products.
Introduction
Electrochemical CO2 reduction (CO2RR) into carbon-based fuels and value-added chemicals is considered a forward-looking strategy to alleviate energy and environmental crisis and advance the goals of carbon neutrality.1–4 With the merits of fully exposed, highly unsaturated coordination metal sites, atomically dispersed transition metals anchored on N-doped carbon supports (M-N-C) are considered as promising and efficient catalysts for reducing CO2 into CO, which has been evaluated as one of the most economical and feasible products.5–8 However, M-N-C catalysts still suffer sluggish kinetics resulting in high overpotential and low current density, which seriously hinders the large-scale application of catalysts.9–11Significant efforts have been made to optimize the intrinsic activity for CO production or enrich the density of the single metal atomic sites on M-N-C catalysts (e.g., tuning the element of central metal atoms, regulating the coordination environment, etc.) in the past few years.12–16 For instance, Hu et al. conducted single-atom Fe3+ sites coordinated with pyrrolic nitrogen to promote the conversion of CO2 to CO. The high catalytic activity was attributed to the Fe3+ sites, enhancing CO2 adsorption and weakening CO absorption compared to conventional Fe2+ sites.12 Li et al. regulated the nitrogen coordination numbers over single Co sites through changing annealing conditions to enhance the selectivity toward CO.14 Recently, the local microenvironment of the catalyst–electrolyte interface, including the local pH, the CO2 concentration, and the cations and anions in electrolytes, have also been demonstrated as critical factors affecting the CO2 activation and the CO2RR reaction kinetics.17–20 For instance, Alexis et al. revealed that the alkali metal cations in electrolytes could affect the local pH on the catalytic interface. Alkali metal cations with larger radius exhibit a stronger buffering effect, thus decreasing the local pH value and consequently enhancing the CO2RR performance.20 As given in eqn (1), previous studies have revealed that the 2e− transfer pathway of CO production on M-N-C catalysts usually starts with a proton-coupled electron transfer (PCET) step to form the *COOH intermediate, which is considered pH-dependent due to the proton participation.21–25 Therefore, the kinetics of the PCET process could be accelerated by applying an appropriate low local pH.18,26,27
| *CO2 + H+ + e− → *COOH | (1) |
In addition, according to buffer equilibrium, the CO2 concentration on the catalytic interface could increase with the decrease of local pH, which is also beneficial to the activation of CO2.17,20,28 However, as the cathode reaction proceeds, the consumption of protons and the OH− species generated with the output of CO or H2 would significantly increase the local pH value,29 thus hindering the activation of CO2 and the reaction kinetics of the PCET steps.30 Therefore, building catalytic interfaces to buffer the locally generated OH− and hold the local pH value stable could be a promising approach to maintaining the electrocatalytic activity of M-N-C catalysts.
Herein, we developed an effective strategy for regulating local microenvironment to boost CO2 to CO conversion. By constructing efficient Ni/Mn diatomic catalysts (Ni/Mn–N–C) anchored on N-doped carbon supports, it was discovered that the introduction of single atomic Mn–N sites could efficiently buffer the locally generated OH− on the catalytic interface of the Ni–N–C catalyst, thus accelerating PCET steps to enhance the CO2 electroreduction to CO. Atomic dispersed Ni–N and Mn–N sites were uncovered by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and X-ray absorption spectroscopy (XAS). The as-prepared Ni/Mn–N–C catalyst exhibited excellent electrochemical performance with a CO faradaic efficiency of 96.6% and a CO current density of 13.3 mA cm−2 at −0.76 V (vs. reversible hydrogen electrode (RHE), hereinafter), exceeding that of the sole Ni–N–C and Mn–N–C samples. The selectivity of CO could be maintained above 90% after 20 hours. Furthermore, the electrode kinetics experiments revealed that the introduction of atomic Mn–N sites buffered the local pH near the catalyst–electrolyte interface, promoting the kinetics of CO production on Ni–N sites. These findings provided new insights into tuning the surface microenvironment for enhancing CO2RR performance by constructing diverse and synergistic catalytic sites.
Results and discussion
As illustrated in Fig. 1a, the atomically dispersed Ni/Mn–N–C catalyst was synthesized via a two-step process. In brief, the NiMn complex–C was firstly formed by the co-adsorption of inorganic Ni, Mn precursors, and glucose on porous carbon. The Ni/Mn–N–C catalyst was then obtained through the pyrolysis of the Ni/Mn complex–C with melamine at 800 °C (see detail in ESI†). The content of Ni and Mn was evaluated by inductively coupled plasma optical emission spectroscopy (ICP-AES), indicating 3.14 wt% Ni and 2.68 wt% Mn in the as-synthesized Ni/Mn–N–C catalyst (Table S1†). The detailed structure of the Ni/Mn–N–C was first characterized by high-resolution transmission electron microscopy (HRTEM, Fig. 1b). No aggregates were observed in the HRTEM image, indicating the absence of Ni particles or Mn particles. The HAADF-STEM image in Fig. 1c exhibited the presence of abundant atomic-scale isolated bright dots, indicating the atomic dispersion of metal species. No characteristic peak of Ni or Mn was observed in the powder X-ray diffraction (XRD) patterns in Fig. 1d, confirming the absence of obvious Ni particles or Mn particles. The energy-dispersive X-ray spectroscopy (EDS) in Fig. 1e further confirmed that the Ni/Mn were uniformly dispersed on the N-doped carbon supports without obvious aggregation. In addition, the atomically dispersed Ni–N–C and Mn–N–C were prepared in parallel by similar procedures with the same total metal feeding for comparison (Fig. S1 and S2†). | ||
| Fig. 1 (a) Scheme for the synthesis of Ni/Mn–N–C catalyst, (b) HRTEM image, (c) HAADF-STEM image, (d) XRD patterns, and (e) TEM and corresponding elemental mapping images of Ni/Mn–N–C catalyst. | ||
Advanced spectral characterizations were performed to investigate the detailed structures of the as-prepared catalysts. X-ray photoelectron spectroscopy (XPS) was first conducted to analyze the surface chemical states. The survey spectrum demonstrated the presence of C, N, O, Ni, and Mn elements on the Ni/Mn–N–C surface (Fig. S3†). As presented in Fig. 2a and b, the peak at 854.7 eV of Ni 2p spectra (Fig. 2a) and 641.3 eV of Mn 2p spectra (Fig. 2b) showed that both Ni and Mn exhibited +2 valence on the Ni/Mn–N–C surface, which were also observed on the Ni–N–C and Mn–N–C samples (Fig. S4 and S5†).31,32 The peak deconvolution of the high-resolution N 1s spectrum (Fig. 2c) exhibited the existence of four forms of N species on the Ni/Mn–N–C surface (pyridinic at 398.4 eV, pyrrolic N at 399.5 eV, graphitic N at 400.5 eV and N–O at 401.6 eV)12,33–35 with pyridine N as the major N species,7 which was similar in Ni–N–C and Mn–N–C (Fig. S6†).
 | ||
| Fig. 2 (a) The Ni 2p XPS spectra, (b) the Mn 2p XPS spectra, (c) the high-resolution N 1s XPS spectra, (d) Ni K edge XANES spectra, (e) Ni K edge EXAFS spectra, (f) Mn K edge XANES spectra, (g) Mn K edge EXAFS spectra of Ni/Mn–N–C, (h) wavelet transform of the k3-weighted Ni K edge and Mn K edge EXAFS data of Ni/Mn–N–C and reference samples (Ni foil, NiPc, Mn foil and MnPc). | ||
Moreover, the chemical states and local structures of Ni and Mn in Ni/Mn–N–C were characterized by X-ray absorption spectroscopy (XAS). The Ni X-ray absorption near edge structures (XANES) of both Ni/Mn–N–C and Ni–N–C was different from Ni foil but close to NiPc, indicating that the average oxidation states of Ni in the two catalysts are close to +2 (Fig. 2d and S7a†). The Ni extended X-ray absorption fine structure (EXAFS) spectra of both Ni–N–C and Ni/Mn–N–C shown in Fig. 2e and S7b† revealed the presence of Ni–N bonds located at ∼1.4 Å and the absence of Ni–Ni bonds located at ∼2.2 Å, confirming that Ni existed in form of isolated single atomic site instead of metal–metal bonding. Similarly, it was speculated that the average oxidation state of Mn atoms is close to +2 in both Mn–N–C and Ni/Mn–N–C (Fig. 2f and S8a†). The presence of Mn–N bonds located at ∼1.4 Å and the absence of Mn–Mn bonds located at ∼2.3 Å in both Mn–N–C and Ni/Mn–N–C confirmed that the Mn atoms also existed in isolated single atomic state instead of metal–metal bonding (Fig. 2g and S8b†). Furthermore, the wavelet transform contour plots of Ni and Mn in Ni/Mn–N–C showed that the k values with the greatest intensity were close to the Ni–N coordination in NiPc and the Mn–N coordination in MnPc (Fig. 2h),36 respectively, which demonstrated that almost no metal–metal (Ni–Ni, Ni–Mn, Mn–Mn) bonding existed in the Ni/Mn–N–C sample. The results suggested that the Ni/Mn atoms in the as-described Ni/Mn–N–C catalyst were mainly coordinated with N, consistent with the above TEM and XPS analysis results.
The CO2RR performances of as-prepared catalysts were performed in an H-type cell using 0.5 M KHCO3 aqueous solution as electrolytes. As shown in the linear sweep voltammetry curves (Fig. 3a), the higher current density after −0.50 V in CO2-saturated electrolytes (the solid lines) than in Ar (the dashed lines) suggested a higher activity of CO2RR than hydrogen evolution on the Ni/Mn–NC catalyst. Furthermore, the current density observed on Ni/Mn–N–C (red line) was significantly larger than that observed on Ni–N–C (green line) and Mn–N–C (blue line), indicating that Ni/Mn–N–C has an appreciably higher CO2RR response. The potential-dependent selectivity for CO2RR products of the as-prepared catalysts was analyzed by gas chromatography (GC) and hydrogen nuclear magnetic resonance (1H NMR). As shown in Fig. 3b and c, all Ni/Mn–N–C, Ni–N–C, and Mn–N–C catalysts exhibited excellent CO2RR selectivity from −0.56 to −0.96 V, where CO was the main product, with H2 as the only by-product (Fig. S9–S12†). Ni/Mn–N–C outperformed Ni–N–C and Mn–N–C across the entire potential window, reaching a maximum CO faradaic efficiency (FECO) of 96.6% at −0.76 V (Fig. 3b). Moreover, Ni/Mn–N–C achieved a CO current density (jCO) of 13.3 mA cm−2 at −0.76 V, 1.6 and 7.8 times higher than that of Ni–N–C and Mn–N–C, respectively (Fig. 3c). These results demonstrated that the introduction of Mn–N sites significantly enhanced the CO2RR performance of the Ni/Mn–N–C catalyst. The negligible catalytic performance of metal-free N-doped carbon support indicated that the high catalytic activity should be attributed to the metal sites (Fig. S13†). In addition, no appreciable decays of CO selectivity (maintained at above 90%) (Fig. 3d) and no structure changes (Fig. S14†) of Ni/Mn–N–C were observed after a 20 hours stability test at −0.76 V, suggesting the excellent stability of the Ni/Mn–N–C catalyst.
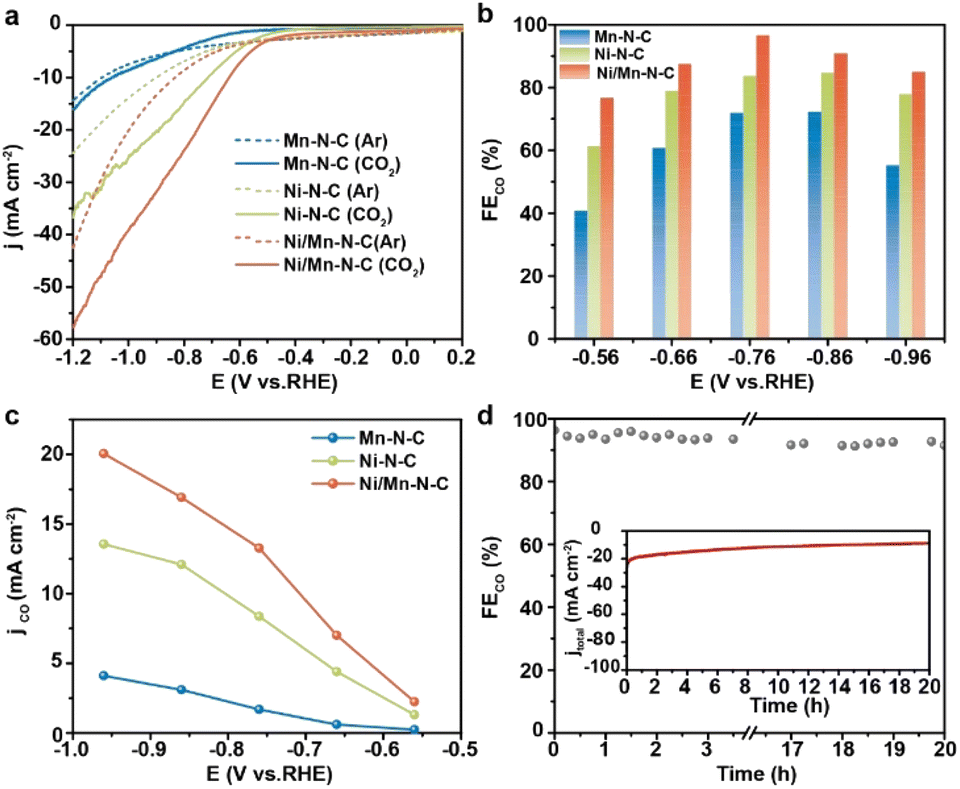 | ||
| Fig. 3 (a) LSV curves performed in Ar-saturated and CO2-saturated 0.5 M KHCO3 solution of Ni/Mn–N–C, Ni–N–C, and Mn–N–C; (b) potential-dependent FECO, (c) potential-dependent jCO of the Ni/Mn–N–C, Ni–N–C and Mn–N–C catalyst for CO2RR, (d) long-term stability of Ni/Mn–N–C for CO2RR at −0.76 V. | ||
To further explore the origin of the high catalytic activity, the electrochemical properties on the catalytic interface of Ni/Mn–N–C catalysts were systematically studied. The electrochemically-active surface areas (ECSA) of as-prepared catalysts were compared using electric double-layer capacitance measurements (Fig. S15†). All as-prepared catalysts displayed similar Cdl (0.0147–0.0174 F cm−2), suggesting the similar ECSAs. Tafel slopes were calculated to reveal reaction kinetics at different sites. Tafel slopes of Ni/Mn–N–C are 169 mV decade−1, less than 192 mV decade−1 of Ni–N–C, and 175 mV decade−1 of Mn–N–C (Fig. S16†), indicating that the introduction of Mn–N sites enhances the kinetics of CO2RR process. Further, we utilized a rotating ring-disk electrode (RRDE) assembly to evaluate the local pH changes on the catalytic interface of CO2RR as in previous reports.37 In brief, the as-prepared catalysts (Ni/Mn–N–C, Ni–N–C, and Mn–N–C) were deposited on the disk electrode and performed the CO2 reduction process (eqn (2)). Then the generated CO and OH− could directly diffuse onto the Pt ring electrode with a rotating speed of 1600 rpm. With the cyclic voltammetry measurements on the Pt ring electrode, the CO oxidation peak was detected for probing the local pH on the catalyst surface (eqn (3)). According to the Nernst relationship, the shift of the CO oxidation peak could proportionally reflect the local pH increase when the generated OH− overcomes the CO2(aq)–HCO3− buffer within the Nernst layer with the electrolysis current density increases.37
| CO2 + 2e− + H2O → CO + 2OH− (on the disc) | (2) |
| CO + 2OH−–2e− → CO2 + H2O (on the Pt ring) | (3) |
The cyclic voltammograms of CO oxidation under different disc potentials of the as-prepared Ni/Mn–N–C, Ni–N–C, and Mn–N–C catalysts are shown in Fig. 4a–c. With the applied CO2RR potential decreased, the oxidation peak potential of CO shifted negatively, indicating the increase of local pH caused by the generated OH−, consistent with the previous study.37 Moreover, both Ni/Mn–N–C and Ni–N–C catalysts exhibited a negative logarithmic relationship between the CO oxidation peak potential and the electrolysis current density under a high current density region (Fig. 4d) with slopes of −0.08 V dec−1 and −0.11 V dec−1, respectively. The more gentle slope of the Ni/Mn–N–C sample than Ni–N–C indicates that the introduction of Mn–N sites may buffer the local pH on the catalytic interface, leading to faster CO2RR kinetics than the Ni–N sites.37
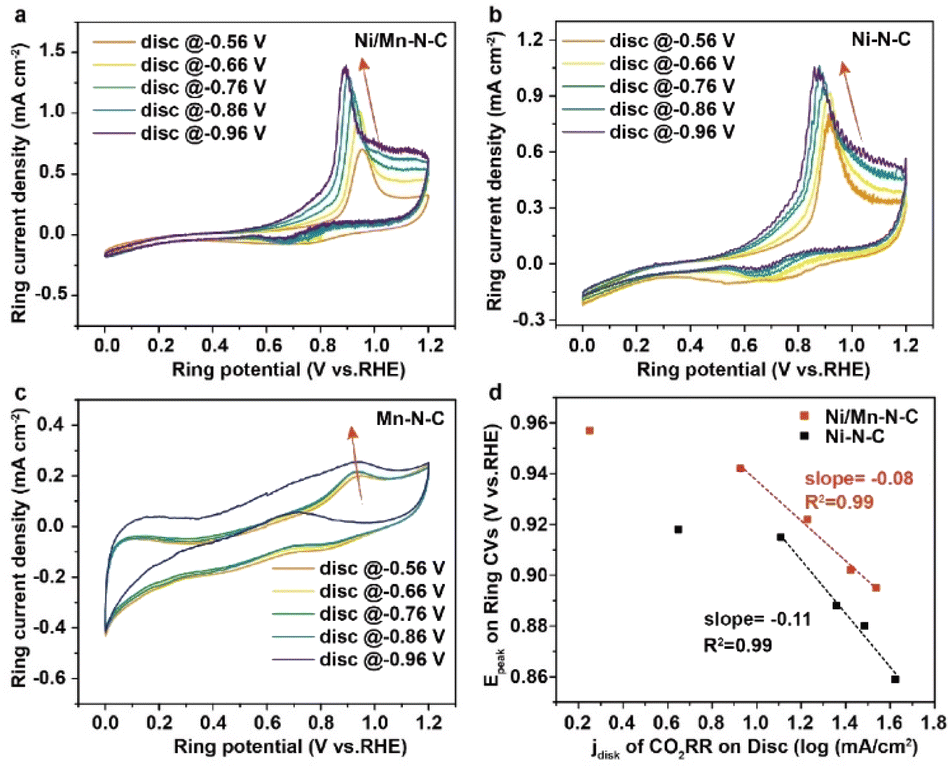 | ||
| Fig. 4 Cyclic voltammograms of CO oxidation on Pt ring electrode on the (a) Ni/Mn–N–C, (b) Ni–N–C, (c) Mn–N–C disk electrode under different CO2RR potentials, (d) the dependence of CO oxidation peak potential on the log value of electrolysis current for Ni/Mn–N–C and Ni–N–C catalysts. | ||
Conclusions
In conclusion, we successfully developed a Ni/Mn–N–C diatomic catalyst and found that single atomic Mn–N auxiliary sites could effectively regulate the local microenvironment of single atomic Ni–N–C sites for accelerating CO2 to CO conversion. Systematic characterizations confirmed that the Ni and Mn were atomically dispersed on the N-doped carbon substrate in form of Ni–N–C and M–N–C coordinate structures. The as-described Ni/Mn–N–C catalyst performed excellent CO2RR activity with the highest CO faradaic efficiency of 96.6% and the partial current density of 13.3 mA cm−2 at −0.76 V, exceeding that of the monometallic Ni–N–C and Mn–N–C counterparts. Further, the detailed electrochemical analyses revealed a buffered local pH on the Ni/Mn–N–C surface, indicating that the introduction of Mn–N sites dynamically regulated the local pH, accelerating the kinetics of the PCET step and thus promoting the CO2 electroreduction to CO. These findings suggested that constructing various synergistic catalytic sites to modulate the surface local microenvironment might be an attractive strategy for promoting CO2-to-CO conversion and providing insights into advanced catalyst design for optimizing the local microenvironment in CO2RR and other electrocatalytic reactions.Data availability
The data that support the plots within this paper and other finding of this study are available from the corresponding authors upon reasonable request.Author contributions
Y. Y., J. F. and J.-S. H. conceived the project. Y. Y. performed experiments. Z. H. Lyu helped do some characterization. Y. Y., J. F., T. T. did data analysis. L.-R. Z. analyzed the XAS data. Q.-H. Z. performed the HAADF-STEM characterizations experiments. Y. Y., J. F., and J.-S. H. cowrote the paper. J.-S. H. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the National Key Research and Development Program of China (2020YFB1505801), the National Natural Science Foundation of China (22025208, 22075300, 22102191), the DNL Cooperation Fund, CAS (DNL202008) and the Chinese Academy of Sciences. They greatly thank Yang Sun for XRD analysis; Dr Zhi-Juan Zhao and Xiao-Yu Zhang for their help in XPS data analysis; and Professor Li-Rong Zheng for his help with the XAS experiments.Notes and references
- S. Lin, S. Diercks Christian, Y.-B. Zhang, N. Kornienko, M. Nichols Eva, Y. Zhao, R. Paris Aubrey, D. Kim, P. Yang, M. Yaghi Omar and J. Chang Christopher, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- L.-P. Yuan, W.-J. Jiang, X.-L. Liu, Y.-H. He, C. He, T. Tang, J. Zhang and J.-S. Hu, ACS Catal., 2020, 10, 13227–13235 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- C. He, Y. Zhang, Y. Zhang, L. Zhao, L.-P. Yuan, J. Zhang, J. Ma and J.-S. Hu, Angew. Chem., Int. Ed., 2020, 59, 4914–4919 CrossRef CAS PubMed.
- J. Fu, Y. Yang and J.-S. Hu, ACS Mater. Lett., 2021, 3, 1468–1476 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang, J.-S. Hu and L.-J. Wan, Acta Phys.-Chim. Sin., 2020, 36, 1906085 Search PubMed.
- T. Tang, L. Ding, Z. Jiang, J.-S. Hu and L.-J. Wan, Sci. China: Chem., 2020, 63, 1517–1542 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, M. Chen Hao and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 1022–1032 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- L.-P. Yuan, T. Tang, J.-S. Hu and L.-J. Wan, Acc. Mater. Res., 2021, 2, 907–919 CrossRef CAS.
- L. Zhao, S.-Q. Wang, S. Liang, Q. An, J. Fu and J.-S. Hu, Coord. Chem. Rev., 2022, 466, 214603 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. C. Bui, C. Kim, A. J. King, O. Romiluyi, A. Kusoglu, A. Z. Weber and A. T. Bell, Acc. Chem. Res., 2022, 55, 484–494 CrossRef CAS PubMed.
- H. Hashiba, L.-C. Weng, Y. Chen, H. K. Sato, S. Yotsuhashi, C. Xiang and A. Z. Weber, J. Phys. Chem. C, 2018, 122, 3719–3726 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS.
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- H. Li, J. Wei, X. Zhu, L. Gan, T. Cheng and J. Li, J. Phys. Chem. C, 2022, 126, 9665–9672 CrossRef CAS.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- M. Dunwell, X. Yang, B. P. Setzler, J. Anibal, Y. Yan and B. Xu, ACS Catal., 2018, 8, 3999–4008 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- M. R. Singh, E. L. Clark and A. T. Bell, Phys. Chem. Chem. Phys., 2015, 17, 18924–18936 RSC.
- I. Katsounaros, J. C. Meier, S. O. Klemm, A. A. Topalov, P. U. Biedermann, M. Auinger and K. J. J. Mayrhofer, Electrochem. Commun., 2011, 13, 634–637 CrossRef CAS.
- L. Zhao, Y. Zhang, Z. Zhao, Q.-H. Zhang, L.-B. Huang, L. Gu, G. Lu, J.-S. Hu and L.-J. Wan, Natl. Sci. Rev., 2020, 7, 27–36 CrossRef CAS PubMed.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef PubMed.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, ACS Catal., 2022, 12, 1216–1227 CrossRef CAS.
- Y. Yang, J. Fu, Y. Ouyang, T. Tang, Y. Zhang, L.-R. Zheng, Q.-H. Zhang, X.-Z. Liu, J. Wang and J.-S. Hu, Natl. Sci. Rev., 2022 DOI:10.1093/nsr/nwac248.
- H. Funke, M. Chukalina and A. C. Scheinost, J. Synchrotron Radiat., 2007, 14, 426–432 CrossRef CAS PubMed.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, supporting figures and tables. See DOI: https://doi.org/10.1039/d2sc04776d |
| This journal is © The Royal Society of Chemistry 2022 |