
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormal nucleophilic pyrrolylmethylation via palladium-based auto-tandem catalysis: switchable regiodivergent synthesis and remote chirality transfer†
Zhi
Chen
a,
Yu-Fan
Li
a,
Shun-Zhong
Tan
a,
Qin
Ouyang
 b,
Zhi-Chao
Chen
*a,
Wei
Du
a and
Ying-Chun
Chen
*ab
b,
Zhi-Chao
Chen
*a,
Wei
Du
a and
Ying-Chun
Chen
*ab
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry and Sichuan Province, Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: chenzhichao@scu.edu.cn; ycchen@scu.edu.cn; Fax: +86 28 85502609
bCollege of Pharmacy, Third Military Medical University, Shapingba, Chongqing 400038, China
First published on 6th October 2022
Abstract
Although nucleophilic benzylation-type reaction to introduce various aromatic systems into molecules has been widely explored, the related pyrrolylmethylation version remains to be disclosed. Reported herein is a palladium-catalysed multiple auto-tandem reaction between N-Ts propargylamines, allyl carbonates and aldimines in the presence of an acid, proceeding through sequential allylic amination, cycloisomerisation, vinylogous addition and aromatisation steps. A diversity of formal pyrrolylmethylated amine products were finally furnished efficiently. In addition, switchable regiodivergent 3-pyrrolylmethylation and 4-pyrrolylmethylation were realised by tuning catalytic conditions. Moreover, remote chirality transfer with readily available enantioenriched starting materials was well achieved with an achiral ligand, relying on diastereoselective generation of η2-Pd(0) complexes between Pd(0) and chiral 1,3-diene intermediates in the key vinylogous addition step. A few control experiments were conducted to elucidate the palladium-involved tandem reaction and regiodivergent synthesis.
Introduction
Nucleophilic benzylation-type reaction provides an effective approach to introduce an aromatic system into molecules. In comparison with many protocols available for benzylic functionalisation of six-membered aromatic compounds [Scheme 1a(i)],1 reactions involving five-membered heteroaromatic benzylic nucleophiles or relevant precursors have been significantly underdeveloped. You and Newhouse independently made an important breakthrough relying on the formation of Lewis acid stabilised deprotonated methyl azaarenes in the presence of excess strong Brønsted bases (TMPZnX) [Scheme 1a(ii)].2 However, electron-rich heterocyclic aromatic compounds, such as methyl-substituted pyrroles and furans, were not applicable, because of the lack of an appropriate activation strategy to overcome the intrinsic weak acidity of benzylic protons. On the other hand, a few methylene-substituted heterocycles could perform as nucleophilic benzylation precursors to couple with electrophiles through an Alder-ene reaction, which provided an alternative access to diverse heteroarylmethylated products [Scheme 1a(iii)].3 Unfortunately, these preformed electron-rich dienes, especially pyrrole-derived ones, were liable to aromatise, which not only limited their application scope, but also rendered formal pyrrolylmethylation reaction unsuccessful.3a,4 As a result, a new synthetic strategy remains to be developed to accomplish the efficient construction of pyrrolylmethylated products.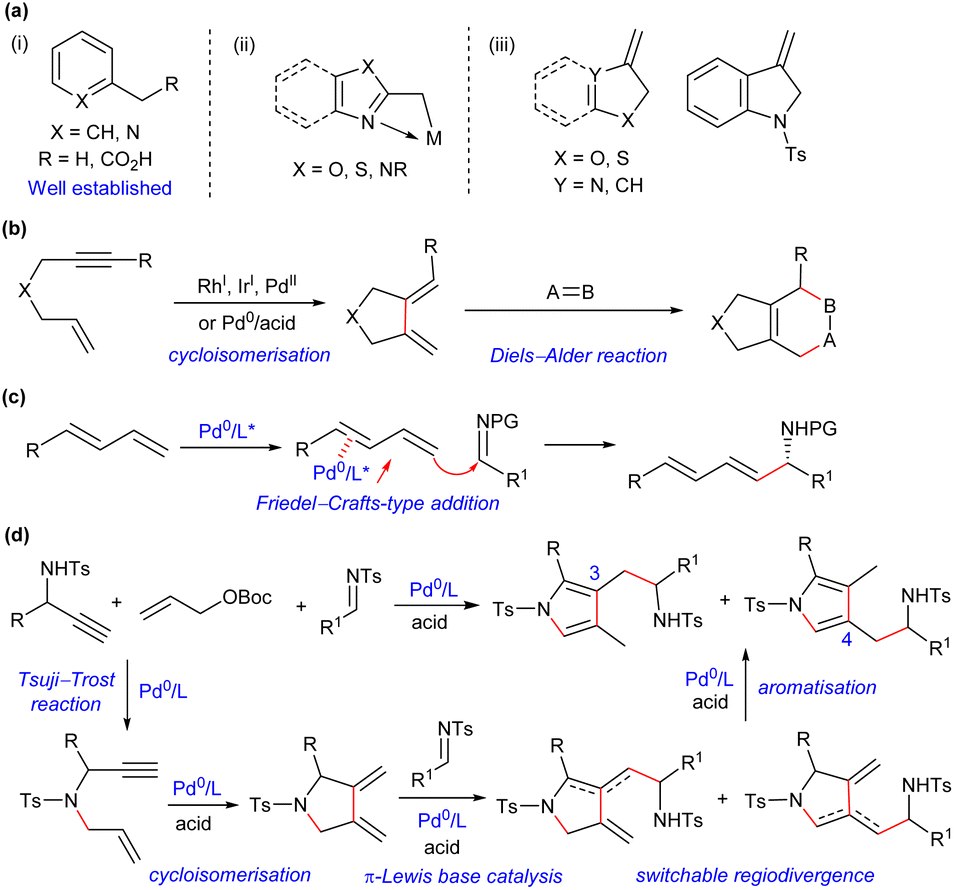 | ||
| Scheme 1 Reported nucleophilic benzylation-type strategies and our design for formal pyrrolylmethylation. (a) Typical nucleophilic benzylation precursors. (b) Tandem cycloisomerisation/Diels–Alder reaction of 1,6-enynes. (c) Coupling of 1,3-dienes and imines via π-Lewis base catalysis of Pd(0). (d) This work: formal pyrrolylmethylation via palladium-based auto-tandem catalysis. | ||
Enynes are readily accessible and versatile synthons, and the transition metal-catalysed cycloisomerisation reaction of 1,6-enynes,5 pioneered by Trost,6 provides a powerful access to exocyclic conjugated 1,3-dienes, which can be efficiently trapped by various dienophiles in a tandem Diels–Alder cycloaddition reaction (Scheme 1b).7 However, other types of derivation reactions of 1,3-dienes initiated from cycloisomerisation of 1,6-enynes have been significantly underdeveloped, probably because such 1,3-dienes are thermally unstable, which should be prepared freshly and used immediately. Recently, our group disclosed a Friedel–Crafts-type coupling reaction of linear 1,3-dienes and imines via Pd(0)-based π-Lewis base catalysis (Scheme 1c).8 We further noticed that the expected 1,3-dienes, though in an exocyclic form, would be readily obtained from simple propargyl amines and allyl carbonates via a palladium-catalysed tandem allylic amination and cycloisomerisation process. We envisioned that such 1,3-dienes would be feasibly assembled with imines under auto-tandem Pd(0) catalysis, and the unprecedented formal pyrrolylmethylation products, which are not readily accessible by other means, would be finally furnished after isomerisation, as proposed in Scheme 1d. Moreover, potential regiodivergent vinylogous additions of the unsymmetric 1,3-diene intermediates might be achievable by properly tuning the catalytic conditions,9 which would enrich the structural diversity of densely substituted pyrrole frameworks. Consequently, such a multicomponent reaction, starting from readily available building blocks and combining palladium-mediated four consecutive transformations, would finely demonstrate the synthetic efficacy and versatility of auto-tandem catalysis (ATC).10
Results and discussion
Condition optimisation
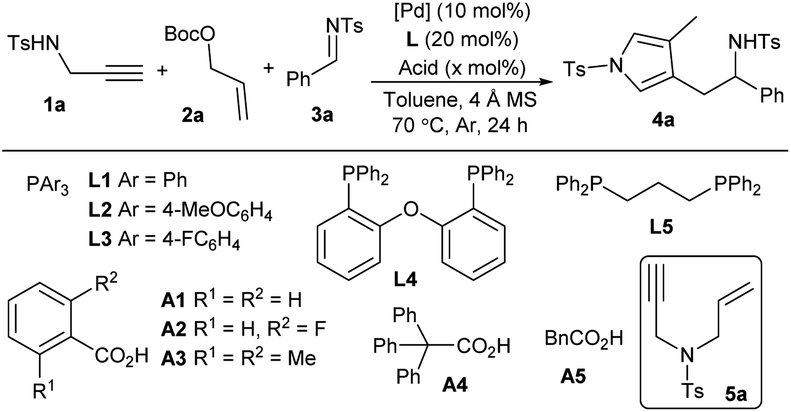
tWe began our exploration by examining the reaction with N-Ts propargyl amine 1a, allylic carbonate 2a and N-Ts aldimine 3a in toluene at 70 °C in the presence of catalytic amounts of Pd(PPh3)4 and benzoic acid A1. Pleasingly, the proposed tandem reaction occurred as expected, and the aromatic pyrrole product 4a, proceeding through an N-allylation, cycloisomerisation, vinylogous addition and aromatisation sequence, was obtained in a moderate yield after 24 h (Table 1, entry 1). The combination of Pd2dba3 with PPh3L1 demonstrated to be less efficient (entry 2). Electron-richer ligand L2 delivered comparable results (entry 3), but electron-deficient ligand L3, bisphosphine L4 and L5 could not promote the allylic amination to produce 5a (entries 4–6). An acid additive was found to be crucial for the tandem conversion, as only N-allylation intermediate 5a was isolated quantitatively in the absence of A1, indicating the necessity of both palladium and acid for subsequent cycloisomerisation (entry 7). A slight effect was observed for different acid loadings (entries 8–10). More acids were examined (entries 11–14), and stronger o-fluorobenzoic acid A2 improved the yield significantly (entry 11). In addition, temperature and solvents were further evaluated, but no better results were obtained (entries 15 and 16). Moreover, a slightly reduced yield was obtained in the absence of 4 Å MS, probably due to the partial decomposition of imine 3a (entry 17).11|
|
|||||
|---|---|---|---|---|---|
| Entry | [Pd] | L | Acid | x | Yieldb (%) |
| a Unless noted otherwise, reactions were performed with 1a (0.1 mmol), 2a (0.1 mmol), 3a (0.05 mmol), [Pd] source (10 mol%), L (20 mol%), acid (x mol%) and 4 Å MS (20 mg) in degassed dry toluene (0.5 mL) at 70 °C for 24 h under Ar. b Yield of isolated product 4a. c At 80 °C. d In xylene (0.5 mL). e Without 4 Å MS. | |||||
| 1 | Pd(PPh3)4 | — | A1 | 20 | 58 |
| 2 | Pd2dba3 | PPh3 | A1 | 20 | 54 |
| 3 | Pd2dba3 | L2 | A1 | 20 | 53 |
| 4 | Pd2dba3 | L3 | A1 | 20 | NR |
| 5 | Pd2dba3 | L4 | A1 | 20 | NR |
| 6 | Pd2dba3 | L5 | A1 | 20 | NR |
| 7 | Pd(PPh3)4 | — | A1 | 0 | 5a, 95 |
| 8 | Pd(PPh3)4 | — | A1 | 10 | 50 |
| 9 | Pd(PPh3)4 | — | A1 | 40 | 68 |
| 10 | Pd(PPh3)4 | — | A1 | 80 | 68 |
| 11 | Pd(PPh3)4 | — | A2 | 40 | 86 |
| 12 | Pd(PPh3)4 | — | A3 | 40 | 54 |
| 13 | Pd(PPh3)4 | — | A4 | 40 | 65 |
| 14 | Pd(PPh3)4 | — | A5 | 40 | 66 |
| 15c | Pd(PPh3)4 | — | A2 | 40 | 80 |
| 16d | Pd(PPh3)4 | — | A2 | 40 | 41 |
| 17e | Pd(PPh3)4 | — | A2 | 40 | 80 |
Substrate scope of three-component ATC reaction
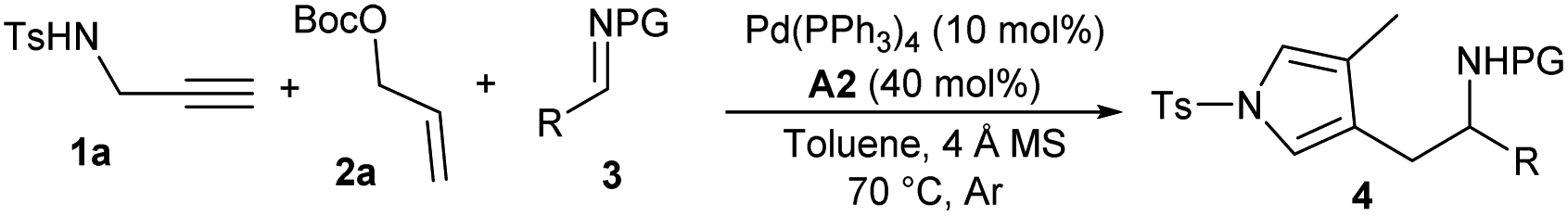
Consequently, the substrate scope and limitations of the multicomponent auto-tandem reaction were investigated under the catalysis of Pd(PPh3)4 and o-fluorobenzoic acid A2. As summarised in Table 2, an array of aryl aldimines 3 bearing either electron-donating or -withdrawing groups performed well in the reactions with propargyl amine 1a and allylic carbonate 2a, and corresponding products 4b–4g were isolated in moderate to good yields (Table 2, entries 2–7). Similar results were obtained with naphthyl and heteroaryl derived aldimines (entries 8–10). Nevertheless, alkyl-tethered aldimines proved to be less reactive, and a longer reaction time was required to obtain better results (entries 11 and 12). Besides, aldimines with other N-protecting groups were also applicable, affording 4m–4o with comparable yields (entries 13–15). In addition, the reaction took place efficiently on a 1.0 mmol scale (entry 16).|
|
||||
|---|---|---|---|---|
| Entry | R | PG | t (h) | Yieldb (%) |
| a Unless noted otherwise, reactions were performed with 1a (0.2 mmol), 2a (0.2 mmol), imine 3 (0.1 mmol), Pd(PPh3)4 (10 mol%), A2 (40 mol%) and 4 Å MS (40 mg) in degassed dry toluene (1.0 mL) at 70 °C under Ar. b Yield of isolated product. c With 5a (0.2 mmol) instead of 1a and 2a. d On a 1.0 mmol scale. | ||||
| 1 | Ph | Ts | 24 | 4a, 86 |
| 2 | 2-MeC6H4 | Ts | 24 | 4b, 93 |
| 3 | 3-MeC6H4 | Ts | 36 | 4c, 70 |
| 4 | 4-MeC6H4 | Ts | 24 | 4d, 80 |
| 5 | 4-PhC6H4 | Ts | 24 | 4e, 79 |
| 6 | 4-FC6H4 | Ts | 24 | 4f, 68 |
| 7 | 4-ClC6H4 | Ts | 24 | 4g, 74 |
| 8 | 2-Naphthyl | Ts | 24 | 4h, 66 |
| 9 | 2-Furyl | Ts | 24 | 4i, 80 |
| 10 | 2-Thienyl | Ts | 36 | 4j, 72 |
| 11 | PhCH2CH2 | Ts | 96 | 4k, 62 |
| 12 | c-Hexyl | Ts | 96 | 4l, 23 |
| 13 | Ph | Ns | 96 | 4m, 52 |
| 14c | Ph | Bz | 24 | 4n, 70 |
| 15c | Ph | SO2NMe2 | 24 | 4o, 90 |
| 16d | Ph | Ts | 24 | 4a, 82 |
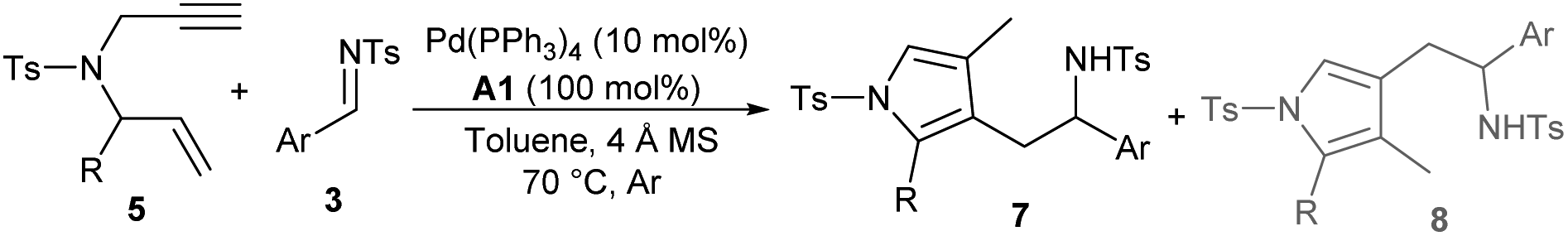
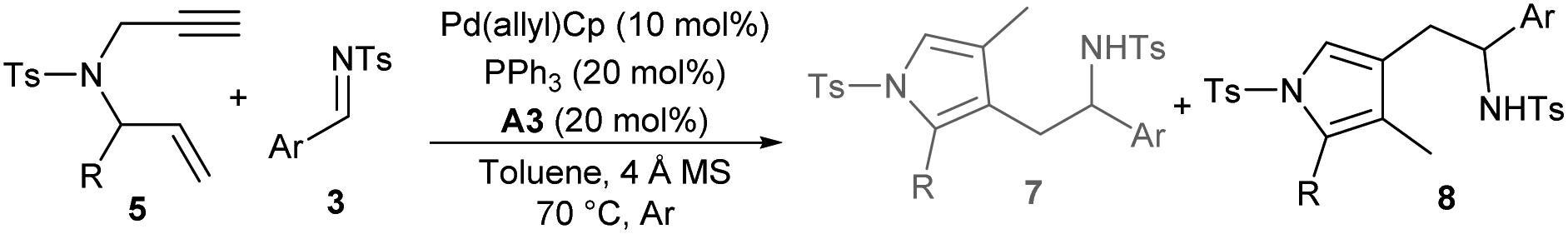
Switchable regiodivergent tandem reaction
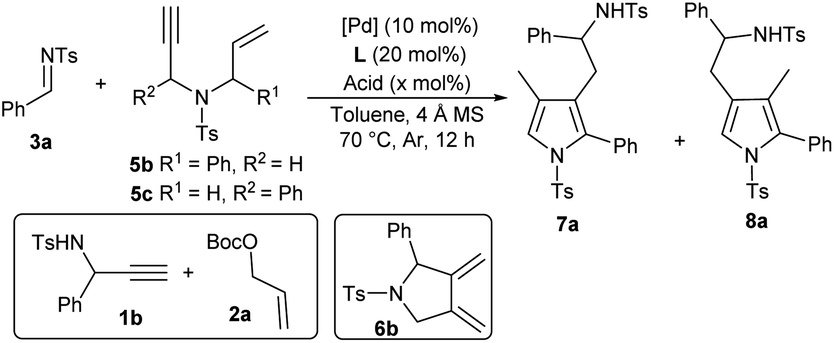
After establishing the auto-tandem catalysis, we turned to explore substrates with more complicated substitutions. Interestingly, the 1,6-enyne 5b, which could lead to the formation of unsymmetric exocyclic 1,3-diene intermediate 6bvia cycloisomerisation, delivered a pair of separable regioselective isomers 7a and 8a efficiently in combination with imine 3a under the catalysis of Pd(PPh3)4 and acid A1 (Table 3, entry 1). While using other acid additives had a marginal effect on the regioselectivity (entries 2 and 3), it was pleasing that increasing the loadings of acid A1 dramatically improved the formation of 3-pyrrolylmethylated product 7a (entry 4), and excellent yield and regioselectivity were obtained by employing stoichiometric A1 (entry 5). In order to switch the regioselectivity, more reaction parameters were investigated. A series of Pd(II) sources were tested. While using Pd(OAc)2 or [Pd(allyl)Cl]2 still favoured the formation of isomer 7a (entries 6 and 7), the combination of Pd(allyl)Cp and PPh3 could promote the production of 4-pyrrolylmethylated adduct 8a dominantly (entry 8).11 A survey of acid additives (entries 9–12) showed that A3 further improved the yield and regioselectivity (entry 10). Notably, the regioselectivity was significantly eroded by using stoichiometric A3 (entry 13). Moreover, employing 1,6-enyne 5c instead of 5b still furnished 7a in a good yield in the presence of Pd(PPh3)4 and acid A1 (entry 14), but poor regioselectivity was observed when the optimal conditions for isomer 8a were utilised (entry 15). Importantly, the catalytic three-component cascade reaction of propargylamine 1b and allylic carbonate 2a as precursors of 5c still proceeded well, giving isomer 7a in a good yield (entry 16).11|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Pd] | L | Acid | x | Yieldb (%) | rrc |
a Unless noted otherwise, reactions were performed with 1,6-enyne 5b (0.1 mmol), imine 3a (0.05 mmol), [Pd] source (10 mol%), L (20 mol%), acid (x mol%) and 4 Å MS (20 mg) in degassed dry toluene (0.5 mL) at 70 °C for 12 h under Ar.
b Isolated yield.
c rr = 7a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8a, determined by 1H-NMR analysis.
d With 5c (0.1 mmol) instead of 5b.
e With 1b (0.1 mmol) and 2a (0.1 mmol) instead of 5b at 80 °C. :8a, determined by 1H-NMR analysis.
d With 5c (0.1 mmol) instead of 5b.
e With 1b (0.1 mmol) and 2a (0.1 mmol) instead of 5b at 80 °C.
|
||||||
| 1 | Pd(PPh3)4 | — | A1 | 20 | 75 | 52:48 |
| 2 | Pd(PPh3)4 | — | A2 | 20 | 65 | 63:37 |
| 3 | Pd(PPh3)4 | — | A3 | 20 | 60 | 54:46 |
| 4 | Pd(PPh3)4 | — | A1 | 50 | 90 | 75:25 |
| 5 | Pd(PPh3)4 | — | A1 | 100 | 91 | 94:6 |
| 6 | Pd(OAc)2 | PPh3 | A1 | 20 | 48 | 60:40 |
| 7 | [Pd(allyl)Cl]2 | PPh3 | A1 | 20 | 36 | 67:33 |
| 8 | Pd(allyl)Cp | PPh3 | A1 | 20 | 71 | 13:87 |
| 9 | Pd(allyl)Cp | PPh3 | A2 | 20 | 80 | 40:60 |
| 10 | Pd(allyl)Cp | PPh3 | A3 | 20 | 82 | 8:92 |
| 11 | Pd(allyl)Cp | PPh3 | A4 | 20 | 47 | 24:76 |
| 12 | Pd(allyl)Cp | PPh3 | A5 | 20 | Trace | — |
| 13 | Pd(allyl)Cp | PPh3 | A3 | 100 | 62 | 44:56 |
| 14d | Pd(PPh3)4 | — | A1 | 100 | 87 | 96:4 |
| 15d | Pd(allyl)Cp | PPh3 | A3 | 20 | 73 | 30:70 |
| 16e | Pd(PPh3)4 | — | A2 | 40 | 80 | 95:5 |
Consequently, we first explored the substrate scope for the construction of 3-pyrrolylmethylated adducts 7 using catalytic Pd(PPh3)4 and stoichiometric benzoic acid A1. As summarised in Table 4, a broad variety of aryl- or heteroaryl substituted aldimines 3 were well tolerated in the reactions with 1,6-enyne 5b. High regioselectivity was generally observed, and the expected regioselective products 7b–7k were smoothly isolated in moderate to excellent yields (Table 4, entries 2–11). On the other hand, 1,6-enynes 5 with diverse aryl or heteroaryl substituents also reacted well with imine 3a, giving products 7l–7o in good yields (entries 12–15). In addition, 2-styryl- or alkyl-substituted 1,6-enynes 5 were also applied to give the desired products 7p–7s with excellent regioselectivity (entries 16–19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | R | t (h) | Yieldb (%) | rrc |
|
a Unless noted otherwise, reactions were performed with 1,6-enyne 5 (0.2 mmol), imine 3 (0.1 mmol), Pd(PPh3)4 (10 mol%), A1 (100 mol%) and 4 Å MS (40 mg) in degassed dry toluene (1.0 mL) at 70 °C under Ar.
b Yield of isolated pure product 7.
c rr = 7:8, determined by 1H NMR analysis.
|
|||||
| 1 | Ph | Ph | 12 | 7a, 90 | 95:5 |
| 2 | 2-MeC6H4 | Ph | 36 | 7b, 76 | >95:5 |
| 3 | 3-MeC6H4 | Ph | 24 | 7c, 88 | >95:5 |
| 4 | 4-MeC6H4 | Ph | 36 | 7d, 71 | >95:5 |
| 5 | 4-PhC6H4 | Ph | 24 | 7e 64 | 91:9 |
| 6 | 2-FC6H4 | Ph | 12 | 7f, 56 | 81:19 |
| 7 | 4-FC6H4 | Ph | 12 | 7g, 75 | 93:7 |
| 8 | 4-ClC6H4 | Ph | 12 | 7h, 82 | 95:5 |
| 9 | 2-Naphthyl | Ph | 36 | 7i, 82 | 90:10 |
| 10 | 2-Furyl | Ph | 36 | 7j, 74 | >95:5 |
| 11 | 2-Thienyl | Ph | 36 | 7k, 51 | >95:5 |
| 12 | Ph | 4-MeC6H4 | 12 | 7l, 81 | 93:7 |
| 13 | Ph | 4-ClC6H4 | 12 | 7m, 76 | 92:8 |
| 14 | Ph | 2-Furyl | 36 | 7n, 90 | >95:5 |
| 15 | Ph | 2-Thienyl | 36 | 7o, 80 | >95:5 |
| 16 | Ph | 2-Styryl | 48 | 7p, 36 | >95:5 |
| 17 | Ph | n-Butyl | 48 | 7q, 87 | >95:5 |
| 18 | Ph | i-Propyl | 48 | 7r, 47 | >95:5 |
| 19 | Ph | c-Hexyl | 48 | 7s, 79 | >95:5 |
Meanwhile, the substrate scope for the synthesis of 4-pyrrolylmethylated isomers 8 was investigated under the catalysis of Pd(allyl)Cp, PPh3 and acid A3. As summarised in Table 5, moderate to good yields were uniformly attained for an array of assemblies of 1,6-enynes 5 and aldimines 3 (Table 5, entries 2–14). It should be noted that for products 8l and 8n with a 2-substituted phenyl group, apparent diastereoselectivity was observed for the newly generated axial chirality because of the hindered rotation (entries 12 and 14). However, the regioselectivity for heteroaryl or alkyl substituted 1,6-enynes was not switched, whereas 3-pyrrolylmethylated products 7n and 7q were produced exclusively (entries 15 and 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | R | t (h) | Yieldb (%) | rrc |
|
a Unless noted otherwise, reactions were performed with 1,6-enyne 5 (0.2 mmol), imine 3 (0.1 mmol), Pd(allyl)Cp (10 mol%), PPh3 (20 mol%), A3 (20 mol%) and 4 Å MS (40 mg) in degassed dry toluene (1.0 mL) at 70 °C under Ar.
b Yield of isolated pure product 8.
c rr = 7:8, determined by 1H NMR analysis.
d Total yield of 7j and 8j, with 20:80 rr as inseparable isomers.
e With 1:1 dr for atropisomers.
|
|||||
| 1 | Ph | Ph | 36 | 8a, 75 | 7:93 |
| 2 | 2-MeC6H4 | Ph | 36 | 8b, 74 | 7:93 |
| 3 | 3-MeC6H4 | Ph | 24 | 8c, 66 | 10:90 |
| 4 | 4-MeC6H4 | Ph | 96 | 8d, 67 | 18:82 |
| 5 | 4-PhC6H4 | Ph | 24 | 8e, 72 | <5:95 |
| 6 | 2-FC6H4 | Ph | 24 | 8f, 54 | <5:95 |
| 7 | 4-FC6H4 | Ph | 12 | 8g, 72 | <5:95 |
| 8 | 4-ClC6H4 | Ph | 12 | 8h, 73 | <5:95 |
| 9 | 2-Naphthyl | Ph | 36 | 8i, 69 | 14:86 |
| 10d | 2-Furyl | Ph | 36 | 7j+8j, 70 | 20:80 |
| 11 | 2-Thienyl | Ph | 36 | 8k, 60 | 26:74 |
| 12e | Ph | 2-MeC6H4 | 36 | 8l, 73 | <5:95 |
| 13 | Ph | 4-MeC6H4 | 12 | 8m, 62 | 18:82 |
| 14e | Ph | 2-ClC6H4 | 36 | 8n, 67 | <5:95 |
| 15 | Ph | 2-Furyl | 36 | 7n, 76 | >95:5 |
| 16 | Ph | n-Butyl | 48 | 7q, 45 | >95:5 |
Asymmetric catalytic exploration
Much effort was dedicated to investigating the asymmetric catalytic version of this new cascade process. A number of commonly used chiral ligands in combination with Pd2dba3 failed to promote the reaction of imine 3a and pre-prepared 1,6-enyne 5a or give poor enantioselectivity.11 To our delight, a bifunctional phosphine ligand L6 exhibited good catalytic activity and moderate enantiocontrol.12 However, poor enantiocontrol was observed for the three component auto-tandem reaction due to the presence of in situ formed t-BuOH, which has been verified by a control experiment (Scheme 2).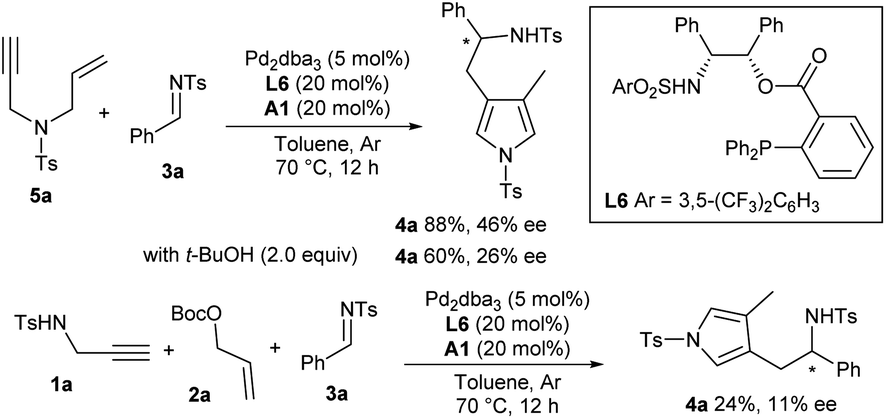 | ||
| Scheme 2 Asymmetric catalytic cascade reaction. | ||
Enantioselective synthesis through chirality transfer
Although satisfactory results were not obtained for the catalytic asymmetric reaction of in situ formed symmetric exocyclic 1,3-diene 6a, we envisioned that the diastereoselective formation of the η2-Pd(0) complex would be expected if an enantioenriched 1,3-diene 6b was involved. As a result, remote chirality transfer would be potentially applicable in the vinylogous addition step,13 which could provide an alternative strategy for constructing chiral pyrrole derivatives. As illustrated in Scheme 3, employing the readily available propargyl amine (S)-1b (99% ee) as the chiral starting material, the asymmetric three-component reaction with carbonate 2a and imine 3a took place efficiently under the catalysis of Pd(PPh3)4 and acid A2. Notably, excellent chirality transfer was achieved, and the 3-pyrrolylmethylated product (R)-7a was furnished with only marginal ee losses. More imines with diverse substitutions also delivered the corresponding chiral adducts with outstanding enantioselectivity. Alkyl substituted propargyl amine (S)-1c (95% ee) could be used to produce chiral (R)-7q in a good yield. In addition, complete chirality transfer was obtained by using 1,6-enyne (R)-5b (88% ee), albeit in a significantly reduced yield.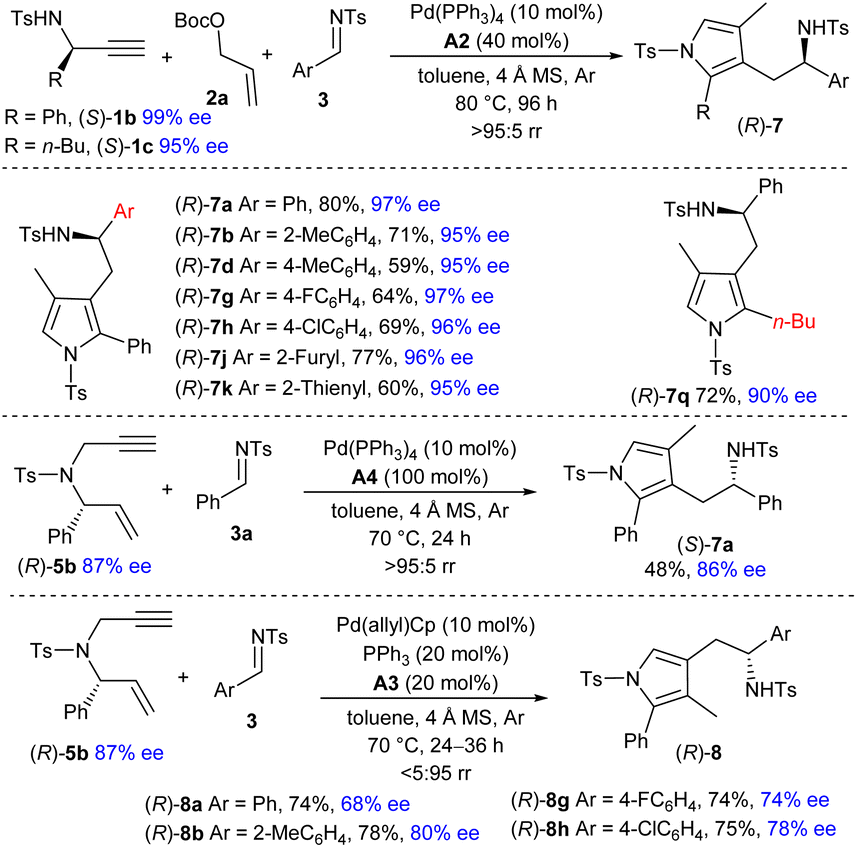 | ||
| Scheme 3 Asymmetric synthesis through remote chirality transfer. | ||
We further investigated the preparation of chiral 4-pyrrolylmethylated adducts (R)-8 by using (R)-5b as the starting reagent under the catalysis of Pd(allyl)Cp, PPh3 and acid A3. Although the addition site is quite far away from the chiral centre, significant chirality was still retained via Pd-mediated π-Lewis base catalysis, and a series of enantioenriched isomers (R)-8 were yielded effectively.14
More substrate exploration and synthetic transformations
Apart from the construction of pyrrole derivatives, the oxygen-tethered 1,6-enyne 9 was applicable to deliver corresponding aromatic furan products 10 and 11 in moderate regioselectivity and fair yield (Scheme 4a). In addition, more types of electrophiles and enynes were investigated, whereas complex reaction profiles or no obvious conversions were generally observed.11 As outlined in Scheme 4b, treating product 4a with N-bromosuccinimide (NBS) in CCl4 afforded dibromo product 12 in an excellent yield, and regioselective Suzuki coupling and debromination were conducted to deliver product 8m in a moderate yield. In addition, a cyano group could be introduced by treating 7a with TMSCN, phenyliodinebis(trifluoroacetate) and BF3·Et2O (product 13).15 On the other hand, 7a could be chemoselectively brominated by NBS in CH3CN, and an intramolecular SN2 reaction of resultant 14 afforded bicyclic framework 15 in a moderate yield. It should be noted that these polysubstituted pyrrole frameworks are widely witnessed in natural products and pharmaceuticals.16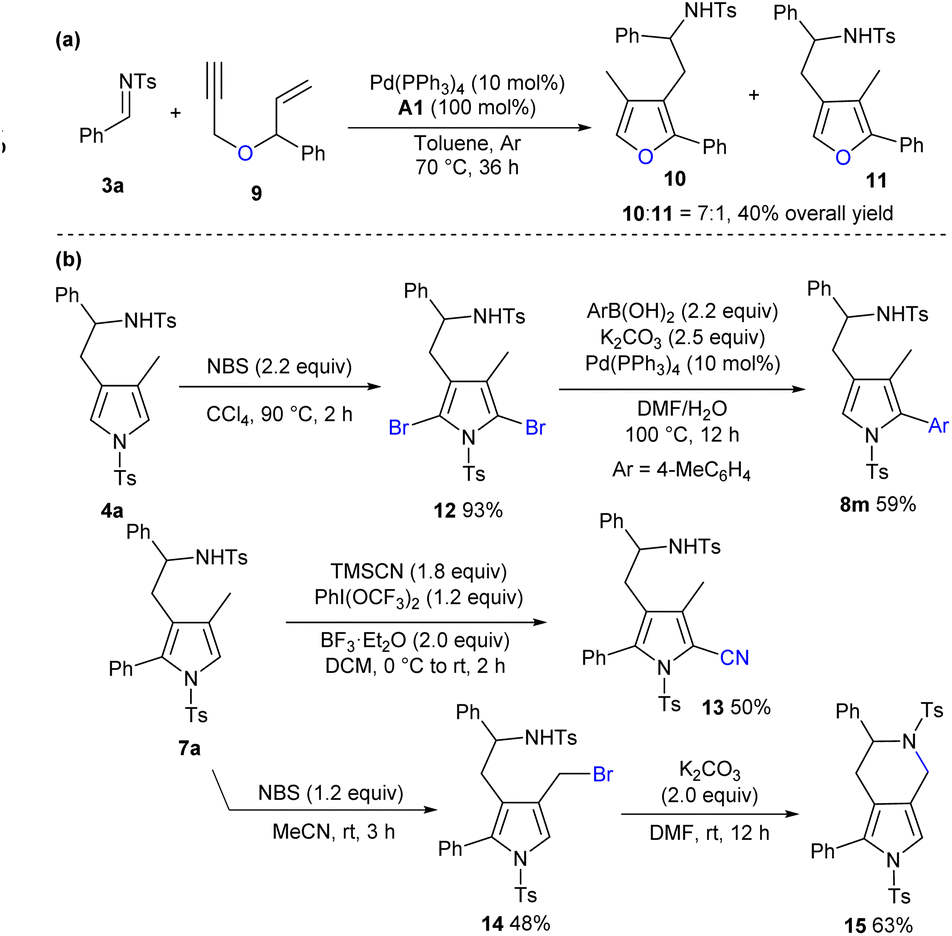 | ||
| Scheme 4 More substrate exploration and synthetic transformations. (a) Construction of trisubstituted furans. (b) Synthetic transformations. | ||
Reaction pathway and regiodivergence proposal
To gain some insight into the reaction process, several control experiments were conducted. As illustrated in Scheme 5a, the reaction of propargyl amine 1a and allylic carbonate 2a efficiently delivered 1,6-enyne 5a in 2 h under Pd(0) catalysis. 1,3-Diene 6a could be isolated in a moderate yield via 5-exo-dig cycloisomerisation of 5a under the same catalytic conditions, and acid was proved to be crucial for this Pd(0)-mediated reaction.17 The subsequent vinylogous addition and aromatic isomerisation process between 3a and 6a also proceeded smoothly, furnishing the desired adduct 4a in a moderate yield. The possible diene intermediate Int-1 was not obtained, and control experiments and Xia's study demonstrated that both palladium and acid were indispensable for the vinylogous addition and aromatisation steps.18 Moreover, Pd(II) or Sc(OTf)3 could not promote the transformations, indicating that an Alder-ene type process would not be involved.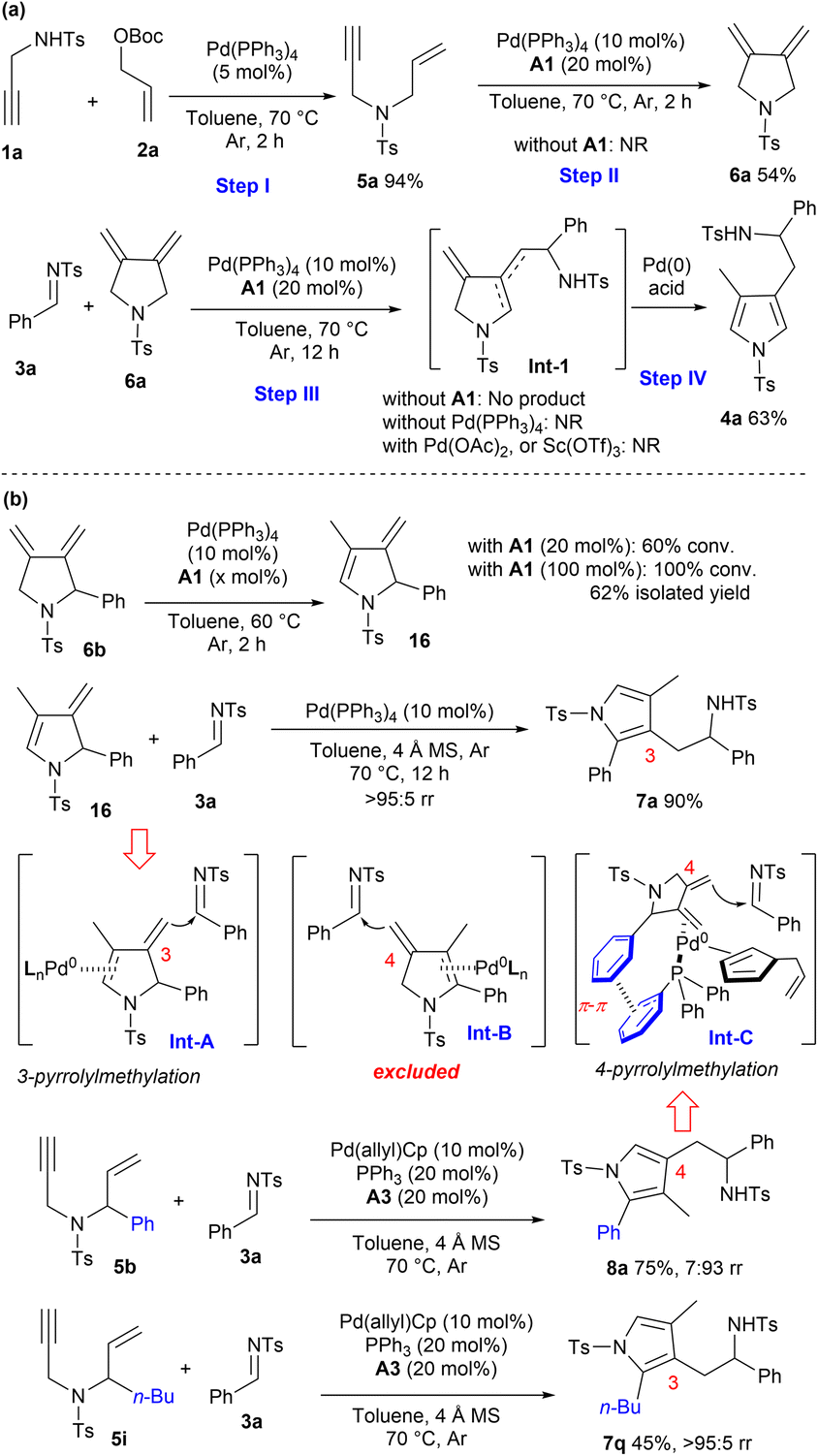 | ||
| Scheme 5 Control experiments for mechanism and regiodivergence elucidation. (a) Elucidation of auto-tandem catalytic reaction. (b) Proposal of regiodivergent pyrrolylmethylation. | ||
Meanwhile, more control experiments were carried out to rationalise the origin of regiodivergent pyrrolylmethylation. It was found that exocyclic 1,3-diene intermediate 6b could gradually isomerise to endocyclic diene 16 in the presence of Pd(0) and benzoic acid A1, and complete conversions were observed within 2 h by using stoichiometric A1. Moreover, the coupling reaction of diene 16 and imine 3a proceeded very efficiently under Pd(0) catalysis, exclusively giving 3-pyrrolylmethylated product 7a in an excellent yield, which well supported that the η2-complex Int-A would be the key intermediate in the vinylogous addition step (Scheme 5b). In contrast, a similar isomerisation process of 1,3-diene 6b to deliver the 4-pyrrolylmethylated product 8aviaInt-B would be excluded, because chirality transfer could not be available even when chiral 6b was applied. As demonstrated in Scheme 5b, a phenyl group on 1,6-enyne 5b was crucial for the formation of regioisomer 8a; in sharp contrast, the n-butyl substituted enyne 5i gave 3-pyrrolylmethylated product 7q exclusively. Accordingly, a π–π stacking between intermediate 6b and ligand PPh3 might help stabilise the η2-Pd(0) complex Int-C,19 which might be responsible for the observed regioselective 4-pyrrolylmethylation reaction. In addition, the in situ formed allyl cyclopentadiene might contribute partially as a potential ligand, as proposed in Int-C.20
Conclusions
We have investigated a palladium-catalysed assembly of N-Ts propargylamines, allyl carbonates and N-Ts aldimines in the presence of an acid additive, chemoselectively proceeding through an auto-tandem allylic amination, 1,6-enyne-cycloisomerisation, vinylogous addition and aromatic isomerisation sequence. A wide spectrum of formal pyrrolylmethylation products with dense substitutions was finally furnished straightforwardly. Importantly, regiodivergent construction of 3-pyrrolylmethylated or 4-pyrrolylmethylated derivatives could be accomplished by tuning catalytic conditions, and a substrate-ligand π–π stacking interaction might play a key role. Although only moderate enantiocontrol was achieved from prochiral precursors via asymmetric catalysis, fine remote chirality transfer could be achieved from enantioenriched 1,6-enynes or even N-Ts propargylamines for both regiodivergent reactions with an achiral ligand, relying on the diastereoselective generation of η2-Pd(0) complexes between Pd(0) and in situ formed chiral 1,3-diene intermediates in the key vinylogous addition step. More results with regard to palladium-based auto-tandem catalysis will be reported in due course.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the NSFC (21901169, 21931006 and 21921002) and 111 project (B18035).Notes and references
- For selected reviews, see: (a) D. S. Latha and S. Yaragorla, Eur. J. Org. Chem., 2020, 2020, 2155 CrossRef CAS; (b) T. Jiang, H. Liu, H. Zhang and H. Huang, Chin. J. Chem., 2021, 39, 1070 CrossRef CAS; (c) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2009, 131, 12056 CrossRef CAS PubMed; (d) J. Zhang, C. Stanciu, B. Wang, M. M. Hussain, C.-S. Da, P. J. Carroll, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2011, 133, 20552 CrossRef CAS PubMed; (e) X.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 4002 CrossRef CAS PubMed; (f) P. J. Moon, Z. Wei and R. J. Lundgren, J. Am. Chem. Soc., 2018, 140, 17418 CrossRef CAS PubMed; (g) D. Pal, T. B. Wright, R. O'Connor and P. A. Evans, Angew. Chem., Int. Ed., 2021, 60, 2987 CrossRef CAS PubMed.
- For selected examples, see: (a) X.-J. Liu, W.-Y. Zhang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2022, 61, e202200164 CAS; (b) P. Zhang, J. Wang, Z. R. Robertson and T. R. Newhouse, Angew. Chem., Int. Ed., 2022, 61, e202200602 CAS.
- For selected examples, see: (a) X.-J. Liu, C. Zheng, Y.-H. Yang, S. Jin and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 10493 CrossRef CAS PubMed; (b) W. Luo, J. Zhao, C. Yin, X. Liu, L. Lin and X. Feng, Chem. Commun., 2014, 50, 7524 RSC; (c) K. S. Nalivela, M. Rudolph, E. S. Baeissa, B. G. Alhogbi, I. A. I. Mkhalid and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2183 CrossRef CAS.
- F. Wu, L. Chen, Y. Wang and S. Zhu, Org. Chem. Front., 2019, 6, 480 RSC.
- For selected reviews, see: (a) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS PubMed; (b) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884 RSC; (c) Y. Hu, M. Bai, Y. Yang and Q. Zhou, Org. Chem. Front., 2017, 4, 2256 RSC.
- B. M. Trost and M. Lautens, J. Am. Chem. Soc., 1985, 107, 1781 CrossRef CAS.
- For selected examples, see: (a) Y. Yamamoto, H. Hayashi, T. Saigoku and H. Nishiyama, J. Am. Chem. Soc., 2005, 127, 10804 CrossRef CAS PubMed; (b) J. Cui, J. Hao, O. A. Ulanovskaya, J. Dundas, J. Liang and S. A. Kozmin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6763 CrossRef CAS PubMed; (c) M. Ishida and K. Tanaka, Org. Lett., 2013, 15, 2120 CrossRef CAS PubMed; (d) H. Zheng, Y. Wang, C. Xu, Q. Xiong, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 5327 CrossRef CAS PubMed.
- B.-X. Xiao, B. Jiang, R.-J. Yan, J.-X. Zhu, K. Xie, X.-Y. Gao, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 4809 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Dong, A. W. Schuppe, B. K. Mai, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2022, 144, 5985 CrossRef CAS PubMed; (b) W. Shao, C. Besnard, L. Guénée and C. Mazet, J. Am. Chem. Soc., 2020, 142, 16486 CrossRef CAS PubMed; (c) Z.-X. Wang and B.-J. Li, Angew. Chem., Int. Ed., 2022, 61, e202201099 CAS; (d) M. Dai, Z. Sun and L.-A. Chen, Angew. Chem., Int. Ed., 2022, 61, e202203835 CAS; (e) Y. Xi, W. Huang, C. Wang, H. Ding, T. Xia, L. Wu, K. Fang, J. Qu and Y. Chen, J. Am. Chem. Soc., 2022, 144, 8389 CrossRef CAS PubMed; (f) W. Chai, Q. Zhou, W. Ai, Y. Zheng, T. Qin, X. Xu and W. Zi, J. Am. Chem. Soc., 2021, 143, 3595 CrossRef CAS PubMed; (g) W.-S. Jiang, D.-W. Ji, W.-S. Zhang, G. Zhang, X.-T. Min, Y.-C. Hu, X.-L. Jiang and Q.-A. Chen, Angew. Chem., Int. Ed., 2021, 60, 8321 CrossRef CAS PubMed.
- For selected reviews and examples, see: (a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365 CrossRef CAS; (b) N. Shindoh, Y. Takemoto and K. Takasu, Chem. – Eur. J., 2009, 15, 12168 CrossRef CAS PubMed; (c) J.-R. Chen, C.-F. Li, X.-L. An, J.-J. Zhang, X.-Y. Zhu and W.-J. Xiao, Angew. Chem., Int. Ed., 2008, 47, 2489 CrossRef CAS PubMed; (d) A. R. O. Venning, M. R. Kwiatkowski, J. E. Roque Peña, B. C. Lainhart, A. A. Guruparan and E. J. Alexanian, J. Am. Chem. Soc., 2017, 139, 11595 CrossRef CAS PubMed; (e) P. Chen, Z.-C. Chen, Y. Li, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2019, 58, 4036 CrossRef CAS PubMed; (f) J. Long, R. Yu, J. Gao and X. Fang, Angew. Chem., Int. Ed., 2020, 59, 6785 CrossRef CAS PubMed; (g) J.-X. Zhu, Z.-C. Chen, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200880 CAS.
- Please see the ESI† for more details.
- X.-X. Yang, R.-J. Yan, G.-Y. Ran, C. Chen, J.-F. Yue, X. Yan, Q. Ouyang, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2021, 60, 26762 CrossRef CAS PubMed.
- (a) J. Buter, D. Heijnen, C. Vila, V. Hornillos, E. Otten, M. Giannerini, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 3620 CrossRef CAS PubMed; (b) S. Gima, I. Nakamura and M. Terada, Eur. J. Org. Chem., 2017, 2017, 4375 CrossRef CAS; (c) J. Li and S. R. Gilbertson, Org. Lett., 2021, 23, 2911 CrossRef CAS PubMed; (d) J.-L. Zhang, J.-Y. Liu, G.-Q. Xu, Y.-C. Luo, H. Lu, C.-Y. Tan, X.-Q. Hu and P.-F. Xu, Org. Lett., 2021, 23, 3287 CrossRef CAS PubMed.
- The absolute configuration of (R)-7a and (R)-8b was assigned based on experimental and calculated electronic CD (ECD) spectra, see the ESI†.
- T. Dohi, K. Morimoto, N. Takenaga, A. Goto, A. Maruyama, Y. Kiyono, H. Tohma and Y. Kita, J. Org. Chem., 2007, 72, 109 CrossRef CAS PubMed.
- For selected reviews, see: (a) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801 RSC; (b) R. Khajuria, S. Dham and K. K. Kapoor, RSC Adv., 2016, 6, 37039 RSC; (c) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233 RSC.
- (a) B. M. Trost, D. L. Romero and F. Rise, J. Am. Chem. Soc., 1994, 116, 4268 CrossRef CAS; (b) A. Mekareeya, P. R. Walker, A. Couce-Rios, C. D. Campbell, A. Steven, R. S. Paton and E. A. Anderson, J. Am. Chem. Soc., 2017, 139, 10104 CrossRef CAS PubMed.
- L. Ding, Y.-H. Deng, T.-Y. Sun, D. Wang, Y.-D. Wu and X.-F. Xia, Org. Chem. Front., 2021, 8, 4785 RSC.
- (a) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637 CrossRef CAS PubMed; (b) Y. Chen, Z. Wang, W. Zhao, S. Sun, L. Yang, J. Zhang, D. Zhang, M. Cheng, B. Lin and Y. Liu, Chem. Commun., 2022, 58, 3051 RSC; (c) G. Zuccarello, J. G. Mayans, I. Escofet, D. Scharnagel, M. S. Kirillova, A. H. Pérez-Jimeno, P. Calleja, J. R. Boothe and A. M. Echavarren, J. Am. Chem. Soc., 2019, 141, 11858 CrossRef CAS PubMed; (d) S. Jerhaoui, J.-P. Djukic, J. Wencel-Delord and F. Colobert, ACS Catal., 2019, 9, 2532 CrossRef CAS.
- (a) M. R. Buchner, B. Bechlars, B. Wahl and K. Ruhland, Organometallics, 2012, 31, 588 CrossRef CAS; (b) D. M. Norton, E. A. Mitchell, N. R. Botros, P. G. Jessop and M. C. Baird, J. Org. Chem., 2009, 74, 6674 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, NMR spectra, HRMS spectra, ECD spectra and HPLC chromatograms, CIF file of racemic products 4a and 7o. CCDC [2184605 and 2184606]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05210e |
| This journal is © The Royal Society of Chemistry 2022 |