
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFemtosecond anisotropy excitation spectroscopy to disentangle the Qx and Qy absorption in chlorophyll a†
Clark
Zahn
*,
Till
Stensitzki‡
and
Karsten
Heyne
 *
*
Department of Physics, Free University Berlin, Arnimallee 14, D-14195 Berlin, Germany. E-mail: lark.zahn@fu-berlin.de; karsten.heyne@physik.fu-berlin.de; clark.zahn@fu-berlin.de; Tel: +49 30 838 56107
First published on 12th October 2022
Abstract
Chlorophyll a (Chl a) belongs to the most important and most investigated molecules in the field of photosynthesis. The Q-band absorption is central for energy transfer in photosystems and the relative orientation of the Qy transitions of interacting chlorophylls governs the energy transfer. Chl a was well investigated, but a quantitative separation of Qx and Qy contributions to the Q-band of the Chl a absorption spectrum is still missing. We use femtosecond Vis-pump – IR-probe anisotropy excitation spectroscopy to disentangle the overlapping electronic Qx and Qy contributions quantitatively. In an anisotropy excitation spectrum we trace the dichroic ratio of a single vibration, i.e. the keto C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1690 cm−1, as a function of excitation wavelength. The change in dichroic ratio reflects altering Qy and Qx contributions. We identified Qx00 (0–0 transition of Qx) and Qx01 transition at (636 ± 1) nm and (607 ± 2) nm, respectively, and the Qy01 and Qy02 at (650 ± 6) nm, and (619 ± 3) nm, respectively. We find that Qx absorption, contributes to 50% to 72% at 636 nm and 49% to 71% at 606 nm to the Chl a absorption at room temperature. The Q band was well modelled by a single vibronic progression for the Qx and Qy transition of (700 ± 100) cm−1, and the energy gap between Qx00 and Qy00 was found to be (820 ± 60) cm−1. This precise description of the hexa-coordinated Chl a absorption spectrum will foster more accurate calculations on energy transfer processes in photosystems, and advance the detailed understanding of the intricate interaction of chlorophyll molecules with the solvent.
O stretching vibration at 1690 cm−1, as a function of excitation wavelength. The change in dichroic ratio reflects altering Qy and Qx contributions. We identified Qx00 (0–0 transition of Qx) and Qx01 transition at (636 ± 1) nm and (607 ± 2) nm, respectively, and the Qy01 and Qy02 at (650 ± 6) nm, and (619 ± 3) nm, respectively. We find that Qx absorption, contributes to 50% to 72% at 636 nm and 49% to 71% at 606 nm to the Chl a absorption at room temperature. The Q band was well modelled by a single vibronic progression for the Qx and Qy transition of (700 ± 100) cm−1, and the energy gap between Qx00 and Qy00 was found to be (820 ± 60) cm−1. This precise description of the hexa-coordinated Chl a absorption spectrum will foster more accurate calculations on energy transfer processes in photosystems, and advance the detailed understanding of the intricate interaction of chlorophyll molecules with the solvent.
1 Introduction
Absorption spectra of many important molecules such as photoreceptors, metal complexes and different kinds of tetrapyrroles, including chlorophylls, show spectrally overlapping contributions of different electronic transitions. A clear identification of the contributions at a given wavelength is typically connected with calculations in combination with measurements at different experimental conditions.1–4 However, the interpretation of the experimental results depends strongly on the calculations, which become increasingly difficult for larger molecules. Here, we demonstrate a method based on polarization resolved femtosecond Vis-pump IR-probe spectroscopy to disentangle spectral contributions of different electronic transitions in the electronic ground state, without requiring theoretical calculations. As an application example we have chosen the decomposition of the Q-band of chlorophyll a (Chl a).Chl a plays a fundamental role in photosynthesis, from the initial energy transfer in the light harvesting complex to the electron transfer in the reaction center.5 In order to understand the different photosynthetic processes, an understanding of Chl a and its photophysical properties is crucial. Chl a shares many properties with other cyclic tetrapyrroles. As described by the Gouterman Model,6 its chlorin macrocycle gives rise to two distinct broad absorption bands: the soret (B)-band at 350 nm to 450 nm and the Q-band at 550 nm to 680 nm. In addition, both the B- and the Q-band are further comprised of two distinguishable electronic states with different geometrical orientations: Bx, By and Qx, Qy.7 Here, x and y indicate the expected directions of the transition dipole moments (tdm) for the transition between the ground-state and the respective excited-state within the chlorophylls macrocycle plane as depicted in Fig. 1 inset.
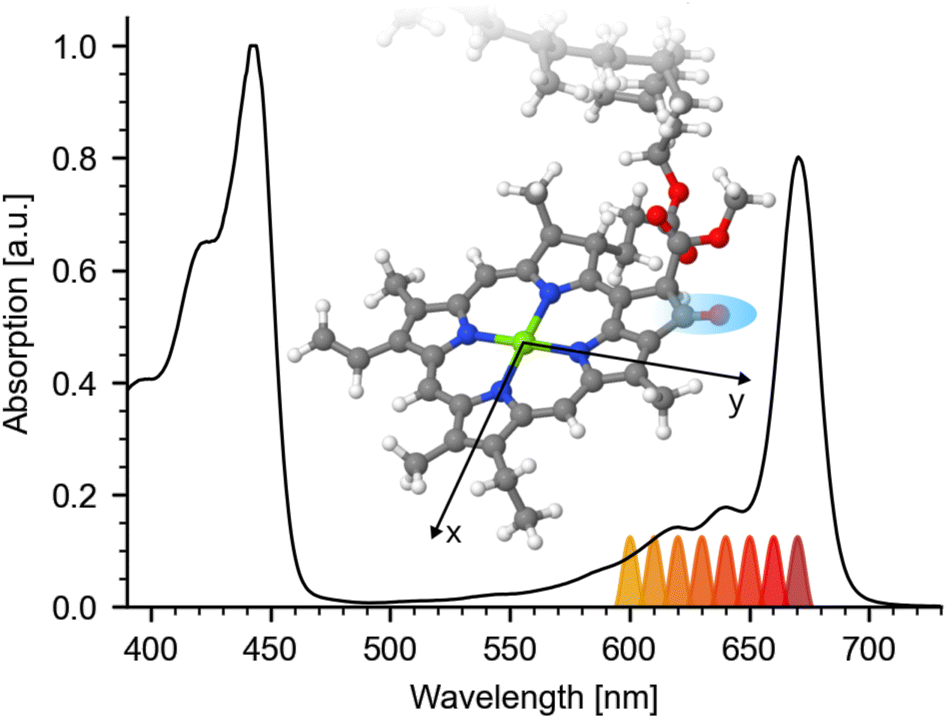 | ||
| Fig. 1 Visible steady state absorption of hexa-coordinated Chl a in pyridine. The spectrum is normalized to its Soret-band at 443 nm. Q-band absorption is exhibiting a maximum at 670 nm followed by a shoulder around 644 nm and high energy side peaks at 619 nm and 585 nm. Different excitation wavelengths for the pump-pulse are indicated below the spectrum. Inset: Geometry optimized structure of Chl a macrocycle with x and y axis (taken from ref. 25). Atoms are color coded: carbon (gray), nitrogen (blue), magnesium (green), oxygen (red), and hydrogen (white). The investigated keto CO group is marked by a blue region. | ||
Particularly, the Q-band plays a key role in the efficient energy transport.5 Thus, its physical properties are widely studied using different experimental techniques, including linear dichroism,8–10 circular dichroism and magnetic circular dichroism (MCD),1,11–17 spectral hole-burning18,19 and fluorescence spectroscopy,20–22 as well as different time-resolved transient absorption23–25 and 2D-absorption measurements.2,26,27 Despite extensive research, the exact composition of the Q-band absorption is not yet fully understood. Even though, the low energetic Qy 0–0 transition (Qy00) could be assigned to the prominent low energy peak of the Q-band at 660 nm to 670 nm early on,8,10–13 particularly the assignment of the Qx transition long remained controversial,1,8,10–17,19,20 with different assignment options being considered. Moreover, changes in the macrocycle conformation, induced by penta- or hexa-coordination, induce a shift of the Qx band position of Chl a,14,17,20 complicating conclusive assignment. Yet, only in the last decade, new qualitative information on the spectral position of Qx and Qy was obtained.17,20 Furthermore, different polarisation resolved studies using femtosecond Vis-pump VIS-probe and 2DES spectroscopy investigated the spectral contributions of Qx and Qy.2,23,24 They show strong polarization dependence as a function of probing and pumping wavelengths. Unfortunately, the broad excited state absorption (ESA) of Chl a with different anisotropic contributions,23 together with the broad bleaching (BL) and stimulated emission contributions makes the interpretation very difficult without simplifications.2 Yet, despite different approaches, up to now, to our knowledge no quantitative disentangling of Qx and Qy contributions was reported, so that the exact Qx and Qy absorption spectrum remains unknown.
Here, we apply anisotropy excitation spectroscopy tracing the impact of electronic excitation on the vibrational anisotropy at different excitation wavelengths. Compared to probing electronic transitions, the much narrower line-width and the multitude of vibrational bands enable a precise measurement of anisotropy changes. However, even though in nature Chl a are largely penta-coordinated,28,29 the two diastereotopic faces (syn and anti) of penta-coordinated Chl a,30,31 make an unambiguous analysis of anisotropy changes difficult. Thus, using hexa-coordinated Chl a in pyridine allows for a clear analysis of its anisotropy excitation spectrum, providing a first clear separation of the Qx and Qy absorption spectra and a giving a more in-depth understanding of the electronic structure of Chl a.
2 Materials and methods
2.1 Sample preparation
Chl a from spinach was purchased from Sigma-Aldrich. All samples were dissolved in pyridine and prepared in sample cells with a thickness of 100 μm, with a sample concentration of 1.5 mM to 3 mM (corresponding to an absorption of ∼2.5 OD at 671 nm). In pyridine, Chl a is hexa-coordinated and has a flat macrocycle.14,20,32,332.2 Transient absorption
Femtosecond laser pulses were generated starting from pulses delivered by a 1.088 kHz Ti:Sa laser system (Coherent Legend USP, 80 fs pulses at 808 nm). Spectrally tunable visible pump pulses were generated by cascaded OPAs generating fs output pulses of ∼6 μJ and (130 ± 20) fs. In detail, a collinear BBO OPA pumped at 400 nm generated near-IR pulses at 1200 nm to 1350 nm, which were further amplified by two KTA amplification stages pumped with the fundamental at 800 nm. The near-IR pulses were then frequency doubled in a BBO-crystal, generating desired pump wavelengths between 600 and 670 nm with a spectral width of FWHM (10 ± 3) nm. A filter (Schott KG4) and a polarizer removed the remaining near-IR frequencies. To ensure photoselection conditions, the pump-pulses were further attenuated to 0.2 μJ to 0.5 μJ, exciting less than 20% of the sample. A λ/2-plate was used to set the polarization of the pump-beam, alternating between perpendicular and parallel pump – probe configuration. The diameter of the pump beam on the sample was (220 ± 40) μm (FWHM), the diameter of the two probe beams on the sample was (150 ± 30) μm (FWHM).IR probe beams with energies 50 nJ were generated as reported elsewhere.34 Two reflections of the fs mid-IR pulse were taken, one as probe beam and one as reference. Both beams pass through the same sample volume, with the reference pulse arriving 1.5 ns before the probe, further reducing the shot-to-shot noise.35 The system response duration was about (300 ± 50) fs. Both IR pulses are detected by dispersing the beams with an imaging spectrograph and recording both beams simultaneously with a 2 × 32 element MCT-array from Infrared Associates. The spectral resolution was better than 3 cm−1. The sample was moved with a Lissajous-scanner to ensure a fresh sample volume between consecutive pump pulses. The probe pulses were delayed using a mechanical translation stage.
We excited the isotropic Chl a solution with linear polarized excitation pulses introducing a sub-set of oriented excited Chl a molecules (photoselection). This generates a transient anisotropy in the sub-set of excited molecules, which decays through rotational diffusion with τ ∼ 90 ps.25 On a time-scale between 5 to 15 ps the change in anisotropy of the keto CO stretching vibration is negligible within the S/N (see ESI Fig. SI 1†).
In Vis-pump IR-probe experiments, only signals from the excited sub-sets are detected. With polarization resolved mid-IR probing, the dichroic ratio D is given by 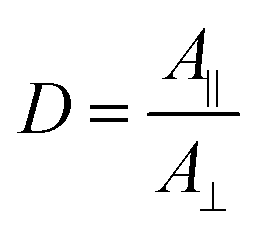 for signal amplitudes of parallel A‖ and perpendicular A⊥ polarizations with respect to pump-pulse polarization. The ratio of the probed amplitudes reflect the projection of the vibrational tdm onto the excited electronic tdm. In particular, the relative angle θ between the excited and probed tdm is given by
for signal amplitudes of parallel A‖ and perpendicular A⊥ polarizations with respect to pump-pulse polarization. The ratio of the probed amplitudes reflect the projection of the vibrational tdm onto the excited electronic tdm. In particular, the relative angle θ between the excited and probed tdm is given by 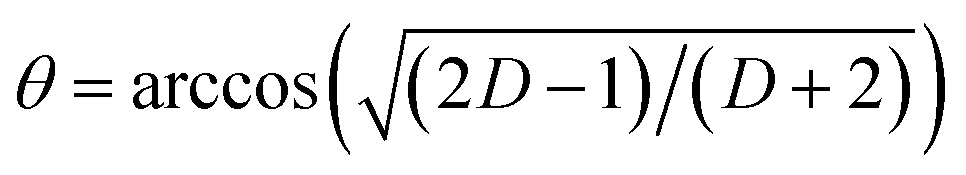 .
.
3 Results and discussion
Steady state absorption of Chl a in pyridine is shown in Fig. 1, displaying Soret-band absorption at 390 nm to 460 nm and Q-band absorption at 570 nm to 670 nm. We performed photoselection experiments creating an oriented sub-set of excited Chl a molecules and probed their dynamics by polarization resolved mid-IR spectroscopy. Photoexcitation of the Q-band initiates ultrafast 100 fs to 200 fs dynamics assigned to internal conversion from Qx to Qy,2,23,24,26 followed by population of the Qy state, decaying on a nanosecond time-scale,5 with negligible amplitude changes from 0.4 ps to 15 ps (see ESI Fig. SI 1†). Absorption spectra for different excitation wavelengths averaged over delay times from 5 ps to 15 ps are displayed in Fig. 2. Parallel and perpendicular polarizations with respect to the pump-polarization are shown in black and red, respectively. The spectral region of the keto CO stretching vibration shows a prominent bleaching (BL) signal at 1690 cm−1 and an associated excited state absorption (ESA) at 1650 cm−1.36 Upon visible excitation at the low energy side of the Q-band at 670 nm, a pure Qy contribution is observed.8,10–13 In this case, the observed dichroic ratio D (670 nm) is around 2.59, with a 1σ range from 2.47 to 2.73. This value corresponds to a relative angle θ of (18 ± 3)° between the keto CO vibrational tdm and the Qy tdm in agreement with earlier findings.37
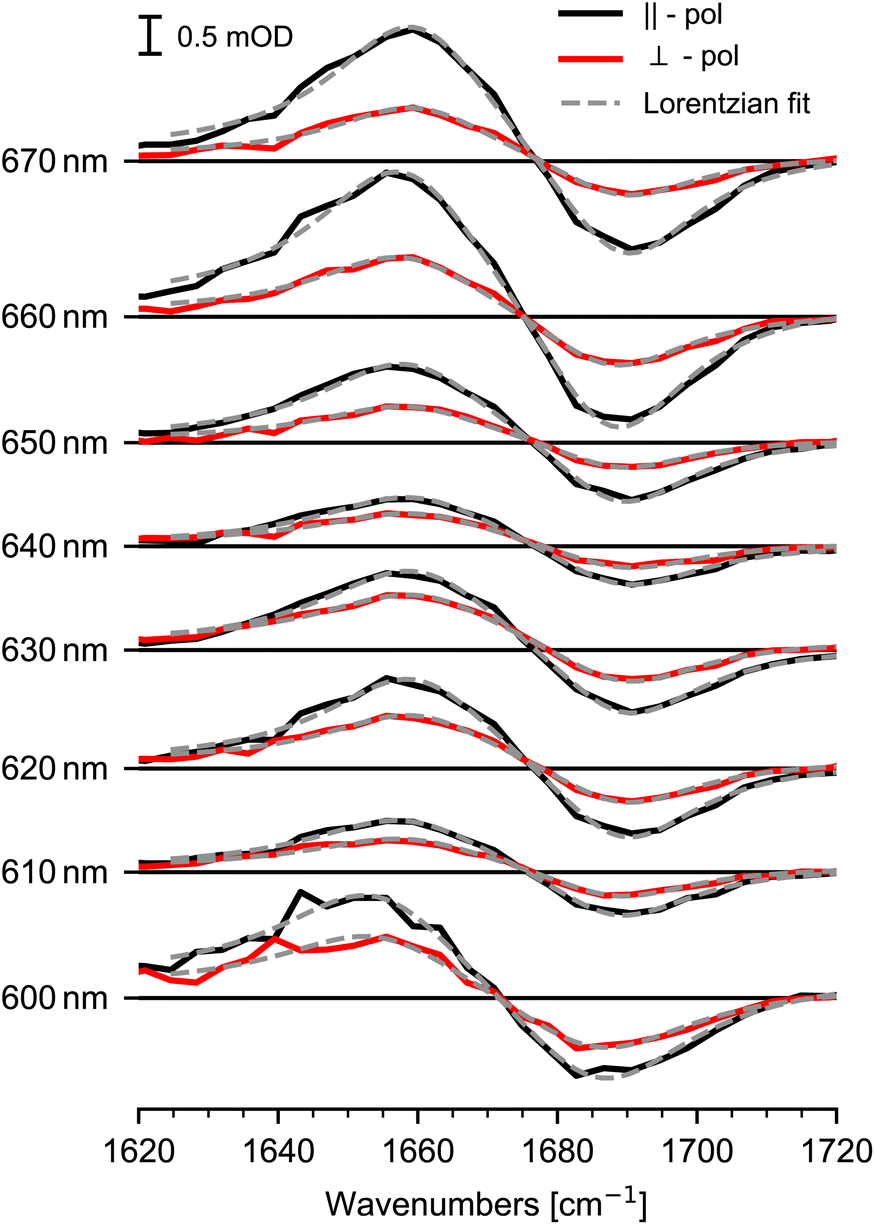 | ||
| Fig. 2 Transient mid-IR absorption spectra of the keto CO stretching vibration for different excitation wavelengths averaged over delay times from 5 ps to 15 ps for parallel (black) and perpendicular (red) polarization orientation. Bleaching (negative) signals at 1690 cm−1 reflect the keto CO stretching vibration in the electronic ground state and (positive) signals at 1650 cm−1 display excited state signals in the Qy state. The orientation of the detected sub-set is induced by the effective tdm given by the mixture of excited Qx and Qy tdms. Lorentzian fits are presented by gray dashed lines. | ||
Upon excitation at shorter wavelengths the dichroic ratio decreases from 2.4 at 660 nm to 1.6 at 640 nm. Going to even shorter wavelengths, the ratio increases until 620 nm, then decreases again at wavelength of 600 nm. Since, the dichroic ratio is fixed for a given electronic transition,38 this twofold decrease indicates two distinct contributions of another electronic transition, i.e. the Qx transition. The dichroic ratio changes by about 1 as a function of excitation wavelengths, presenting a high anisotropy contrast for the keto CO stretching vibration in connection to Q-band transitions. Thus, the keto CO stretching vibration provides a well suited marker band for anisotropy excitation spectroscopy of Chl a.
We modelled the keto CO ESA and BL signals for both polarizations simultaneously with a sum of Lorentzians, as depicted in Fig. 2. Since the dichroic ratio of the vibrational transition is expected to be identical for the BL and ESA band in a rigid molecule,39 we further constrained the fit by modeling both BL and ESA band with the same dichroic ratio. Errors are calculated using exhaustive search analysis.39,40 The resulting changes in the dichroic ratio or anisotropy of the keto CO vibration as a function of excitation wavelength are displayed in the excitation spectrum in Fig. 3a.
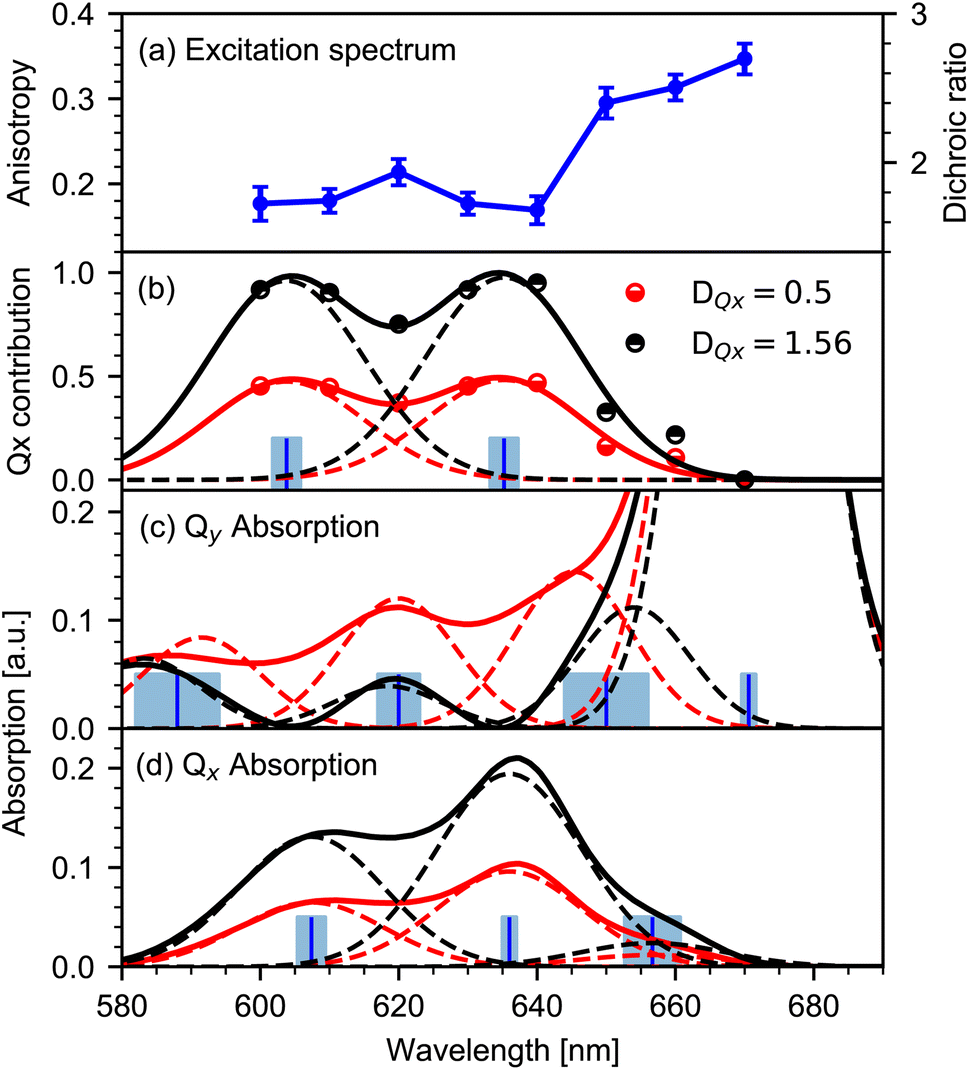 | ||
| Fig. 3 Mapping the Qx and Qy absorption spectrum from the anisotropy excitation spectrum. (a) Excitation spectrum of the keto CO stretching vibration (blue dots) as a function of excitation wavelength. Left scale anisotropy, right scale dichroic ratio. Errors (1σ ranges) are presented for the dichroic ratio. (b) Contribution c(λ) of the Qx-transition to the Chl a absorption spectrum at different wavelengths (dots) calculated from (a). Modelling of c(λ) (solid lines) by two Gaussian peaks (dashed lines) for two limiting DQx values of 1.56 (black) and 0.5 (red). Blue bars represent the center of the Gaussians. Error ranges for DQx values of 1.56 and 0.5 are indicated in light blue. (c) Qy absorption spectrum (product of Chl a absorption spectrum with (1 − c(λ)), solid lines) for the two limiting DQx values (same color code as in b). Modelled spectrum by four Gaussians (dashed lines) with identical widths, and peaks Qy00 at (671 ± 1) nm, Qy01 at (650 ± 6) nm and Qy02 at (619 ± 3) nm. Note, that the last peak at (588 ± 6) nm is not covered by the anisotropy excitation experiment. (d) Qx absorption spectrum (product of c(λ) with the Chl a absorption spectrum, solid lines) for the two limiting DQx values (same color code as in b). Modelled spectrum with three Gaussians (dashed lines) with identical widths and peaks Qx00 at (636 ± 1) nm, Qx01 at (607 ± 2) nm, and Qx10 at (657 ± 4) nm. | ||
3.1 Qx contribution in the Chl a spectrum
Changes in anisotropy directly mirror altering contributions of excited electronic transitions. The dichroic ratio D and anisotropy r can be used interchangeably (r = (D − 1)/(D + 2)). Here, the dichroic ratio is directly obtained from modelling the data (see Fig. 3a). Thus, in order to unravel the Qx and Qy absorption, it is convenient to work with the dichroic ratio. For two contributing transitions Qx and Qy, the dichroic ratio for different excitation wavelengths D(λ) is a linear combination of the corresponding Qx and Qy dichroic ratios DQx and DQy:| D(λ) = (1 − c(λ))DQy + c(λ)DQx | (1) |
with c(λ) being the relative contribution of the Qx transition between zero and one, and 1 − c(λ) being the relative contribution of the Qy transition. The relative contribution c(λ) of the Qx transition to the absorption spectrum is given by:
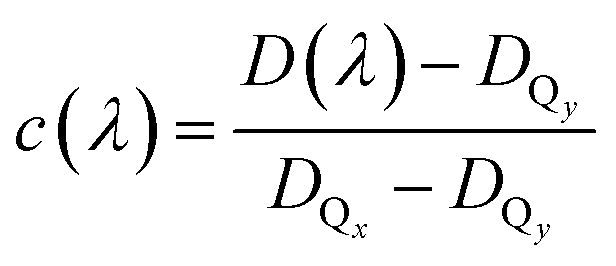 | (2) |
In general, the dichroic ratio of a band can vary between 0.5 and 3. However, since the Qx contribution cannot exceed 100%, the largest possible value of DQx is 1.56 (see Fig. 3a), therefore limiting the range of DQx to 0.5 to −1.56.
For the limiting values of DQx, the relative Qx contribution c(λ) is presented in Fig. 3b (dots). In the observed range, the relative contribution c(λ) exhibits two peaks at around 635 nm and 605 nm. We modelled the relative contribution c(λ) of Qx with two Gaussians centered at (635 ± 2) nm and (604 ± 2) nm, as depicted in Fig. 3b (solid lines). Contributions below 600 nm are needed to model the data. Note that for wavelengths below 600 nm the significance of the modelled contribution c decreases rapidly to shorter wavelengths.
3.2 Extracting pure Qx and Qy absorption spectra
The individual Qy and Qx absorption spectra are calculated by the product of the Chl a absorption spectrum (normalized to the Q-band maximum at 670 nm) with the modelled contributions of Qy (1 − c(λ)) and Qx as shown in Fig. 3b. The resulting absorption spectra of the pure Qy and Qx transitions for the limiting DQx values of 0.5 and 1.56 are depicted in Fig. 3c and d, respectively.3.3 Analysis of the Qy absorption spectrum
Modelling the Qy absorption spectrum with Gaussians of the same spectral widths yields peaks at (670 ± 2) nm, (648 ± 5) nm and (617 ± 2) nm (dashed lines in Fig. 3c). We assign the peak at 671 nm to the 0–0 transition Qy00 and the remaining two peaks to one vibronic progression with 0–1 transition Qy01 at 650 nm and 0–2 transition Qy02 at 619 nm. The frequency differences between adjacent Qy peaks is (700 ± 200) cm−1.In order to model the data, an additional peak at shorter wavelength is needed, yielding a peak position at and (588 ± 6) nm. As the peak is out of the spectral range of our measured anisotropy excitation spectrum, it is not possible to determine its exact contribution to the absorption spectrum. However, the spectral position matches the vibronic progression of (700 ± 200) cm−1, suggesting possible 0–3 transition Qy03 at (588 ± 6) nm.
Spectroscopic investigations24,26 on penta-coordinated Chl a revealed coherent oscillations with a frequency of about 700 cm−1, showing a substantial coupling between vibrational modes and electronic excitations in this spectral range. Moreover, hole-burning fluorescence spectroscopy revealed vibrational contributions around 700 cm−1 for penta- and hexa-coordinated Chl a.20 The peak around (648 ± 5) nm was observed before, but was associated either to the Qx transition20 or to the Qy transition.10 With our method we can assign this peak to the Qy transition.
3.4 Analysis of the Qx absorption spectrum
The Qx absorption spectrum is shown in Fig. 3d, displaying peaks at (657 ± 4) nm, (636 ± 1) nm and (607 ± 2) nm (dashed lines). We assign the peaks (636 ± 1) nm and (607 ± 2) nm to 0–0 transition Qx00, and 0–1 transition Qx01 of the Qx absorption, respectively. The very small peak around 657 nm is tentatively assigned to the 1–0 transition Qx10. This contribution should be strongly temperature dependent. Spectral position and assignment of both Qx and Qy bands is summarized in Table 1.The frequency difference between Qx00 and Qx01 is (700 ± 100) cm−1, and between Qx10 and Qx00 is (500 ± 200) cm−1. Thus, a vibronic progression in the Qx absorption spectrum with a frequency around 700 cm−1 seems plausible.
3.5 Vibronic progressions
The vibronic progression frequency of Qx match the one from the Qy transitions. Thus, we propose a single vibronic progression of about 700 cm−1 in the Qx and Qy absorption spectra. Other more complex interpretations are possible, but our simple model explains the spectroscopic findings in the absorption spectrum reasonably. From our analysis, the Qy − Qx energy gap is (820 ± 60) cm−1, matching the reported value of about 875 cm−1,20 and compatible with 970 cm−1.1 The energy gap is close to the frequency of the vibronic progression, making it difficult to reliably determine the contributions from the visible spectra alone. In this context we interpret the reported value of 1294 cm−1 for the vibronic progression.103.6 Refined absorption spectra
Up to this point, no additional information on the relative orientations of the electronic tdms were taken into account. Following the generally accepted Gouterman Model,6 the relative angle between the Qx and Qy tdm is expected to be close to 90°. This is confirmed by experimental studies on hexa- and penta-coordinated Chl a reporting angles between Qy and Qx tdm of about 70° to 74°.2,10 If we assume the angle between Qy and Qx tdm to be in the range of 70° to 90°, and combine this information with a maximal angle of 20° between the keto CO tdm and Qy tdm (D+1σ(670 nm) = 2.47 ≙ 20°), we get a maximal relative angle range of 50° to 90° between keto CO tdm and Qx tdm. This correspond to a value range of DQx = 0.5–1.15. Taking this into account, the disentangled Qx and Qy absorption spectra of Chl a in pyridine and their possible variation due to different DQx values are presented in Fig. 4. The sum of Qx and Qy spectra of corresponding DQx values give the measured Chl a absorption spectrum. We find that the Qx contribution to the Chl a Q-band absorption spectrum between 590 nm and 700 nm is always smaller than (72 ± 5)%, and is in the range of 50% to 72% at 636 nm and of 49% to 71% at 606 nm.
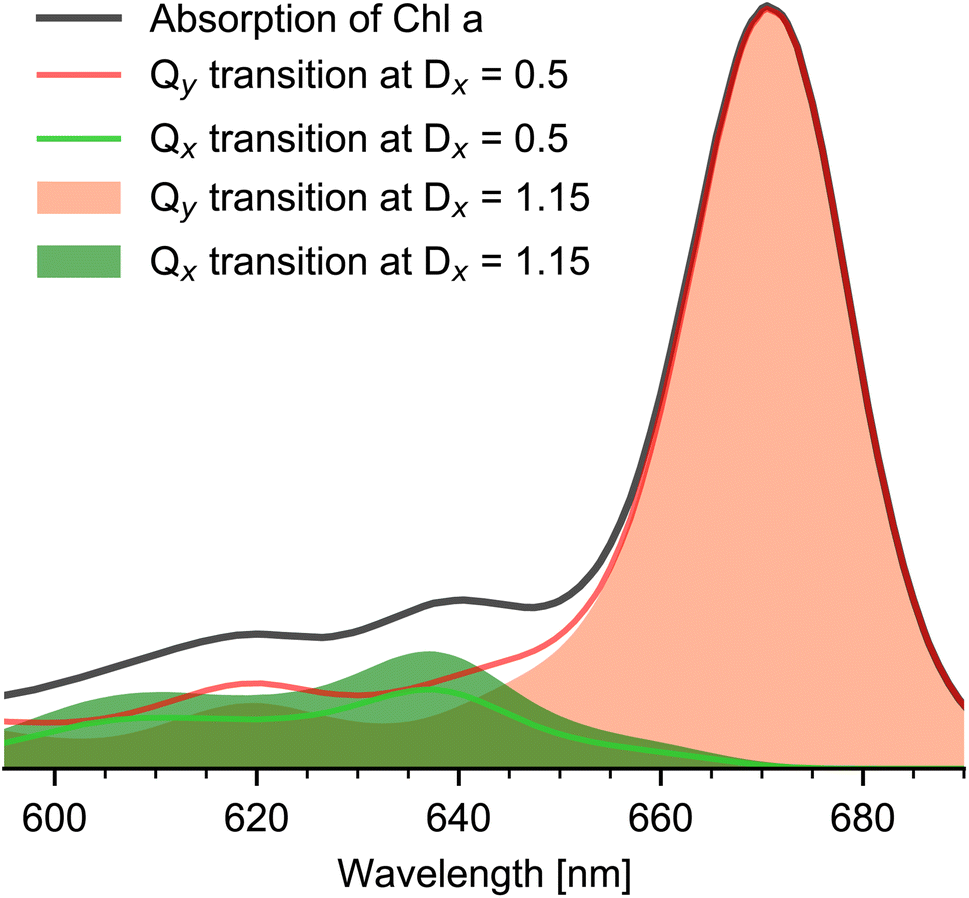 | ||
| Fig. 4 Chl a absorption spectrum and Qx and Qy contributions. Hexa-coordinated Chl a absorption spectrum (black line) in pyridine solution and Qy (orange) and Qx (green) absorption spectra for the two limiting DQx values 0.5 (minimal, solid lines) and 1.15 (maximal, filled area). The sum of Qx and Qx spectra at the same DQx value gives the experimental Chl a absorption spectrum. | ||
Using the disentangled absorption spectrum, we can determine the oscillator strength ratio between f(Qx) and f(Qy). With normalized Qy00 absorption at 671 nm and Qx00 absorption strength at f(636 nm) = 0.14 ± 0.04, the ratio f(Qx)/f(Qy) is between 5.6–10. This is in agreement with a value of 5.9 reported for Chl a in pyridine1 and with a value of 7.5 for penta-coordinated Chl a in ethanol.24 In addition, from the relative intensities of the Qx and Qy absorption bands, we calculated the relative Franck–Condon factors (see Table 2) in agreement with earlier findings.10
| Qx | Qy |
|---|---|
(Qx00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Qx01) nm = 1.5 ± 0.2 :Qx01) nm = 1.5 ± 0.2 |
(Qy00:Qy01) nm = 7 ± 2 |
| — | (Qy00:Qy02) nm = 9 ± 3 |
3.7 Discussion
The optical properties of Chl a are strongly affected by temperature, macrocycle coordination, and interaction with solvent molecules.14,20,32,41,42 In the gas phase recent calculations report a Qx − Qy energy gap of 2400 cm−1.43 In condensed phase the energy gap was reported to be around 900 cm−1,1,20 matching our findings of (820 ± 60) cm−1. The fraction of Qx contribution to the Chl a absorption spectrum was estimated between 10% to 25% (ref. 1) in agreement with our findings of 14% to 27% in the spectral region from 590 nm to 720 nm.Moreover, a strong vibronic coupling was predicted that inseparably mixes Qx and Qy transitions.1 From our measurements we can state that such a mixing has no or negligible effect on the orientation of the electronic tdms, since the tdms are well distinguishable and are the cause for our observed anisotropy contrast. Hence, the orientation of the excited electronic Qx tdm is preserved on a hundred picosecond time-scale. Upon Qx excitation, ultrafast energy relaxation to Qy is reported, with a time constant of 150 fs to 200 fs.2,23,24,26 We found that excitation of Chl a in the spectral range between 606 nm and 636 nm leads to an approximate 1:1 excitation of Qx and Qy transitions. Thus, excitation with linear polarized light generates populated Qy states along the x- and y-axis direction. This is essential for simulating energy transfer processes at various excitation wavelengths in photosystems, e.g. in LHC II.37,44,45
We localized the Qx00 transition at 636 nm. For wavelength longer than 650 nm only the Qy transition contributes to the Chl a spectrum, revealing a shoulder at (650 ± 6) nm. Up to now, this shoulder remained largely unnoticed, leading prior studies to locate the Qy01 at 613 nm to 619 nm.1,10,20 We assign the Qy01 to the peak at (650 ± 6) nm.
Consequently, in contrast to earlier findings, we explain the substructure of the absorption spectrum of the hexa-coordinated Chl a between 600 nm to 720 nm by a single vibronic progression with a frequency of about 700 cm−1.
4 Conclusion
Femtosecond Vis-pump – IR-probe anisotropy excitation spectroscopy is an efficient tool to identify spectral positions and contributions of different electronic transitions within the electronic absorption spectrum. Tracing the pump wavelength dependence of vibrational anisotropy allows for separation of electronic transitions with distinct tdm orientations.Applying this method on hexa-coordinated Chl a, we separated Qx and Qy contributions to the Q-band absorption quantitatively, with 0–0 transition of Qy (Qy00) at (671 ± 1) nm, and Qx00 at (636 ± 1) nm. The investigated spectral Q-band range is well modelled by a single vibronic progression with a single frequency of 700 cm−1 for both the Qx and the Qy transition. We clearly identified the Qy01 and Qy02 vibronic progression peaks at (650 ± 6) nm, and (619 ± 3) nm, respectively, and the Qx01 peak at (607 ± 2) nm. The Qx and Qy transition tdms are well separated and not mixed. The energy gap between Qx00 and Qy00 was specified to (820 ± 60) cm−1. We identified distinct relative Franck–Condon factors for the Qy and Qx transitions. The ratio of Qy00 to Qy01, and Qy02 is 7 ± 2 and 9 ± 3, respectively. In contrast the ratio of Qx00 to Qx01 is 1.5 ± 0.2, reflecting a substantially stronger vibronic coupling in the Qx transition. The foundation for this contrast may be attributed to the difference in symmetry, but further studies need to explore the details of this contrast. We expect that extending the studies to multiple vibrational transitions may increase accuracy and provide more detailed information.
Our results provide an essential framework for further calculations of energy transfer processes on photosystems and a starting point for comparing Chl a properties with penta-coordinated macrocycle. Anisotropy excitation spectroscopy is a powerful and straight-forward tool to unravel different electronic ground-state transitions and their vibronic sub-structure in a variety of important photoactive molecules such as porphyrins, corroles, linear tetrapyrroles, metal complexes and photoreceptors.
Data availability
Data for this paper, including transient polarization resolved data are available at https://box.fu-berlin.de/s/YbB4AamN4JZiYLB.Author contributions
K. H. designed the experiment; C. Z. performed the Vis-IR spectroscopy and analyzed the data, T. S. and K. H. contributed to analyzing the data, C. Z. and K. H. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft DFG 169703910 and SFB-1078 for financial support.References
- J. Reimers, Z.-L. Cai, R. Kobayashi, M. Rätsep, A. Freiberg and E. Krausz, Sci. Rep., 2013, 3, 2761 CrossRef PubMed.
- Y. Song, A. Schubert, E. Maret, R. K. Burdick, B. D. Dunietz, E. Geva and J. P. Ogilvie, Chem. Sci., 2019, 10, 8143–8153 RSC.
- F. Santoro, A. Lami, R. Improta, J. Bloino and V. Barone, J. Chem. Phys., 2008, 128, 224311 CrossRef PubMed.
- M. S. Katelyn, J. Dasgupta, J. C. Lagarias and R. A. Mathies, J. Am. Chem. Soc., 2009, 131, 13946 CrossRef PubMed.
- R. E. Blankenship, Molecular mechanisms of photosynthesis, Wiley-Blackwell, London, 2nd edn, 2014 Search PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- M. Gouterman and L. Stryer, J. Chem. Phys., 1962, 37, 2260–2266 CrossRef CAS.
- L. L. Shipman, T. M. Cotton, J. R. Norris and J. J. Katz, J. Am. Chem. Soc., 1976, 98, 8222–8230 CrossRef CAS PubMed.
- D. Bauman and D. Wrobel, Biophys. Chem., 1980, 12, 83–91 CrossRef CAS PubMed.
- M. Fragata, B. Norden and T. Kurucsev, Photochem. Photobiol., 1988, 47, 133–143 CrossRef CAS.
- C. Houssier and K. Sauer, J. Am. Chem. Soc., 1970, 92, 779–791 CrossRef CAS.
- C. Weiss, J. Mol. Spectrosc., 1972, 44, 37–80 CrossRef CAS.
- M. V. Belkov and A. P. Losev, Spectrosc. Lett., 1978, 11, 653–669 CrossRef CAS.
- M. Umetsu, Z.-Y. Wang, M. Kobayashi and T. Nozawa, Biochim. Biophys. Acta, Bioenerg., 1999, 1410, 19–31 CrossRef CAS.
- M. Umetsu, Z.-Y. Wang, K. Yoza, M. Kobayashi and T. Nozawa, Biochim. Biophys. Acta, Bioenerg., 2000, 1457, 106–117 CrossRef CAS.
- J. L. Hughes, R. Razeghifard, M. Logue, A. Oakley, T. Wydrzynski and E. Krausz, J. Am. Chem. Soc., 2006, 128, 3649–3658 CrossRef CAS PubMed.
- J. R. Reimers and E. Krausz, Phys. Chem. Chem. Phys., 2014, 16, 2315–2322 RSC.
- R. Avarmaa and K. Rebane, Spectrochim. Acta, Part A, 1985, 41, 1365–1380 CrossRef.
- J. L. Hughes, B. Conlon, T. Wydrzynski and E. Krausz, Phys. Procedia, 2010, 3, 1591–1599 CrossRef CAS.
- M. Rätsep, J. Linnanto and A. Freiberg, J. Chem. Phys., 2009, 130, 194501 CrossRef PubMed.
- M. Rätsep, Z.-L. Cai, J. R. Reimers and A. Freiberg, J. Chem. Phys., 2011, 134, 024506 CrossRef PubMed.
- M. Rätsep, J. M. Linnanto and A. Freiberg, J. Phys. Chem. B, 2019, 123, 7149–7156 CrossRef PubMed.
- P. Martinsson, J. A. Oksanen, M. Hilgendorff, P. H. Hynninen, V. Sundström and E. Åkesson, Chem. Phys. Lett., 1999, 309, 386–394 CrossRef CAS.
- Y. Shiu, Y. Shi, M. Hayashi, C. Su, K. Han and S. Lin, Chem. Phys. Lett., 2003, 378, 202–210 CrossRef CAS.
- M. Linke, A. Lauer, T. von Haimberger, A. Zacarias and K. Heyne, J. Am. Chem. Soc., 2008, 130, 14904–14905 CrossRef PubMed.
- E. Meneghin, C. Leonardo, A. Volpato, L. Bolzonello and E. Collini, Sci. Rep., 2017, 7, 11389 CrossRef PubMed.
- N. H. C. Lewis and G. R. Fleming, J. Phys. Chem. Lett., 2016, 7, 831–837 CrossRef CAS PubMed.
- P. Jordan, P. Fromme, H. Witt, O. Klukas, W. Saenger and N. Krauß, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- A. Ben-Shem, F. Frolow and N. Nelson, Nature, 2004, 426, 630–635 CrossRef PubMed.
- T. S. Balaban, P. Fromme, A. R. Holzwarth, N. Krauß and V. I. Prokhorenko, Biochim. Biophys. Acta, Bioenerg., 2002, 1556, 197–207 CrossRef CAS.
- M. O. Senge, A. A. Ryan, K. A. Letchford, S. A. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef.
- S. Krawczyk, Biochim. Biophys. Acta, Bioenerg., 1989, 976, 140–149 CrossRef CAS.
- K. Saito, T. Suzuki and H. Ishikita, J. Photochem. Photobiol., A, 2018, 358, 422–431 CrossRef CAS.
- R. A. Kaindl, M. Wurm, K. Reimann, P. Hamm, A. M. Weiner and M. Woerner, J. Opt. Soc. Am. A, 2000, 17, 2086–2094 CrossRef CAS.
- T. Stensitzki, Y. Yang, V. Kozich, A. A. Ahmed, F. Kössl, O. Kühn and K. Heyne, Nat. Chem., 2018, 10, 126–131 CrossRef CAS PubMed.
- A. S. Holt and E. E. Jacobs, Plant Physiol., 1955, 30, 553–559 CrossRef CAS PubMed.
- M. Kaucikas, K. Maghlaoui, J. Barber, T. Renger and J. Thor, Nat. Commun., 2016, 7, 13977 CrossRef CAS PubMed.
- Y. Yang, D. Jones, T. von Haimberger, M. Linke, L. Wagnert, A. Berg, H. Levanon, A. Zacarias, A. Mahammed, Z. Gross and K. Heyne, J. Phys. Chem. A, 2012, 116, 1023–1029 CrossRef CAS PubMed.
- M. Theisen, M. Linke, M. Kerbs, H. Fidder, M. E.-A. Madjet, A. Zacarias and K. Heyne, J. Chem. Phys., 2009, 131, 124511 CrossRef PubMed.
- M. Newville, T. Stensitzki, D. B. Allen and A. Ingargiola, LMFIT: Non-Linear Least-Square Minimization and Curve-Fitting for Python, 2014 Search PubMed.
- S. B. Broyde and S. S. Brody, J. Chem. Phys., 1967, 46, 3334–3340 CrossRef CAS PubMed.
- I. Renge and R. Avarmaa, Photochem. Photobiol., 1985, 42, 253–260 CrossRef CAS.
- A. Sirohiwal, R. Berraud-Pache, F. Neese, R. Izsák and D. A. Pantazis, J. Phys. Chem. B, 2020, 124, 8761–8771 CrossRef CAS.
- L. O. Pålsson, M. D. Spangfort, V. Gulbinas and T. Gillbro, FEBS Lett., 1994, 339, 134–138 CrossRef.
- Y. Shibata, S. Nishi, K. Kawakami, J.-R. Shen and T. Renger, J. Am. Chem. Soc., 2013, 135, 6903–6914 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03538c |
| ‡ Present address: Institute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam-Golm, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |