
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electrochemical multicomponent reaction toward C–H tetrazolation of alkyl arenes and vicinal azidotetrazolation of alkenes†
Yi
Yu
,
Xiao-Bin
Zhu
,
Yaofeng
Yuan
 and
Ke-Yin
Ye
*
and
Ke-Yin
Ye
*
Key Laboratory of Molecule Synthesis and Function Discovery (Fujian Province University), College of Chemistry, Fuzhou University, Fuzhou 350108, China. E-mail: kyye@fzu.edu.cn
First published on 9th November 2022
Abstract
The widespread use of tetrazoles in medicine, biology, and materials science continuously promotes the development of their efficient and selective syntheses. Despite the prosperous development of multicomponent reactions, the use of the most abundant and inexpensive chemical feedstocks, i.e., alkanes and alkenes, toward the preparation of diverse tetrazoles remains elusive. Herein, we developed an electrochemical multicomponent reaction (e-MCR) for highly efficient and selective C–H tetrazolation of alkyl arenes. When applied to alkenes, the corresponding vicinal azidotetrazoles were readily obtained, which were further demonstrated to be versatile building blocks and potential high-energy materials.
Tetrazoles constitute an important pharmacophore in medicinal chemistry thanks to their bioisosterism to carboxylic acid, furan, and amide moieties, as well as other beneficial physicochemical properties.1 In addition, their nitrogen-rich character also makes tetrazoles one of the most popular explosophores in high-energy materials.2 Therefore, the development of efficient, straightforward, and ideally modular synthetic approaches toward diverse novel tetrazole scaffolds would significantly accelerate the relevant research in pharmaceutical, biology, and materials science.
The last few decades have witnessed tremendous advances in the highly efficient and selective C(sp3)–H functionalization of saturated hydrocarbons. Specifically, oxidative cross-dehydrogenative coupling reactions3 between alkyl arenes and tetrazoles have been intensively investigated.4 We reasoned that a multicomponent reaction (MCR), when combined with state-of-the-art C(sp3)–H functionalization, is an appealing approach to quickly construct complex tetrazoles in a highly modular manner ideally starting from the readily available chemical feedstock.5 However, direct use of the most abundant and inexpensive unsaturated hydrocarbons in the MCR reaction for the synthesis of tetrazoles remains elusive.
Electrosynthesis provides new opportunities for the functionalization of C(sp3)–H bonds6 owing to its unique capability to tune reactivity and selectivity via the precise control of applied potentials.7 Therefore, we envisaged that an electrochemical multicomponent reaction (e-MCR) of saturated hydrocarbons, nitriles, and azides, may constitute a highly straightforward and modular approach for the preparation of diverse tetrazoles (Scheme 1A, left).
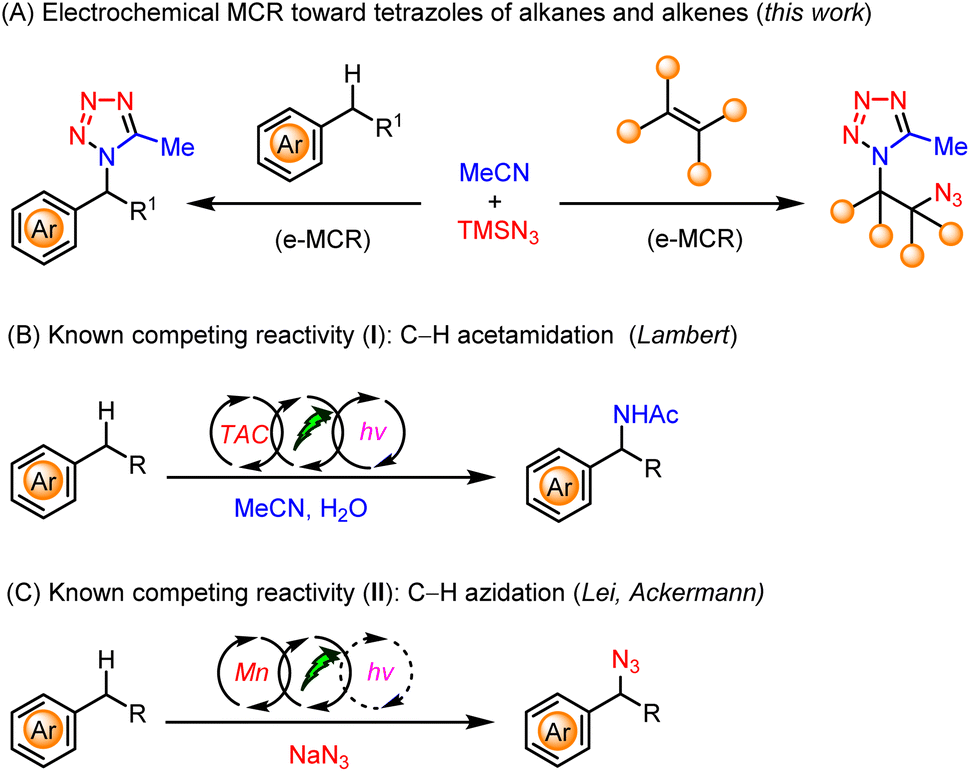 | ||
| Scheme 1 Tetrazoles via electrochemical multicomponent reactions. | ||
However, the realization of such a hypothesis needs to surpass several competing reaction pathways of alkyl arenes, i.e., electrochemical C–H acetamidation8 with acetonitrile (Scheme 1B), and C–H azidation9 with azide (Scheme 1C), respectively. In addition, we aim to develop an operationally simple and practical e-MCR, which would mitigate the sophisticated reaction setup as well as the requisite presence of irradiation and a transition-metal catalyst or organocatalyst in the realm of electrophotocatalysis.10
Herein, we report our development of a highly efficient e-MCR for the facile and modular synthesis of tetrazoles, directly using readily available alkanes, acetonitrile, and azidotrimethylsilane. In addition, the same strategy could be further applied to electrochemical difunctionalization of alkenes,11 from which the thus obtained vicinal azidotetrazoles (Scheme 1A, right) were further demonstrated to be versatile building blocks as well as potential high-energy materials.
After extensive optimization of reaction conditions (see the ESI†), we found that the constant-current electrolysis (I = 75 mA) of ethylbenzene (1) and azidotrimethylsilane (2) in an undivided cell, with a graphite anode and a platinum cathode, readily afforded the C–H tetrazolation product (3) in 68% yield (Scheme 2). The use of excess amounts of TMSN3 was to compromise its unproductive consumption of N2 during electrolysis.9a Note that while a relatively low current electrolysis (I = 20 m) was also amenable to achieving this reactivity, the high current density further rendered this reaction complete within 20 min, providing a fast and straightforward approach to access the tetrazole moiety directly from alkyl arenes. Particularly, the addition of HFIP (hexafluoroisopropanol) was pivotal to achieving the anticipated reactivity.12
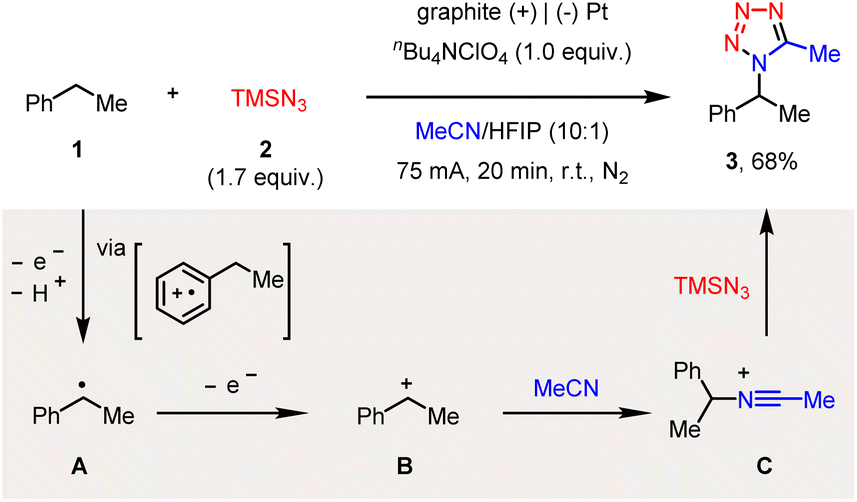 | ||
| Scheme 2 Electrochemical C–H tetrazolation of ethylbenzene. | ||
Regarding the possible mechanism, ethylbenzene (1) is likely to first undergo single-electron oxidation and then deprotonation, via a well-established phenyl radical cation intermediate, to generate a benzyl radical (A), which is followed by second electrochemical oxidation to generate the benzyl carbocation (B).13 The nucleophilic interception by MeCN then forms a Ritter-type nitrilium ion intermediate (C),8 which finally reacts with azide to afford the C–H tetrazolation product (3).14 Another possible route for the formation of the benzyl radical (A), i.e., via the hydrogen-atom transfer (HAT) of the benzylic C–H bond by the electrochemically generated azido radical, is less likely accordingly to our mechanistic investigations (vide infra).
The substrate scope of this electrochemical C–H tetrazolation of alkyl arenes was then investigated (Scheme 3). This reaction tolerated a multitude of substituted ethylbenzenes (4–6), toluene (7), and other alkyl-substituted benzenes (8, 9). Diverse functionalities, such as ketones (10), sulfonyl benzamide (11), trifluoroacetamide (12), and succinimide (13) well survived under the current reaction settings. Despite their high propensity toward nucleophilic substitution by TMSN3, the aliphatic chloride (14) and protected alcohols (15, 16) remained intact. In addition, functionalized cyclic alkyl arenes (17, 18), and thiophene derivatives (19) also proceeded with the anticipated electrochemical C–H tetrazolation smoothly. The current electrochemical C–H tetrazolation could be also extended to the late-stage functionalization of a variety of alkyl arenes derived from biologically relevant compounds and pharmaceuticals, such as FKGK 11 analogue (20), ibuprofen (21), celestolide (22), and retinoic acid receptor agonist analogue (23). Unfortunately, acetonitrile is currently the only amenable organonitrile in this electrochemical C–H tetrazolation.
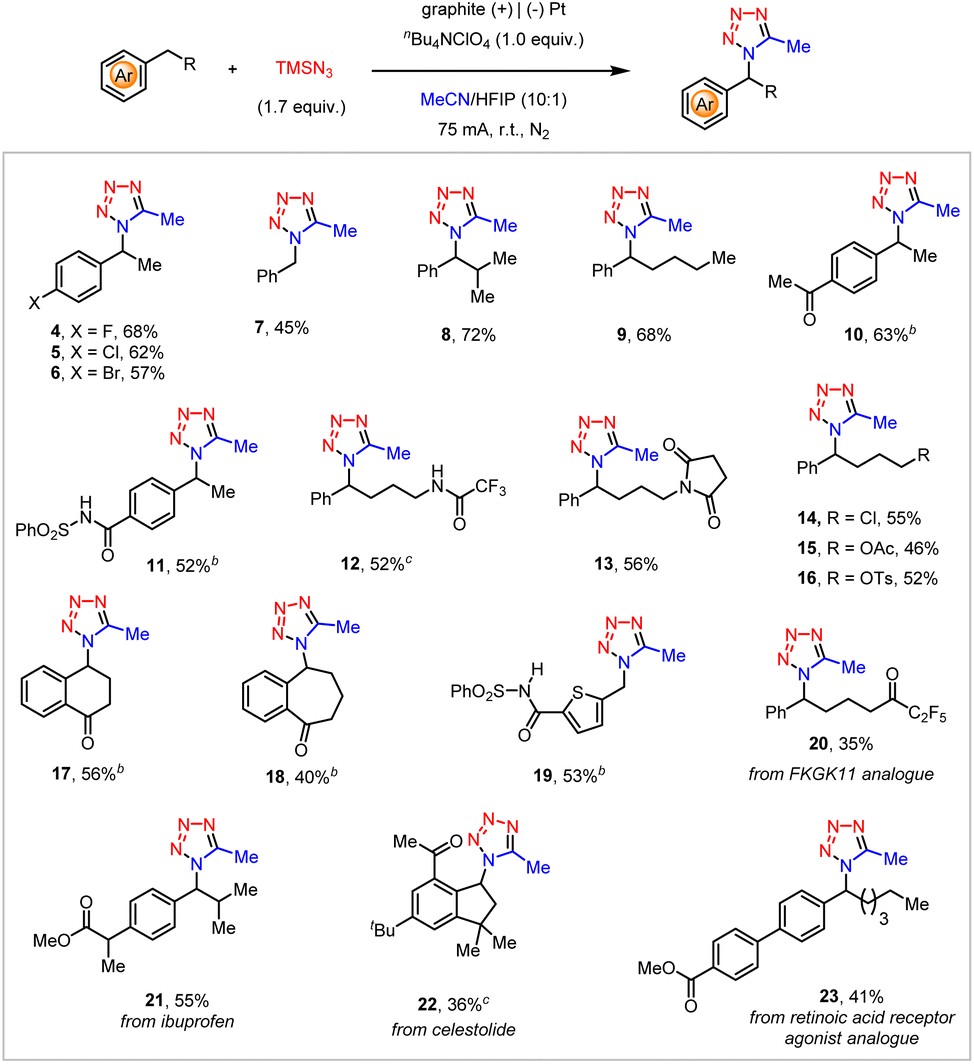 | ||
Scheme 3 Substrate scope of the electrochemical C–H tetrazolation.a aConditions: undivided cell, graphite rod anode (ϕ 6 mm, about 20 ± 1 mm immersion depth in solution), Pt cathode, alkyl arenes (0.3 mmol), TMSN3 (0.51 mmol, 1.7 equiv.), nBu4NClO4 (0.3 mmol), MeCN/HFIP = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 75 mA, 20 min, (F = 3.11 F mol−1) at room temperature under an N2 atmosphere, isolated yield; bLiClO4 (0.3 mmol), 75 mA, 23 min (F = 3.58 F mol−1); c20 mA, 90 min (F = 3.73 F mol−1). :1, 75 mA, 20 min, (F = 3.11 F mol−1) at room temperature under an N2 atmosphere, isolated yield; bLiClO4 (0.3 mmol), 75 mA, 23 min (F = 3.58 F mol−1); c20 mA, 90 min (F = 3.73 F mol−1). | ||
In addition, the e-MCR strategy is readily applicable to the vicinal difunctionalization of alkenes. While the constant-current electrolysis only afforded a moderate yield of the desired product, we found the constant-cell potential electrolysis (Ecell = 2.8 V) of styrene (24) and TMSN3 (2) in a MeCN/HFIP mixed solvent resulted in the formation of a vicinal azidotetrazole in a higher yield (25, 68%, Table 1, entry 1). In this case, the graphite plate anode afforded a slightly higher yield than that of the graphite rod as previously established.15 Again, the choice of HFIP as the co-solvent was crucial since the concomitant formation of the competing azidoacetamide (26) occurred when HOAc or H2O was used instead (entries 2 and 3). The electrolyte is another important parameter to tune product selectivity. For instance, though the use of nBu4NBF4 afforded both azidotetrazole (25) and azidoacetamide (26) almost unselectively (entry 4), nBu4NHSO4 completely suppressed the azidotetrazolation but promoted the azidoacetamidation albeit in a low yield (entry 5).
|
|
||||
|---|---|---|---|---|
| Entry | Electrolyte | Co-solvent | 25 (%) | 26 (%) |
|
a Reaction conditions: undivided cell, graphite plate anode, Pt cathode, 24 (0.3 mmol), TMSN3 (0.75 mmol, 2.5 equiv.), electrolyte (0.3 mmol), MeCN/co-solvent = 12.5:1, at room temperature under an N2 atmosphere, the yield was determined by 1H NMR with 1,3,5-trimethoxybenzene as the internal standard.
b Isolated yield.
|
||||
| 1 | n Bu4NClO4 | HFIP | 68b | 0 |
| 2 | n Bu4NClO4 | HOAc | 28 | 13 |
| 3 | n Bu4NClO4 | H2O | 21 | 12 |
| 4 | n Bu4NBF4 | H2O | 15 | 18 |
| 5 | n Bu4NHSO4 | H2O | 0 | 31 |
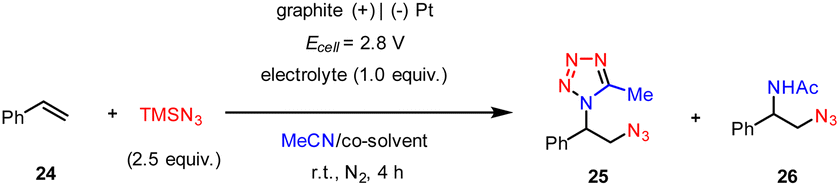
We then investigated the substrate scope of the electrochemical vicinal azidotetrazolation. As illustrated in Scheme 4, a wide array of substituted styrenes were well tolerated to afford the anticipated vicinal azidotetrazoles in moderate to good yields (27–31). In addition, α-alkyl-substituted styrenes generally served as good substrates for the preparation of vicinal azidotetrazoles (32–37). The 1,2-disubstituted linear alkenes (38, 39) proceeded with the anticipated vicinal azidotetrazolation smoothly, too. Notably, the sterically hindered trisubstituted (40, 41) and tetrasubstituted alkenes (42), as well as heteroaryl alkenes (43, 44) were all amenable substrates. Besides acetonitrile, other aliphatic nitriles, such as isobutyronitrile (45) and butyronitrile (46), were also positively engaged in the electrochemical syntheses of vicinal azidotetrazoles. Unfortunately, the current protocol does not tolerate benzonitrile, probably owing to its relatively weak nucleophilicity. Again, this protocol can be successfully applied to late-stage vicinal azidotetrazolation of complex alkenes derived from drug derivatives, including probenecid (47), fenofibric acid (48), and febuxostat (49).
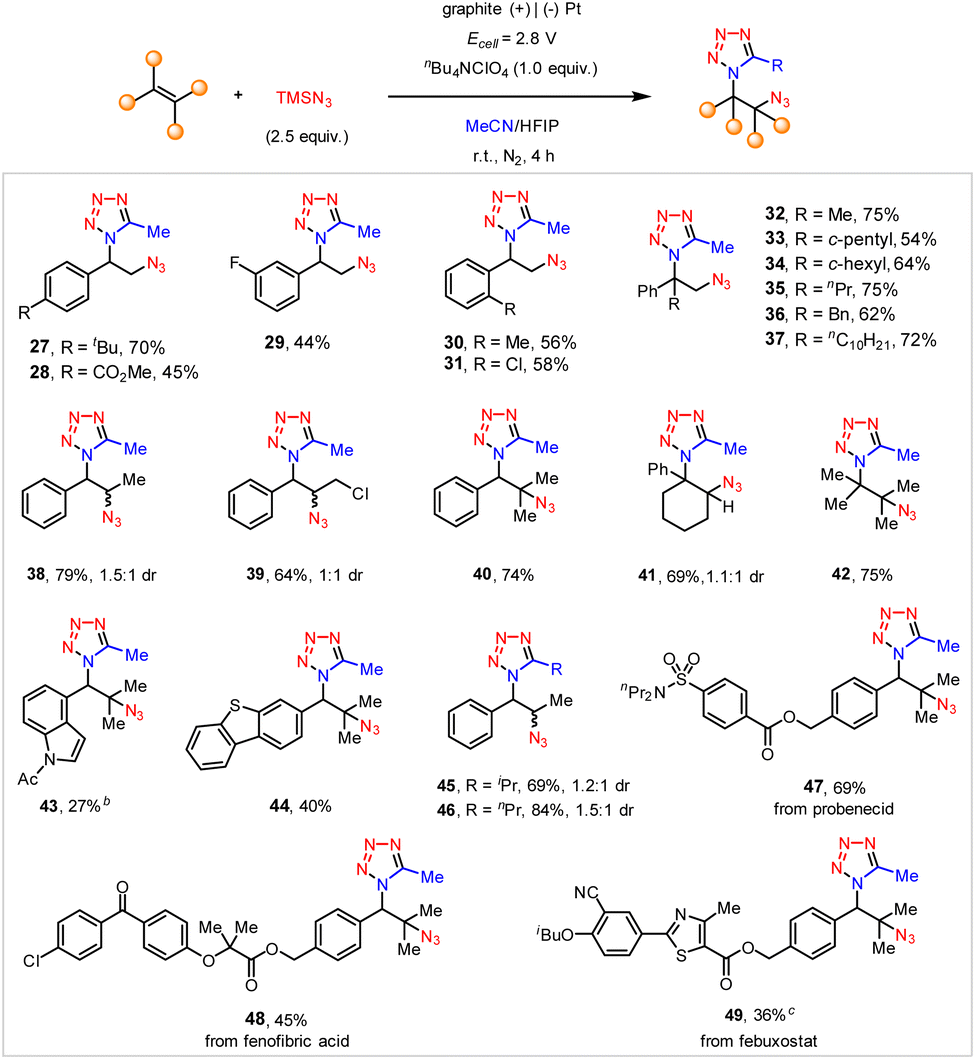 | ||
| Scheme 4 Substrate scope of the electrochemical vicinal tetrazolation.a aReaction conditions: undivided cell, graphite plate anode, Pt cathode, alkene (0.3 mmol), 2 (0.75 mmol, 2.5 equiv.), nBu4NClO4 (0.3 mmol), MeCN/HFIP = 12.5:1, at room temperature under an N2 atmosphere, isolated yield; b2.5 V, 4.5 h; cMeCN/DCM/HFIP = (8.75:3.75:1) as the co-solvent. | ||
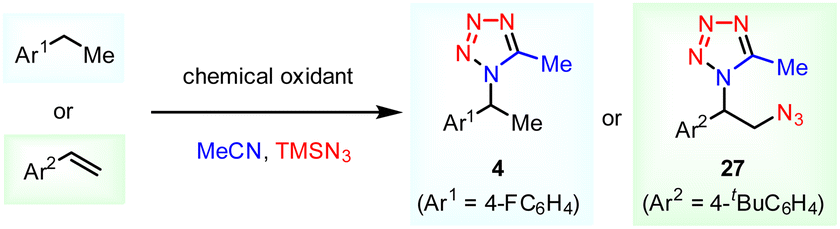
Without electrical input, this e-MCR reactivity could not be replicated by using other common strong oxidants (Table 2). For the C–H tetrazolation (4), low conversions were generally observed and a substantial amount of starting materials were recovered. Similarly, no desired vicinal azidotetrazolation product (27) could be detected even though high conversions were realized. Instead, only the formation of a trace amount of aldehyde was detected. The direct comparison between electrosynthesis and chemical synthesis in the formation of targeted tetrazoles further substantiates the unique potential of electrochemistry in the expansion of novel chemical space and reactivity.
|
|
|||
|---|---|---|---|
| Entry | Oxidant | 4, yielda (conv., %) | 27, yielda (conv., %) |
| a Yield and conversion were determined by NMR of the crude reaction mixture with PhOCF3 or CH2Br2 as the internal standard; n.d. = not detected. | |||
| 1 | Anode | 68 (>95) | 70 (>95) |
| 2 | NFSI | n.d. (11) | n.d. (51) |
| 3 | TBHP | n.d. (20) | n.d. (73) |
| 4 | CAN | n.d. (36) | n.d. (35) |
| 5 | m-CPBA | n.d. (14) | n.d. (22) |
| 6 | DDQ | n.d. (15) | n.d. (91) |
| 7 | PhI(OAc)2 | n.d. (30) | n.d. (>95) |
| 8 | Mn(OAc)3 | n.d. (69) | n.d. (>95) |
| 9 | AgNO3 | n.d. (>95) | n.d. (>95) |
The nitrogen-rich character of the obtained vicinal azidotetrazoles raises safety concerns about their thermal stability since both the azido and tetrazole moieties are frequently present as explosophores in high-energy materials.16 Differential scanning calorimetry (DSC) and thermogravimetric (TG) analyses were then performed to elucidate their thermal sensitivity (Fig. 1).17 Indeed, the azidotetrazole (25) bears a high value of decomposition enthalpy (−1270 J g−1 or −291 kJ mol−1), implying that special attention should be paid to its scaled preparation and usage. However, the high onset temperature value (233 °C) also suggests that this azidotetrazole is relatively less sensitive to heat and would release its energy only at high temperatures. This is further substantiated by its TD24 data, which typically would serve as a consecutive prediction of a reasonable safety margin.18 In addition, the TG analyses indicated that the major decomposition mechanism of this azidotetrazole is the thermal extrusion of the tetrazole moiety at Tm, probably initiated via the loss of molecular nitrogen (Fig. 1, right).19
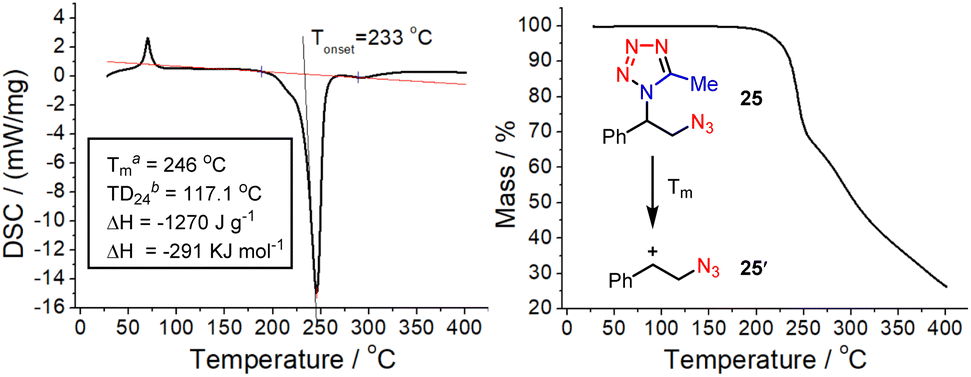 | ||
| Fig. 1 DSC (left) and TG (right) curves of the azidotetrazole 25. (a) Maximum temperature in the DSC curve; (b) temperature at which the time of maximum rate is 24 h. | ||
The azido group of these vicinal azidotetrazoles provides a good platform for their further synthetic elaborations (Scheme 5). For instance, compounds vicinally functionalized both by a tetrazole and a triazole were conveniently prepared via click reactions between the azidotetrazoles and alkynes, such as phenylacetylene (50), and 5-ethynyl-2′-deoxyuridine (51). When treated with trimethyl phosphite, the azido moiety was readily transformed into its corresponding phosphoramidate (52). Finally, a base-promoted dehydroazidation of the vicinal azidotetrazole afforded the vinyltetrazole (53) in a quantitative yield. Note that the tetrazole moiety remained intact during all these derivatizations.
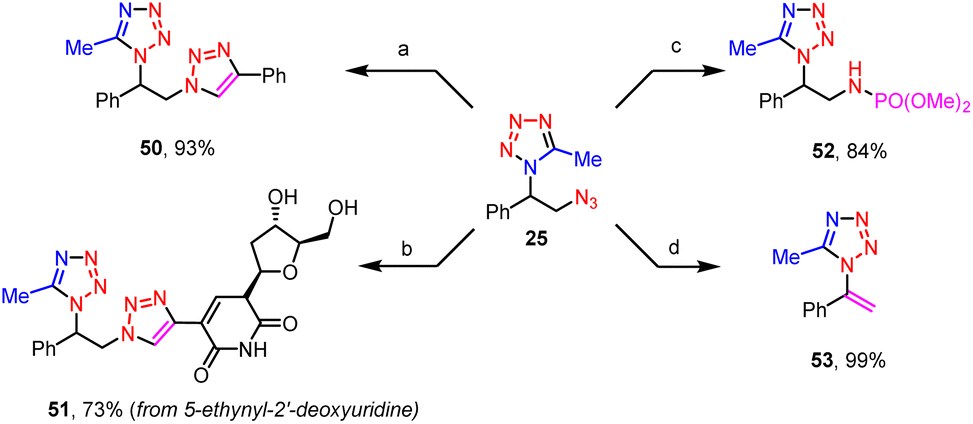 | ||
| Scheme 5 Derivatization of azidotetrazoles. Reaction conditions: (a) phenylacetylene (3 equiv.), CuI (30 mol%), THF, r.t., 4 h; (b) 5-ethynyl-2′-deoxyuridine (3 equiv.), CuI (30 mol%), THF, 60 °C, 5 h; (c) P(OMe)3 (1.3 equiv.), toluene, 80 °C, 3 h; (d) tBuOK (2 equiv.), THF, r.t., 1 min. | ||
To gain some more insights into the reaction mechanism, several mechanistic experiments were performed. Cyclic voltammetry studies indicated that while the oxidation potential of ethyl benzene was rather high (Ep/2 = 2.43 V vs. Ag/AgCl), the addition of HFIP dramatically facilitated its oxidation (Ep/2 = 2.01 V vs. Ag/AgCl). This positive effect of HFIP was attributed to its unique capability to stabilize radical cation intermediates in the anodic oxidation of the substrate (Fig. 2A).12 As illustrated in Fig. 2B, the relatively low oxidation potential of TMSN3 (Ep/2 = 0.92 V vs. Ag/AgCl) should favor its prior oxidation to ethyl benzene or styrene (Ep/2 = 2.21 V vs. Ag/AgCl). Indeed, the involvement of an azido radical was supported by the radical clock experiment (Fig. 2C).
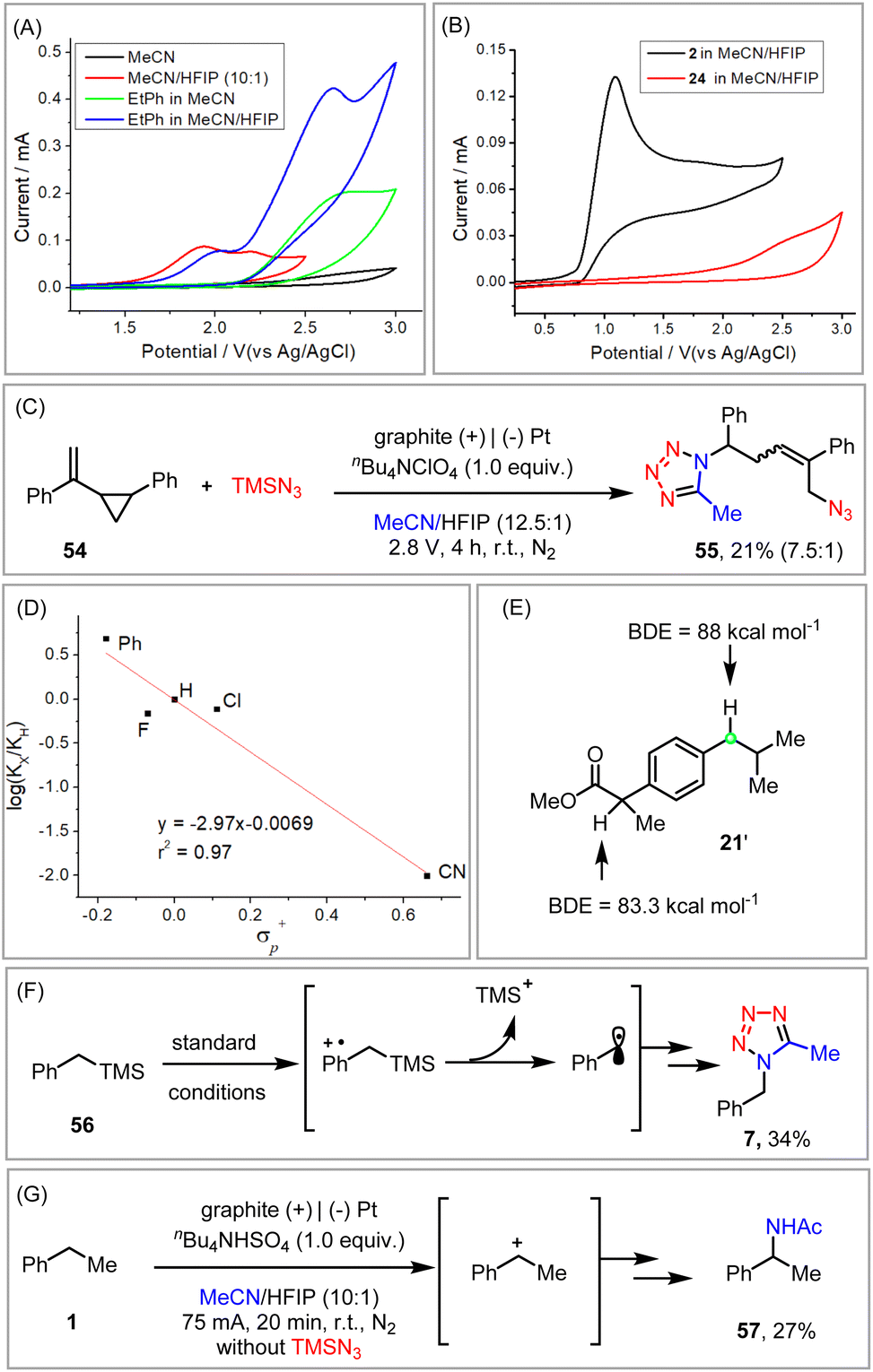 | ||
| Fig. 2 Mechanistic experiments. | ||
In the electrochemical C–H tetrazolation, the Hammett studies (ρ = −2.97) of various p-substituted ethyl benzenes exhibiting a much more negative ρ than that of a typical HAT process (ρ > −1),20a revealed that a positive charge was built up in the transition state of the rate-limiting step (Fig. 2D).20b The possible formation of the benzyl radical via HAT was further disapproved by the fact that benzylic C–H bonds of similar bond dissociation energies but different electronic properties were still well distinguished (21′, Fig. 2E).21 Parallel to the observations in Xu's photoelectrochemical C–H cyanation,22 benzyltrimethylsilane (56) proceeded via the cleavage of the C–Si bond to afford tetrazole (7) exclusively, which again disproved the one-step HAT process but favored a radical cation mechanism (Fig. 2F). Taken together, the formation of the benzyl radical from ethylbenzene is more likely via a stepwise electron transfer/proton transfer process but not HAT.22,23 The involvement of a carbocation intermediate was further substantiated by the formation of the Ritter-type product in the absence of TMSN3 (Fig. 2G).
A proposed mechanistic rationale for the vicinal azidotetrazolation reaction is shown in Scheme 6. Similar to the C–H tetrazolation of alkyl arenes, the β-azido carbocation (II), generated via the sequential addition of the azido radical  and single-electron oxidation, is the key intermediate in the azidotetrazolation reaction (IV, path a).24 Notably, the competing diazidation (III)25 is disfavoured since a radical pathway requires the cross-coupling of two transient radicals (path b).26 On the other hand, a polar pathway to the diazidation product (path c) is mitigated owing to the much excess of MeCN as the carbocation trapping agent on the anode surface, out-competing the low-concentrated TMSN3 because of its easy oxidation character (see the ESI†). This rationale of product selectivity is also applicable to the electrochemical C–H tetrazolation since both reactions share similar Ritter-type nitrilium ion intermediates toward the formation of the tetrazole moiety.
and single-electron oxidation, is the key intermediate in the azidotetrazolation reaction (IV, path a).24 Notably, the competing diazidation (III)25 is disfavoured since a radical pathway requires the cross-coupling of two transient radicals (path b).26 On the other hand, a polar pathway to the diazidation product (path c) is mitigated owing to the much excess of MeCN as the carbocation trapping agent on the anode surface, out-competing the low-concentrated TMSN3 because of its easy oxidation character (see the ESI†). This rationale of product selectivity is also applicable to the electrochemical C–H tetrazolation since both reactions share similar Ritter-type nitrilium ion intermediates toward the formation of the tetrazole moiety.
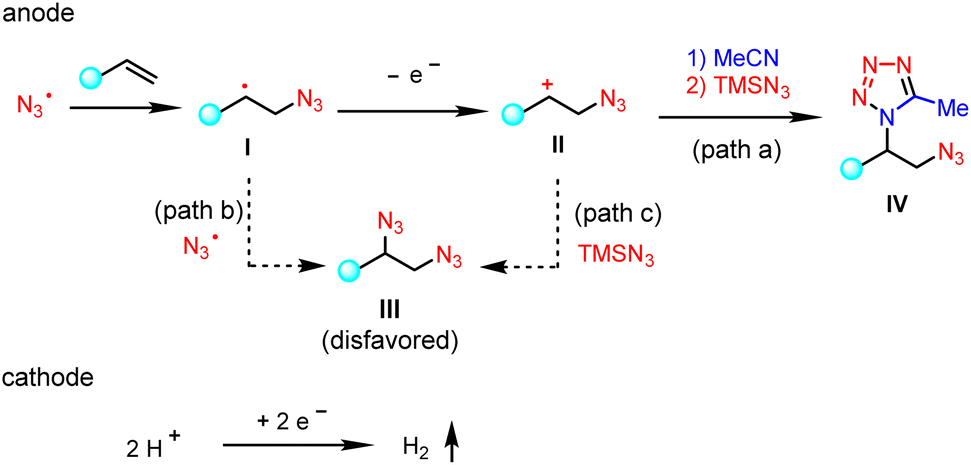 | ||
| Scheme 6 Proposed mechanism. | ||
In summary, we have developed an electrochemical multi-component reaction for the modular assembly of diverse tetrazoles from readily available alkanes, acetonitrile, and azidotrimethylsilane. The fine-tuning of electrolytic conditions detoured other competing pathways but afforded the anticipated benzylic tetrazoles highly selectively. When applied to alkenes, the corresponding vicinal azidotetrazoles were readily obtained, which have been demonstrated to be versatile building blocks and potential high-energy materials.27 The current work indicates that the e-MCR might be broadly applicable for the facile syntheses of diverse nitrogen-containing heterocycles.
Data availability
Detailed synthetic procedures and complete characterization data for all new compounds can be found in the ESI.†Author contributions
K. Y. Y. conceived the idea and supervised the project. Y. Y. conducted the majority of the experimental work. K. Y. Y., Y. Y., X. B. Z., and Y. F. Y. analyzed the data and contributed to the preparation of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (22171046, 21901041, and 22071025), State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University (202008), Hundred-Talent Project of Fujian (50012742), and Fuzhou University (510841) is gratefully acknowledged.Notes and references
- (a) L. V. Myznikov, A. Hrabalek and G. I. Koldobskii, Chem. Heterocycl. Compd., 2007, 43, 1–9 CrossRef CAS; (b) N. Dhiman, K. Kaur and V. Jaitak, Bioorg. Med. Chem., 2020, 28, 115599 CrossRef CAS PubMed; (c) P. Patowary, B. Deka and D. Bharali, Malar. Control Elimin., 2021, 10, 167 Search PubMed.
- (a) A. A. Dippold, D. Izsák, T. M. Klapötke and C. Pflüger, Chem.–Eur. J., 2016, 22, 1768–1778 CrossRef CAS PubMed; (b) T. Wang, H. Gao and J. n. M. Shreeve, Z. Anorg. Allg. Chem., 2021, 647, 157–191 CrossRef CAS.
- (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS.
- (a) J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS; (b) Z. Ruan, Z. Huang, Z. Xu, S. Zeng, P. Feng and P.-H. Sun, Sci. China: Chem., 2021, 64, 800–807 CrossRef CAS.
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970–2042 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS.
- (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (b) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (c) K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed; (d) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (e) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- (a) T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS; (b) T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef CAS PubMed; (c) L.-H. Li, Z.-J. Niu, Y.-X. Li and Y.-M. Liang, Chem. Commun., 2018, 54, 11148–11151 RSC; (d) K. Ishihara, T. Shioiri and M. Matsugi, Org. Lett., 2020, 22, 6244–6247 CrossRef CAS.
- (a) T. H. Meyer, R. C. Samanta, A. Del Vecchio and L. Ackermann, Chem. Sci., 2021, 12, 2890–2897 RSC; (b) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS.
- (a) L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS PubMed; (b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS; (c) Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC; (d) J. Liu, L. Lu, D. Wood and S. Lin, Sci. China: Chem., 2020, 6, 1317–1340 CAS.
- (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS; (b) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed.
- (a) L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Chem. Commun., 1996, 2105–2112 RSC; (b) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef; (c) J. L. Röckl, M. Dörr and S. R. Waldvogel, ChemElectroChem, 2020, 7, 3686–3694 CrossRef.
- Z.-W. Hou, D.-J. Liu, P. Xiong, X.-L. Lai, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef.
- S. J. Wittenberger, Org. Prep. Proced. Int., 1994, 26, 499–531 CrossRef CAS.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- (a) C. B. Storm, J. R. Stine and J. F. Kramer, in Chemistry and Physics of Energetic Materials, ed. S. N. Bulusu, Springer Netherlands, Dordrecht, 1990, pp. 605–639 Search PubMed; (b) J. Rein, J. M. Meinhardt, J. L. Hofstra Wahlman, M. S. Sigman and S. Lin, ChemRxiv, 2021, preprint, DOI:10.33774/chemrxiv-2021-16f6w.
- (a) S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed; (b) T. M. Klapötke, C. Miró Sabaté and M. Rusan, Z. Anorg. Allg. Chem., 2008, 634, 688–695 CrossRef.
- F. Stoessel, Thermal Stability, in Thermal Safety of Chemical Processes: Risk Assessment and Process Design, Wiley, 2008, pp. 279–309 Search PubMed.
- (a) V. G. Kiselev, P. B. Cheblakov and N. P. Gritsan, J. Phys. Chem. A, 2011, 115, 1743–1753 CrossRef; (b) B. Yuan and E. R. Bernstein, J. Chem. Phys., 2016, 144, 234302 CrossRef PubMed; (c) A. I. Lesnikovich, S. V. Levchik, A. I. Balabanovich, O. A. Ivashkevich and P. N. Gaponik, Thermochim. Acta, 1992, 200, 427–441 CrossRef; (d) V. G. Prokudin, V. S. Poplavsky and V. A. Ostrovskii, Russ. Chem. Bull., 1996, 45, 2094–2100 CrossRef.
- (a) W. D. Totherow and G. J. Gleicher, J. Am. Chem. Soc., 1969, 91, 7150–7154 CrossRef CAS; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- The data of BDEs were obtained using web-based computational tool ALFABET, https://bde.ml.nrel.gov/ Search PubMed.
- C.-Y. Cai, X.-L. Lai, Y. Wang, H.-H. Hu, J. Song, Y. Yang, C. Wang and H.-C. Xu, Nat. Catal., 2022, 5, 943–951 CrossRef CAS.
- (a) M. Freccero, A. Pratt, A. Albini and C. Long, J. Am. Chem. Soc., 1998, 120, 284–297 CrossRef CAS; (b) A. J. Fry, J. M. Porter and P. F. Fry, J. Org. Chem., 1996, 61, 3191–3194 CrossRef CAS PubMed; (c) E. Baciocchi, T. Del Giacco, C. Rol and G. V. Sebastiani, Tetrahedron Lett., 1989, 30, 3573–3576 CrossRef CAS.
- S. Hajra, D. Sinha and M. Bhowmick, J. Org. Chem., 2007, 72, 1852–1855 CrossRef CAS.
- (a) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (b) S. Lin, J. Parry and N. Fu, Synlett, 2018, 29, 257–265 CrossRef; (c) J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140, 12511–12520 CrossRef; (d) J. C. Siu, J. B. Parry and S. Lin, J. Am. Chem. Soc., 2019, 141, 2825–2831 CrossRef; (e) H. M. Nelson, J. C. Siu, A. Saha, D. Cascio, S. N. MacMillan, S.-B. Wu, C. Lu, J. A. Rodríguez, K. N. Houk and S. Lin, Org. Lett., 2021, 23, 454–458 CrossRef PubMed.
- (a) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef CAS; (b) H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS; (c) A. Studer, Chem. Soc. Rev., 2004, 033, 267–273 RSC; (d) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- During the submission of this manuscript, Weng and co-workers reported a conceptually similar electrochemical azidation/heterocyclization of tryptophans. Y. Weng, X. Xu, H. Chen, Y. Zhang and X. Zhuo, Angew. Chem., Int. Ed., 2022, e202206308 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05423j |
| This journal is © The Royal Society of Chemistry 2022 |