
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection and screening of basic amino acids using the luminescence switching of a WS2 nanosheet–Ag2O nanoparticle composite†
Neema Pallikkarathodi
Mani
a,
Karthika S.
Sunil
b,
Ann Mary
Tomy
a,
Bhasha
Sathyan
a and
Jobin
Cyriac
 *a
*a
aDepartment of Chemistry, Indian Institute of Space Science & Technology, Thiruvananthapuram – 695 547, India. E-mail: jobincyriac@iist.ac.in
bDepartment of Chemistry, Indian Institute of Science Education and Research, Berhampur – 760010, India
First published on 22nd March 2022
Abstract
The Förster resonance energy transfer (FRET) pair-based detection has gained considerable attention due to its promising sensitivity and selectivity. Herein, we report a turn-on sensor for detecting basic amino acids utilizing the fluorescence emission properties of WS2 nanosheets (WS2 NSs). The sensor functions based on a FRET dyad. The addition of AgNO3 into an alkaline solution of WS2 NSs leads to the in situ formation of silver oxide nanoparticles (Ag2O NPs). The negative surface charge and the presence of sulphur vacancies, together with the possibility for a multiplexed adsorption platform of WS2 NSs, account for the formation Ag2O NPs on their surface. In the WS2 NS–Ag2O NP system (termed as a 0D–2D composite), the fluorescence emission of the nanosheets became subdued due to the FRET. This 0D–2D composite is proven to be an excellent turn-on sensor for basic amino acids. Histidine, lysine and arginine can induce the aggregation of the Ag2O NPs, which shuts the FRET pathway, along with the regeneration of the fluorescence of WS2 NSs. The aggregation of Ag2O NPs occurs in a sensor solution with pH below the isoelectric points of the amino acids to help to discriminate them. The dynamic range and limit of detection of the sensor have been evaluated. The protein detection capability of the sensor was verified using a lysine containing protein, ubiquitin, and by real sample analysis, using biological fluids.
1. Introduction
One of the salient features of atomically thin, 2D layered materials is the interrelation of their dimensionality and fundamental properties.1,2 The quantum confinement effect that emerged upon scaling down the dimensions is manifested as photoluminescence (PL) with an improved quantum yield, enabling these materials to be used as fluorescent-based chemical sensors.3,4 The natural abundance of transition metal dichalcogenides (TMDs) such as MoS2, WS2, etc., makes them attractive in the research realm of 2D layered materials. The increased demand in the market for miniaturization of electronic devices and their semiconducting nature with a sizeable bandgap catalyzes such studies. Layered TMDs of formula MX2 typically have a thickness of 6–9 Å and consist of a single layer of hexagonally packed metal atoms sandwiched between two layers of chalcogen atoms.5,6 The atoms within the layer are bonded by strong covalent bonds, whereas different layers are held together by weak van der Waals interactions, which enable them to exfoliate into monolayers with the emergence of exotic properties.7 In layered materials, the van der Waals interactions are not limited to interplanar interactions. Generally, any passivated, dangling bond-free surface interacts with another moiety via van der Waals forces. Consequently, any layered 2D material can be integrated with various materials of different dimensionalities to form mixed-dimensional structures. 0D–2D materials are such heterostructures produced by integrating 2D materials with 0D materials, primarily through noncovalent interactions. The 2D components may be graphene or transition metal dichalcogenides (TMDCs) and the 0D components may be small organic molecules or quantum dots (QDs). In contrast to conventional, epitaxially grown heterostructures, the interface in a mixed-dimensional structure is relatively complex and less constrained due to the absence of the need for lattice matching. The usual discontinuity in the band structure and resulting potential energy barriers, the density of states (DOS), also undergoes an abrupt transition, which has several observable implications, such as additional resistance at the junction that results from the change in the number of conductance channels.Amino acids (AAs), being the building units of proteins, are associated with various physiological processes and are vital components of life processes.8,9 For example, one of the basic amino acids, lysine (Lys), is associated with weight gain in animals, polyamine synthesis and the Krebs–Henseleit cycle.10 Histidine (His), another basic AA, is closely related to the growth and repair of tissues, the control of transmission of metal elements in biological bases, etc.11 Arginine (Arg), the third and most basic AA, is known for its association with cell division, immune function, the healing of wounds, the release of hormones, etc.12 The AA detection, in general, is thus significant in terms of valuable information it can render in the fields of nutritional analysis and diagnosis of diseases such as Alzheimer's, pancreatitis, etc. Among AAs, the detection of thiol-containing AAs is relatively easy owing to their unique nucleophilicity,13 whereas discrimination of non-thiol-containing AAs is still challenging. Electrochemical and chromatographic approaches are widely used strategies for the detection and characterization of AAs.14–19 Yet, the demand for sophisticated instruments, operational inconveniences, economic viabilities, choice of various parameters such as response time, detection limit, etc., pave the way for the quest for more desirable and convenient methods. Optical chemosensors are marked with their high sensitivity with notable selectivity along with the possibility of visual discrimination.8 Hence, a potential fluorometric probe for recognizing specific AAs susceptible to high-throughput assays holds a breakthrough in the current scenario.
Several nanomaterials are demonstrated as signal transducers in colourimetric/fluorescence chemical sensors. The ease of surveilling the light signal has made them a potential candidate in surface plasmon resonance (SPR)-based calorimetric sensing. Many reports that exploit the changes in the SPR peak of noble metal nanoparticles upon interaction with AAs are available. For example, p-sulfonatocalix[4]arene thiol modified Au nanoparticles have been used as a colourimetric sensor for His, Lys and Arg in water, based on the principle of broadening and shifting of SPR bands to the red region upon interaction, with linear detection limits of 1 mM, 2 mM and 4 mM, respectively.20 Chiral detection of L-His was reported based on L-arginine-sulfonated-substituted zinc tetraphenylporphyrin modified Ag nanoparticles (L-Arg–ZnTPPS–Ag NPs).21 Many groups reported fluorescence-based detection of AAs with improved sensitivity. Lu et al. utilized the fluorescence emission of a water-soluble fluorescein-bis-acrylate carrying pyridinium moiety having high sensitivity towards biothiols, which can discriminate simultaneously basic amino acids and thiol-containing amino acids from other amino acids.13 A simple fluorescent turn-on sensor, employing two complementary cucurbit[n]uril microarrays, has been demonstrated for the sensitive detection of basic AAs in water.22 A close observation of the literature reveals that the detection strategy depends on either the agglomeration of nanoparticles followed by a change in the SPR band or the change in the fluorescence emission of the probe. A combined approach with the ease of agglomeration of nanoparticles and the sensitivity of the fluorescence turn-on methodology would be attractive in chemical sensor applications.
Here, we report a fluorescence sensor for basic amino acids based on WS2 nanosheet–silver oxide nanoparticle (WS2 NS–Ag2O NP) composite materials. The composite was prepared via the in situ formation of silver oxide nanoparticles (Ag2O NPs) by the combined action of fluorescent WS2 nanosheets (WS2 NSs) in the presence of NaOH. The luminescent WS2 NSs were synthesized using a facile hydrothermal method. The fluorescence of WS2 NSs was quenched upon the formation of Ag2O NPs due to resonance energy transfer. The obvious spectral overlap of WS2 NS emission with that of surface plasmon resonance (SPR) absorption of Ag2O NPs explains the nonradiative energy transfer. The quenched fluorescence was selectively recovered in the presence of three basic amino acids: His, Lys and Arg, at a pH below the isoelectric point of each amino acid. The fluorescence recovery was attributed to the aggregation of Ag2O NPs and thereby the lack of spectral overlap between WS2 NSs and Ag2O NPs. The sensor has an appreciable dynamic range and limit of detection (LOD) for all three AA analytes. The formation of Ag2O NPs, the comprehensive mechanism of quenching and the recovery of fluorescence of WS2 NSs were proposed with experimental support. The real sample analysis was also demonstrated using biological fluids.
2. Experimental
2.1 Synthesis of WS2 nanosheets
The WS2 nanosheets were prepared by hydrothermal reaction of commercially available WS2 powder and NaOH in an aqueous medium by taking a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Briefly, WS2 powder (100 mg) was mixed with NaOH (16.0 mg) in 10 mL of water. The resultant solution was taken in a Teflon-lined autoclave and kept for 24 h at 220 °C. The solution after the reaction was centrifuged at 3000 rpm for 30 min; the supernatant was collected, filtered and dialyzed for 3 h using a dialysis bag (molecular weight cut off 500). These purified, light-yellow solutions were used for all further studies.
:1. Briefly, WS2 powder (100 mg) was mixed with NaOH (16.0 mg) in 10 mL of water. The resultant solution was taken in a Teflon-lined autoclave and kept for 24 h at 220 °C. The solution after the reaction was centrifuged at 3000 rpm for 30 min; the supernatant was collected, filtered and dialyzed for 3 h using a dialysis bag (molecular weight cut off 500). These purified, light-yellow solutions were used for all further studies.
2.2 Synthesis of the WS2 nanosheet–Ag2O NP nanocomposite
To a 2.5 mL of purified WS2 nanosheet solution, aliquots of AgNO3 solution (1, 2, 3 or 4 mL of 0.02 M) were added dropwise by stirring. The colour of the solution turned yellow in situ due to the formation of Ag2O NPs on the surface of the WS2 nanosheets.3. Results and discussion
3.1 Synthesis and characterization of WS2 nanosheets
In parallel with the steadily growing interest in 2D layered materials, different synthetic strategies have been reported for these materials. We propose a simple approach to prepare 2D layered materials in a nanosize regime employing a hydrothermal reaction. The vigorous reaction of WS2 powder with a 1:1 molar ratio of NaOH at 220 °C yielded exfoliated WS2 nanosheets. The optimized reaction time for the best photoluminescence (PL) properties was 24 h. Detailed microscopy and photophysical characterization techniques have been carried out to understand the structural and optical attributes of the material. The transmission electron microscopy (TEM) images (see Fig. 1) show the formation of NSs with a lateral dimension in the range of a few hundreds of nanometers. Notably, the crystallinity of the sheets is maintained even after the vigorous hydrothermal reaction, as evident in the selected area electron diffraction (SAED) pattern. The AFM micrographs confirm the formation of nanosheets with a few layer thicknesses (Fig. S1, ESI†). The thickness measurement from the height profiles drawn at different regions of the AFM micrographs confirms the layer thickness of 2.8 nm, indicating the presence of 2–3 layers of sheets.
 | ||
| Fig. 1 (a) TEM micrographs of WS2 nanosheets. The inset of (a) shows the SAED pattern presenting the crystalline nature of the WS2 nanosheets. (b) Magnified TEM image of the WS2 NSs depicting the Moiré pattern. | ||
Raman spectroscopy can effectively determine the number of layers in the exfoliated TMDs. Generally, there are four major Raman-active modes in bulk WS2, namely A1g, E1g, E12g, and E22g modes. Due to the forbidden selection rule in the back-scattering geometry and the limited rejection against Rayleigh scattering, it is challenging to detect the E1g and E22g modes, respectively.23–26 However, the in-plane vibrational E12g mode and the out-of-plane vibrational A1g mode are more significant as they can be used to identify the number of layers present in the sheets. A thorough study regarding the frequency trends of the A1g and E2g modes with varying layer thicknesses was performed by Molina-Śanchez and Wirtz.27 As the number of layers increases from monolayer to bulk, the A1g mode tends to blue-shift, whereas the E12g mode shows a red-shift. The weak interlayer interactions and strong dielectric screening of the long-range Coulomb interactions are reasons for this behavior. Therefore, a direct correlation between the frequency difference (Δω) of the E12g mode and A1g mode and the sample thickness can be established.28 In the present case, for WS2 nanosheets, the value of Δω is <65 cm−1, which corresponds to monolayer thickness, invariably denoting the exfoliation of the bulk material into nanosheets of a few layers (Fig. 2a).
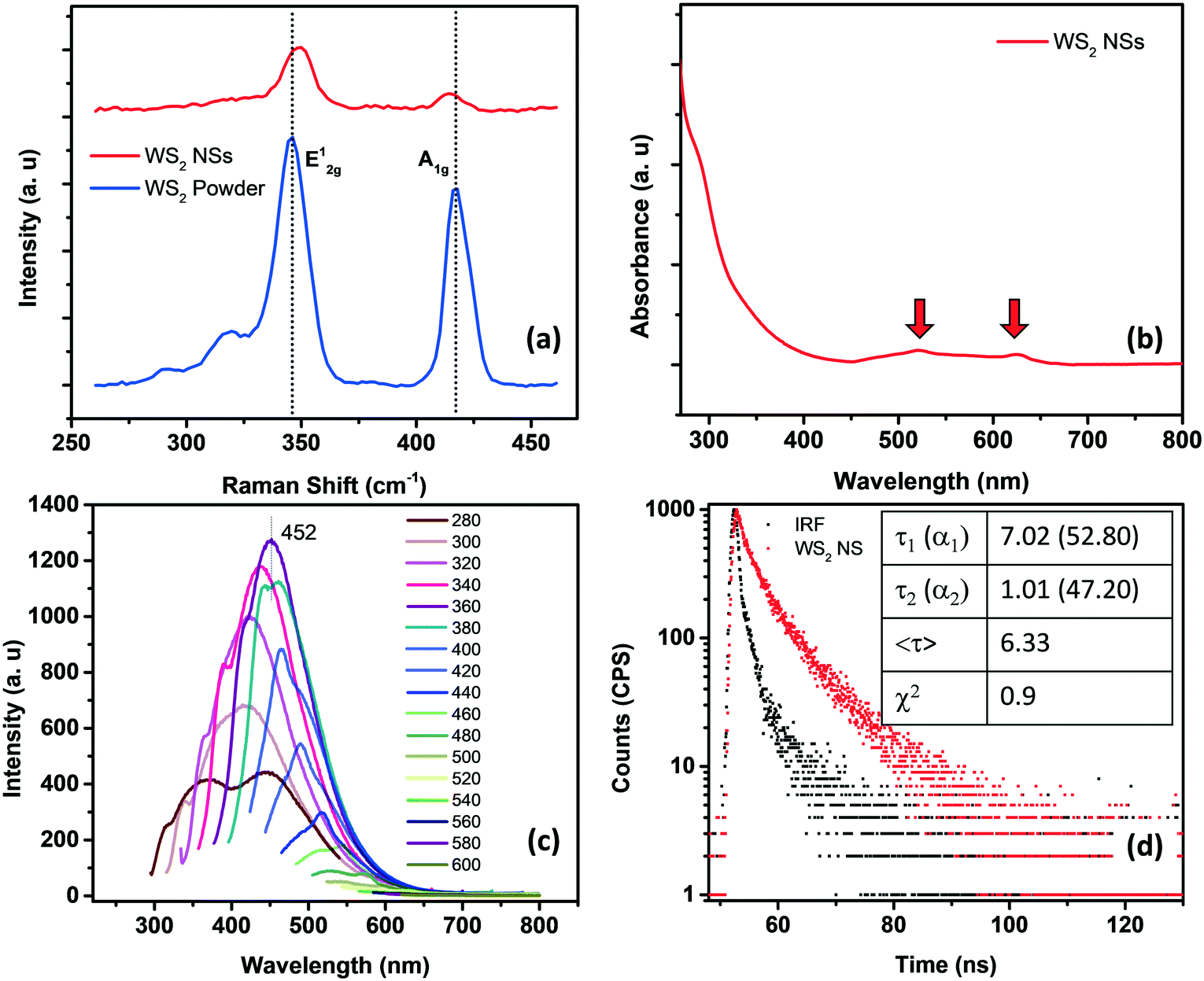 | ||
| Fig. 2 (a) Raman spectra of WS2 powder (blue) and WS2 nanosheets (red). (b) The UV-visible absorption spectrum of WS2 nanosheets, showing the signature peaks of the sheets corresponding to A and B excitons. (c) Excitation dependent emission spectra of WS2 NSs. (d) Lifetime spectrum of WS2 NSs fitted with a bi-exponential function. | ||
The crystalline structure and phase of WS2 NSs were investigated by X-ray diffraction (XRD), using WS2 powder as a reference (Fig. S2, ESI†). The diffraction peaks of the powder sample were indexed using JCPDS Card No. 08-0237, corresponding to the pure hexagonal (P63/mmc space group) WS2 phase. The (002) peak indicates the neat stacking of layers in the powder sample, along with the peaks corresponding to the (004), (100), (101), (102), (103), (006), (105), (106), (110), (008), (112), (107), (114) and (116) planes.29,30 It can be observed that all the diffraction peaks of WS2 NSs can be indexed to the hexagonal 2H phase of bulk WS2, demonstrating the well resolved crystalline structure of WS2 NSs exfoliated under the hydrothermal conditions. The absence of any additional peak indicates neither any noticeable impurity nor any phase change to the 1T phase during the synthesis. Besides, the weak signal from the (002) plane, compared to the WS2 powder, denotes the few-layer structure of WS2 NSs.31 The chemical state and elemental composition of WS2 NSs were examined by X-ray photoelectron spectroscopy (XPS) analysis (Fig. S3, ESI†). The high-resolution core-level XPS peaks corresponding to W4+ 4f7/2 and W4+ 4f5/2 were observed at binding energy values of 33.3 and 35.5 eV, respectively, and a small peak at 39.3 eV for W 5p3/2 indicates the dominance of the (IV) oxidation state for W in WS2 NSs.31–33 The absence of peaks corresponding to the 1T phase (in the range of 31.6–32.2 eV)34 indicates no phase transformation during the hydrothermal synthesis, corroborating the observation in the XRD spectrum. The peaks at 162.0 and 163.2 eV correspond to the S 2p3/2 and 2p1/2 orbitals of divalent S, bonded to W4+. The peaks corresponding to the oxidized form of S are present at binding energy values of 168.0 and 169.2 eV, corresponding to S 2p3/2 and 2p1/2, respectively. We surmise that, during the hydrothermal reaction, the edge S may be oxidized, leading to the formation of S and O containing functionalities. These functional groups describe the substantial aqueous solubility and phenomenal temporal stability. A single peak in the high-resolution XPS spectrum of oxygen at 531.6 eV indicates the S–O bonds (Fig. S3c, ESI†). The absence of a W6+ oxidation state peak and multiple peaks for oxygen in the XPS spectrum depicts that W did not undergo oxidation during the synthesis.
The optical signatures of WS2 NSs have been evaluated using UV-visible and fluorescence spectroscopy. In general, WS2 powder possesses four major absorption bands at around 630, 528, 456 and 417 nm, which are attributed to A, B, C and D excitons, respectively.35 The transitions from the spin-splitting valence band to the conduction band at the K point of the Brillouin zone are responsible for the emergence of peaks A and B. In contrast, the optical transitions from the deep valence band to the conduction band are denoted as C and D peaks.36 As the lateral size of the sheets decreases due to enhanced quantum confinement effects and edge effects, these excitonic positions can be blue-shifted.37 It is known that a continuum of absorption in the excitonic position confirms the phase change of a semiconducting 2H form to a metallic 1T form.38 The A and B excitons present at 625 and 520 nm, respectively, for WS2 NSs confirm the retention of the semiconducting nature of the bulk powder after the hydrothermal reaction in the presence of alkali (Fig. 2b). The quantum confinement effects due to the reduction in lateral dimensions of the sheets to the nano regime are manifested as high PL (see the fluorescence spectra given in Fig. 2c). A typical excitation dependent emission was observed for WS2 NSs, with the best emission centered at 452 nm at an excitation wavelength of 360 nm. The size heterogeneity of the sheets ranging from a few nanometers to micrometers can be one of the reasons behind this behavior. The quantum yield (QY) of the sample calculated using quinine sulphate as a standard has a value of 2.2%, which is comparable with the reported QY value of WS2 nanosheets.37,39 Also, the material shows exceptional temporal stability as well as photostability. This remarkable PL from the nanosheets and its inherent stability open the opportunity to exploit these materials in the field of fluorescence-based chemical sensors. Time-resolved fluorescence spectroscopic analysis of WS2 NSs was performed to unveil the excited state phenomenon occurring within the fluorophore material. The WS2 NSs are reported to have a very short lifetime of 800 ps.40 By shrinking the thickness of the bulk materials, the photo-excited carrier lifetime is generally increased due to the enhanced stability of the excitons. In the present case, the decay curves for WS2 NSs were collected and a bi-exponential fitting was obtained with carrier lifetimes of 2.75 and 9.07 ns by keeping the numerical fitting parameter χ2 near 1 (Fig. 2d).
3.2 Fabrication of the WS2 NS-Ag2O nanoparticle composite
One of the remarkable properties of the present WS2 NSs is their stable PL for several months under laboratory conditions. It was observed that the PL of the nanosheets was quenched instantaneously by the addition of dil. AgNO3. Upon incremental addition of AgNO3 solution (100 μL each of 0.02 M) to a solution of 2.5 mL of WS2 nanosheets, at room temperature, the PL emission is found to plummet accordingly with a change in color of the solution from very light yellow to deep yellow (Fig. 3). The color change, as well as the change in PL, was rapid. This color change was also reflected in the UV-vis spectrum. It has been observed that along with the addition of AgNO3, a new absorption peak below 400 nm emerged (Fig. 3b). The appearance of the yellow color and the new peak at ∼400 nm prompted us to surmise that the in situ formation of nanoparticles, having surface plasmon resonance (SPR) properties, might have taken place at the surface of WS2 NSs.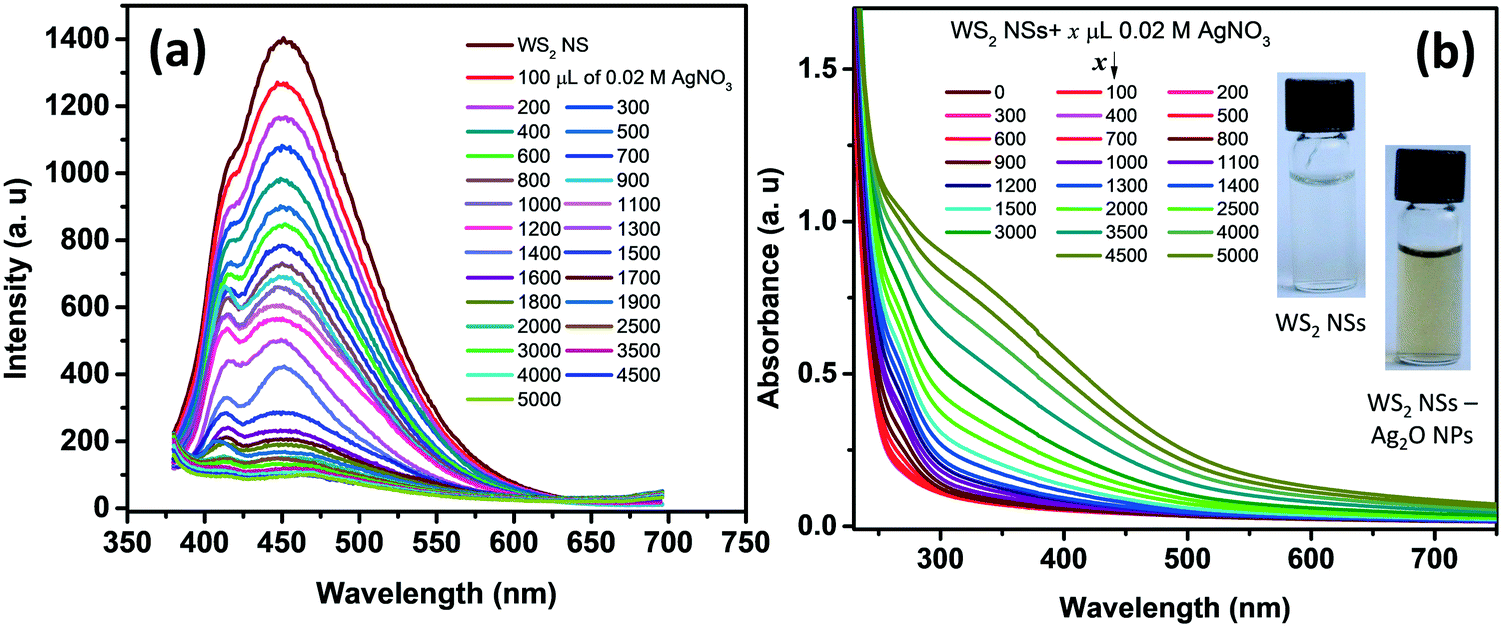 | ||
| Fig. 3 (a) The PL emission spectra of WS2 NSs after adding various concentrations of the AgNO3 solution. (b) Absorption spectra of WS2 nanosheets after adding various concentrations of the AgNO3 solution. The inset of (b) shows the photographs of the WS2 NS solution and WS2 NS–Ag2O NP nanocomposite under visible light. | ||
The formation of silver oxide nanoparticles (Ag2O NPs) from AgNO3 in the presence of NaOH at elevated temperatures is well documented.41,42 Our WS2 NS solution contains NaOH, which was left over from the hydrothermal reactions that anticipated the formation of silver oxide nanoparticles. X-ray diffraction studies unveil the nature of the nanoparticles, as the peak positions for silver nanoparticles (which usually form a face centred cubic lattice) will be different from silver oxide nanoparticles (which form cubic crystals upon reaction between AgNO3 and NaOH). The XRD pattern of the WS2 NS–NP nanocomposite shows remarkable peaks at 2θ values of 27.57°, 31.86°, 38.18°, 46.98°, 54.88°, 64.47° and 69.46°, apart from the peaks corresponding to WS2 NSs (Fig. S4, ESI†). These peaks were indexed to the (110), (111), (200), (211), (220), (311) and (222) crystal planes of the Ag2O NPs, having a cubic phase of silver oxide [JCPDS no. 41-1104].43,44 Therefore, we surmise the formation of Ag2O NPs. We further carried out XPS analysis to confirm the result obtained from the XRD analysis. The high-resolution XPS spectrum corresponding to Ag 3d shows peaks at 367.5 and 373.5 eV, which can be assigned to 3d5/2 and 3d3/2, respectively, of Ag+ ions in the crystal lattice of Ag2O (Fig. S5a, ESI†).43,44 The formation of Ag–O was further confirmed from the XPS spectrum of oxygen 1s. We can observe the emergence of a second peak in the lower energy range of the deconvoluted spectrum of oxygen, corresponding to the Ag–O bond apart from the peak corresponding to the O–S bond (Fig. S5b, ESI†).
Further, TEM analysis confirms the formation of spherical nanoparticles on the surface of WS2 NSs, forming a typical 0D–2D nanocomposite composed of Ag2O NPs on the surface of WS2 NSs (Fig. 4). Optimization of the concentration of WS2 to AgNO3 was carried out to obtain the homogeneous size distribution of Ag2O NPs. From TEM, it was evident that, for a volume ratio of 2.5 mL of WS2 solution, a uniform size distribution of NPs could be seen for 3 mL of 0.02 M AgNO3, and this nanocomposite material is used for further studies. The size distribution of these NPs at various volume ratios along with average diameters is given in Fig. S6.†
 | ||
| Fig. 4 TEM images and the SAED patterns of WS2 NSs solution after adding different volumes of AgNO3 solution. The upper pane shows the TEM images and the lower pane shows the SAED patterns. The volume ratio is written in each image. | ||
One of the significant observations is the quenching of the PL of WS2 nanosheets upon the formation of the WS2–Ag2O NP composite. The general mechanism of quenching involves electron transfer and energy transfer. The spectral overlap of donor emission and acceptor absorption is one of the prerequisites for a celebrated excited-state quenching mechanism, Förster resonance energy transfer (FRET), to materialize. As shown in Fig. 5, there is a remarkable spectral overlap of the absorption of the nanocomposite with the emission of WS2 NSs. Since the Ag2O NPs are dispersed on the surface of the nanosheets, the proximity of donor and acceptor species, the second criterion to realize FRET, is satisfied. Therefore, we ascribe the PL quenching to the FRET mechanism because of the presence of Ag2O NPs with SPR absorption at ∼400 nm.
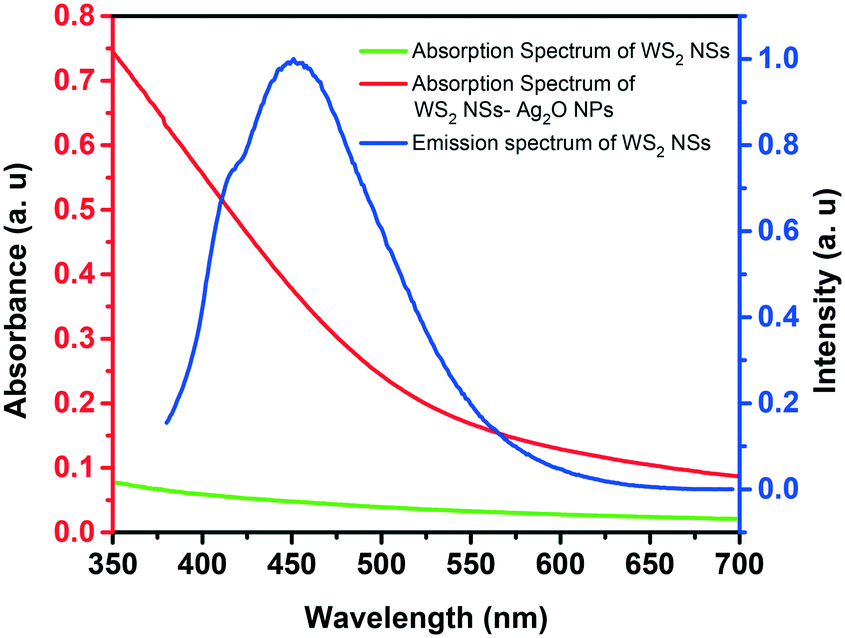 | ||
| Fig. 5 Spectral overlap of the absorption spectrum of the WS2 NS–Ag2O NP composite (red trace) (by the addition of 3.5 mL AgNO3) and the emission spectrum of WS2 nanosheets (blue trace). The absorption spectrum of WS2 NSs alone (green) is given for comparison. | ||
3.3 The nanocomposite as a turn-on sensor for basic amino acids
The quenched fluorescence of WS2 NSs could be recovered by annihilating the nanosheet–nanoparticle dyad, which opens the possibility of creating a turn-on sensor. We have seen that the quenching occurs in the presence of Ag2O NPs. Disruption of these Ag2O NPs can shut the energy transfer pathways, and hence, the PL of WS2 NSs can be recovered. The stability of any colloid is due to the presence of charges on its surface, and in the present case, it can be inferred by zeta potential measurements (Fig. S7, ESI†). Any moiety with a counter charge can destroy the colloidal system and lead to nanoparticle aggregation. Such aggregation of Ag2O NPs leads to the disappearance of SPR peaks, which is responsible for FRET. The biocompatibility and notable aqueous solubility of Ag2O NPs and WS2 NSs prompted us to choose any biologically relevant molecules as analytes. Interestingly, the quenched PL was found to recover with the addition of basic amino acids at a pH value lower than their isoelectric points. We now formulated the WS2 NS–Ag2O NP composite material as a turn-on sensor to detect the three basic amino acids His, Lys and Arg.3.4 Sensor action
Amino acids, though zwitterionic, tend to be positively charged at a pH below their isoelectric points (pI).45 The charges on the surface of AAs can be varied accordingly by changing the pH. The pI values of His, Lys and Arg are 7.59, 9.74 and 10.76, respectively. Hence at a pH of 7, all three AAs will be positively charged, and at pH 9, Lys and Arg will be positively charged, whereas, at pH 10, only Arg will be positively charged. The PL response of the composite towards the three AAs at pH 7 is given in Fig. 6 and S8, ESI.†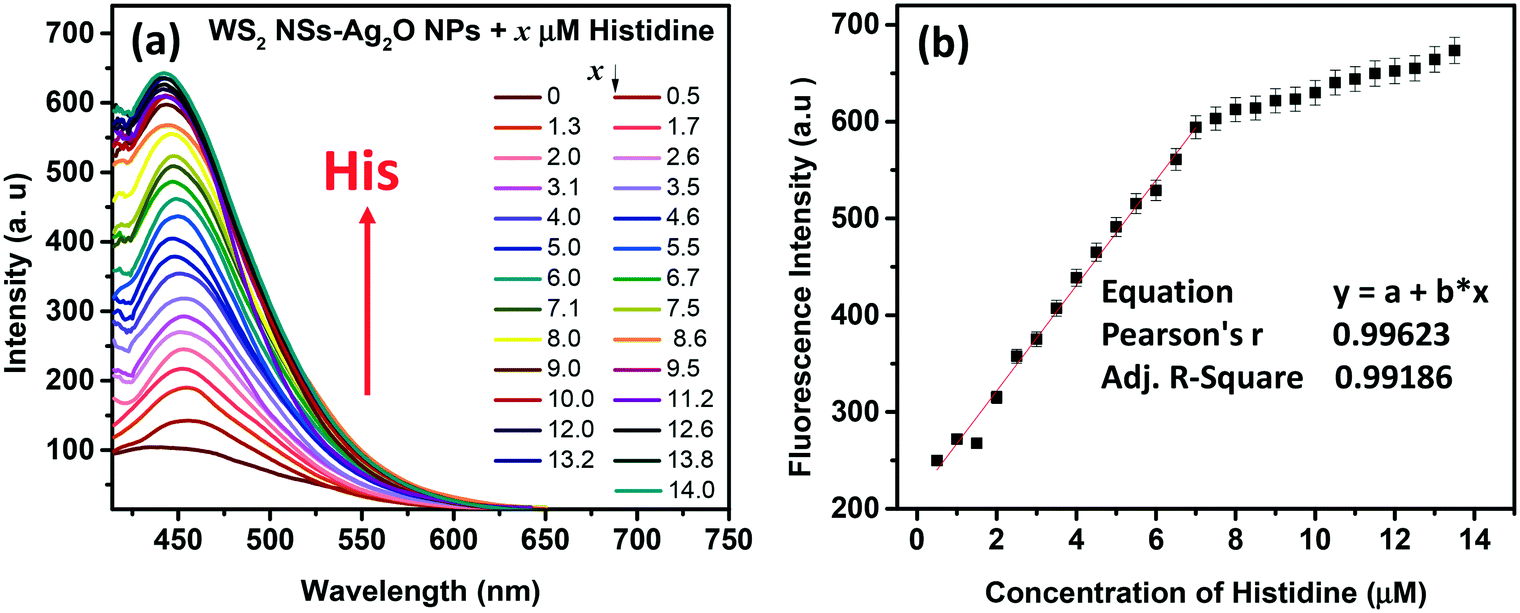 | ||
| Fig. 6 (a) Response of His towards fluorescence emission of the WS2 NS–Ag2O NP nanocomposite at pH 7. The concentration of His in micromolar is given in the figure legend. The excitation wavelength is 360 nm. (b) The calibration curve shows the dynamic range of His. The linear region is considered as the dynamic range of the sensor towards His detection. | ||
It was noted that the PL responses towards these three AAs (His, Lys and Arg) were distinct from each other. We conducted the experiments at three different pH values: at pH 7, 9 and 10. As evident from Fig. 7, at pH 7, all three AAs could turn-on the PL, whereas the recovery effect is negligible for other AAs and potential interfering biomolecules. Thus, the composite is selective towards the detection of all the basic AAs at pH 7. The selectivity is further evaluated by the selectivity coefficient (Karg = Sarg/S0; where Sarg is the response of the sensor towards Arg and S0 is the response of the sensor to other molecules/ions)46,47 using Arg as the standard at different pH values, and is given in Table S1.† The AA structure with a net positive charge below its isoelectric point provides us room for distinction between the basic AAs (Fig. S9, ESI†). For example, by changing pH 7 to pH 9 or 10, the PL response of the nanocomposite has changed. At pH 9 and 10, His cannot turn on the PL, whereas both Lys and Arg could still turn-on the emission of the nanocomposite. And at pH 10, Lys also fails to recover the PL of the composite; only Arg can turn-on the emission. Thus, screening within the basic AAs can be accomplished by the 0D–2D nanocomposite sensor solution, merely by altering the pH of the sensor solution. The fluorescence response of the sensor solution towards Lys and Arg at pH 9 and 10, respectively, is given in Fig. S10, ESI.† The dynamic range of His (at pH 7), Lys (at pH 7 and 9) and Arg (at pH 7, 9 and 10) is summarized in Table S2.† The photographs of the WS2 NSs, WS2 NS–Ag2O NP nanocomposite, and nanocomposite solution containing His, Lys and Arg under visible and UV light show the possibility of a visual screening (Fig. S11, ESI†). The zeta potential values of the WS2 NS–Ag2O NP nanocomposite at various pH values are given in Table S3,† exhibiting its stability at various pH values.
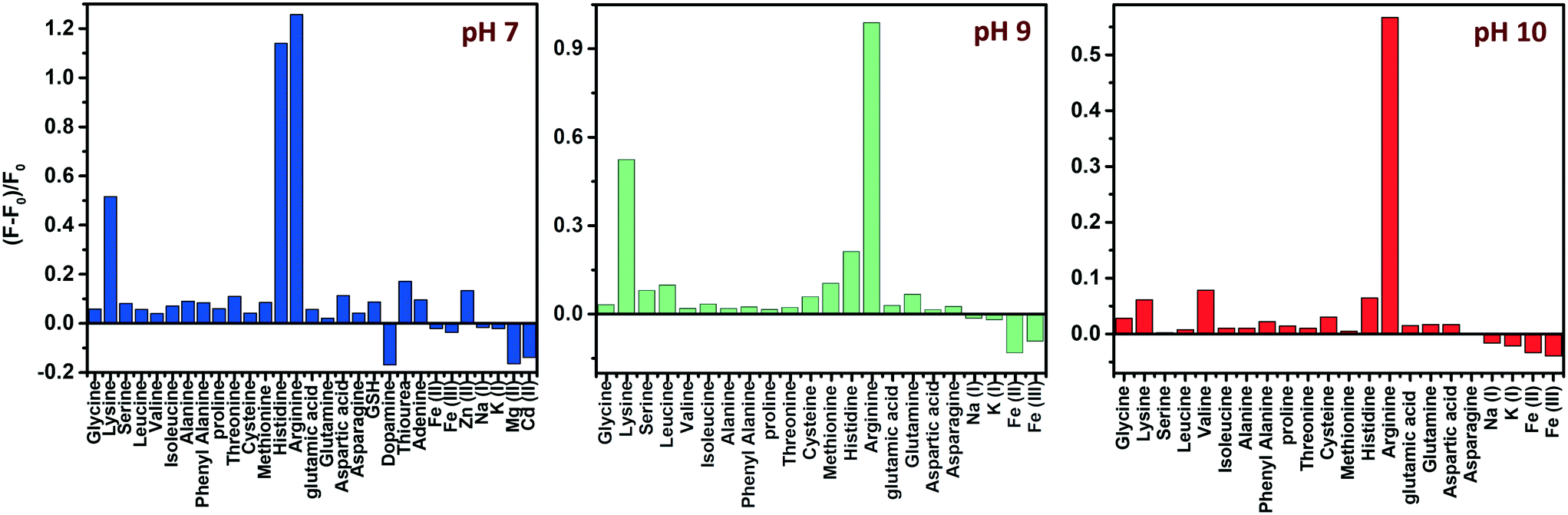 | ||
| Fig. 7 pH-dependent selectivity: effect of different AAs, other biomolecules and metal ions on the fluorescence intensity of the WS2 NS–Ag2O NP nanocomposite at three different pH values. | ||
3.5 Mechanism of formation of Ag2O NPs and fluorescence turn-on sensing
AgNO3 in the presence of NaOH is known to form Ag2O according to the equation given below.41 Thus we propose that in the presence of NaOH, which was used for the hydrothermal reaction, Ag2O particles might have formed.| AgNO3 + NaOH → AgOH + NaNO3 |
| 2AgOH → Ag2O + H2O |
The survey scan XPS spectra of WS2 NSs and the WS2 NS–Ag2O NP composite show peaks corresponding to W, S, and O, along with the peaks of Ag in the case of the composite (Fig. 8a and b). The deconvoluted spectra of W of the WS2 NS–Ag2O NP composite show peaks corresponding to W 4f7/2, 4f5/2 and W 5p3/2 at slightly lower binding energy values (33.23, 35.33 and 39.13 eV, respectively) comparable to WS2 NSs, due to the weaker W–S bond (Fig. 8c). These had binding energy values of 33.3, 35.5 and 39.3 eV for the case of WS2 NSs (see Fig. S3, ESI† and the discussion in section 3.1). The high-resolution XPS peak corresponding to S indicates the chemical reaction possibilities at the nanosheet surface. In both cases, viz. NSs alone and the 0D–2D composite, the XPS peak split to W–S and S–O bonds (Fig. 8d and S3†). The peaks assigned to the metal–S bond (162.0 and 163.2 eV) for the case of WS2 NSs have shifted to 160.7 eV and 161.9 eV in the composite material. This peak shift is attributed to the contribution from the Ag–S bond and subsequent peak broadening.
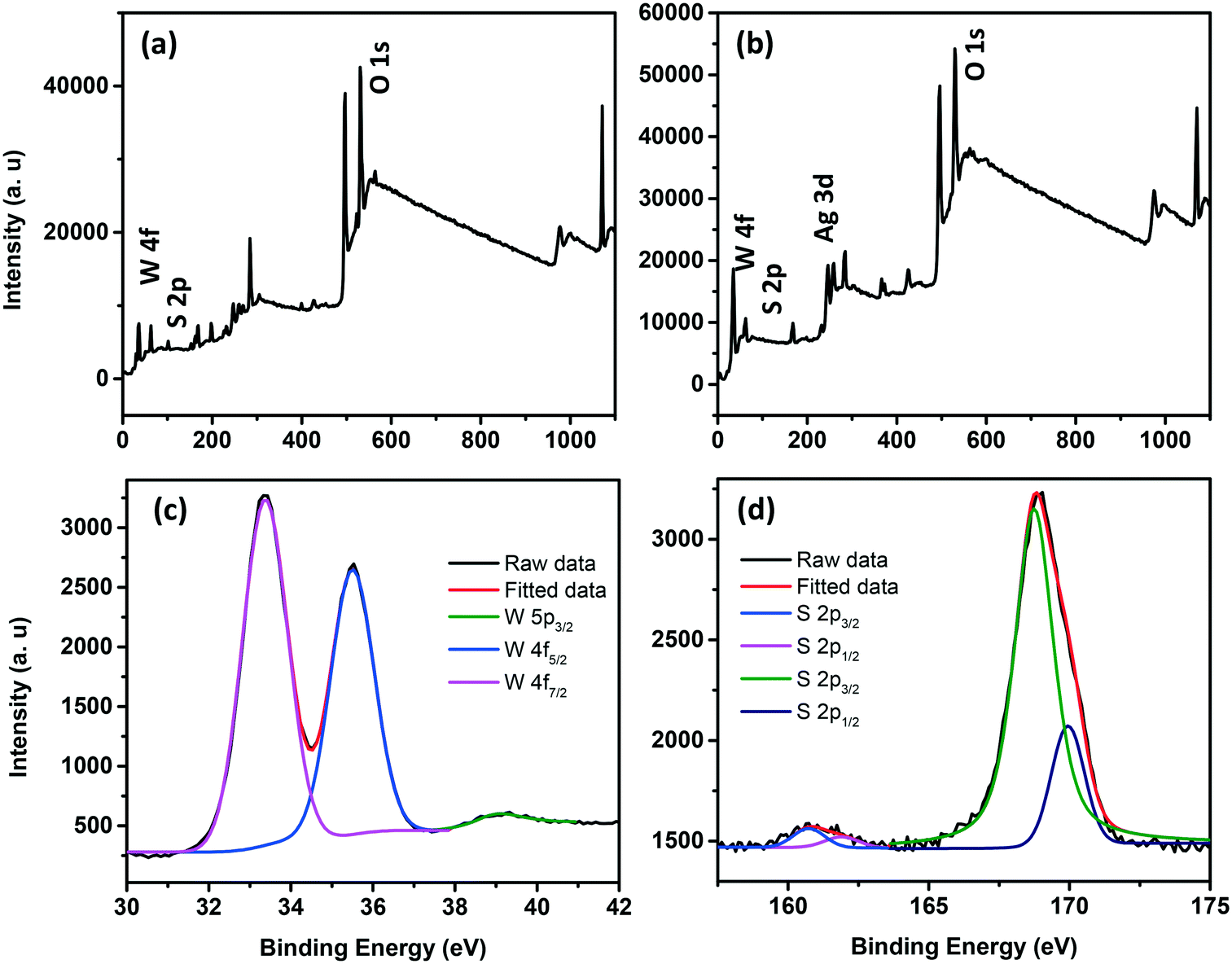 | ||
| Fig. 8 Survey scan XPS spectra of WS2 NSs (a) and the WS2 NS–Ag2O NP nanocomposite (b). High resolution XPS spectra of the W (c) and S (d) regions of the WS2 NS–Ag2O NP nanocomposite. | ||
The crucial part of the turn-on sensing mechanism, on the other hand, was mainly determined by the protonated form of the basic AAs below their pI. In the presence of AAs, the negative charges on the surface of Ag2O NPs are getting nullified, leading to the aggregation of Ag2O NPs and hence the reduction of the SPR peak intensity. This decrease in SPR absorption leads to the shutting off of the FRET pathway and the PL is subsequently recovered. The TEM micrographs, UV-visible absorption studies and lifetime analysis corroborate with these assumptions. As shown in the TEM images given in Fig. 9 and S12, ESI,† His, Lys and Arg induce the aggregation of Ag2O NPs at pH 7. At pH 9, the intact Ag2O NPs are visible in the nanocomposite samples after adding His. At pH 10, addition of His or Lys does not lead to aggregation. This observation further authenticates the involvement of aggregation for the PL recovery (Fig. S13 and S14, ESI†). The absence of SPR peaks in the in the UV-visible absorption spectra (Fig. S15, ESI†) validates the argument.
 | ||
| Fig. 9 TEM images of the WS2 NS–Ag2O NP nanocomposite upon interaction with His, Lys and Arg at pH 7. | ||
Another vital observation of the interaction of AAs with the nanocomposite is the precipitation of Ag2O NPs upon the addition of excess AAs (concentration well above the dynamic range). The yellow color of the solution disappeared with the precipitation of Ag2O as a brown precipitate. The TEM analysis of this colorless supernatant solution shows that Ag2O NPs have been completely precipitated along with some WS2 NSs and the solution contains only the remaining WS2 NSs (Fig. S16, ESI†). The crystallinity and the neat morphology of the sheets were lost after the precipitation of Ag2O NPs. We surmise that the locations with inscribed markings (in Fig. S16, ESI†) are the spots where the Ag2O NPs adsorbed onto the nanosheets. The Ag2O NPs are formed onto the surface of WS2 NSs and may be stabilized by the groups containing S and O. Also, the dangling S atoms of the nanosheets take part in the reaction, eventually forming compounds like Ag2S as well. When the NPs detach from the sheets by aggregation followed by precipitation, the remaining sheets contain several etched areas due to the removal of adsorbed Ag2O NPs along with dangling S groups (as Ag2S). The etched regions are primarily defects due to S or its functional group vacancies. High-resolution XPS of the supernatant solution shows weak S peaks, which are proof of the removal of S from the sensor material by precipitation of nanoparticles. The oxygen region of the XPS spectrum, nonetheless, shows a single peak at 531.5 eV, as in the case of WS2 NSs, which invariably shows the S–O bond. The high-resolution XPS analysis of W indicates no significant peak shift from the nanocomposite values (33.22, 35.32, and 39.02 eV), implying that the oxidation state of W was not affected (Fig. S17, ESI†). The absence of peaks corresponding to W–O and the W6+ state indicate that the dangling bonds created by the removal of S was not satisfied by oxygen. Notably, a full recovery of PL was not attained even after the addition of AAs well above the dynamic range. This could be attributed to the defects created during the removal of Ag2O NPs by the precipitation, which can act as seats for nonradiative transfers (Fig. S18, ESI†).
The excited-state lifetime analysis depicts the complexity of the interactions between the AAs and the nanocomposite (Fig. S19, ESI†). It has been observed that, upon the addition of AgNO3 into WS2 NSs, one of the lifetime components becomes nonradiative with the highest contribution (0.13 ns and 80.38% contribution). We attribute this shift to the energy transfer from WS2 NSs to Ag2O NPs formed on their surface. The absence of FRET upon the addition of AAs is substantiated by the recovery of their lifetime values near that of bare/free WS2 NSs. The emergence of a third lifetime component with a nonradiative pathway for the solutions with His, Lys and Arg might have arisen from the defect created in the sheets during the Ag2O NP formation.
We were interested in knowing whether the aggregation of Ag2O nanoparticles (in the WS2 NS–Ag2O NP nanocomposite) by basic AAs can be modulated by changing the pH and the fluorescence can be quenched reversibly or not. Interestingly, we observed that the increase in the pH of the sensor solution from 7 to 9 for the analyte His led to the fluorescence quenching. This observation is attributed to the separation of aggregated particles due to increased pH (above the isoelectric value of His). We examined the resultant solution using the UV-visible absorption spectra and TEM (see Fig. S20†). After the pH increase, the WS2 NS–Ag2O NP composite shows the presence of a SPR peak at 360 nm, depicting separated Ag2O NPs. The re-appearance of colour was also observed (see the photographs given in Fig. S20†). The same experiment was done for the analyte Lys at pH 7 and 9 and then the pH was increased to 10. Similar results were obtained. These experiments show that the sensor action is reversible and can be modulated by changing the pH.
3.6 Protein sensing and real sample detection
Protein-specific detection is of fundamental importance for diagnosing and detecting protein biomarkers and deciphering biological processes at cellular levels. Most fluorescent probes work under the principle of changes in their photophysical characteristics upon interaction with the protein through chemical reactions or mere physical interactions. Yet, the fluorescence labelling and subsequent detection are invariably laborious, and the development of a mix-and-detect strategy that employs turn-on-based fluorescence detection will be highly appealing.To test our fluorescent probe for protein detection, ubiquitin, a regulatory protein found in eukaryotes tissues, was selected as a target protein, as it contains seven lysine residues in its structure. Two significant functions of ubiquitin are marking proteins to be eliminated from the system and modulating the functions of the substrate protein with which it can conjugate without metabolically destabilizing this acceptor protein.48 The abnormal concentration of ubiquitin is associated with many physiological aberrations. For example, high ubiquitin levels can be observed in the cerebrospinal fluid of Alzheimer's disease patients.49 Therefore, facile detection and precise quantification of ubiquitin are of utmost importance.
We demonstrated a label-free detection of ubiquitin using the present fluorescent probe, the WS2 NS–Ag2O NP composite. The PL intensity is found to be enhanced (6.5 fold) with the ubiquitin concentration from 0 to 61 μg mL−1 linearly (R2 = 0.99) at room temperature without any waiting period (Fig. S21, ESI†). From the UV-vis absorption, the SPR peak of Ag NPs was decreased, suggesting a similar mechanism as we have observed in the case of basic AAs (Fig. S22, ESI†). The lifetime analysis further corroborates these results. As is evident from Fig. S23, ESI,† the lifetime components of WS2 NSs were regenerated in the presence of ubiquitin with similar contributions [9.1 ns (54.4) and 2.8 ns (45.6) for WS2 NSs and 8.5 ns (56.4) and 2.3 ns (43.6) for the WS2 NS–Ag2O NP nanocomposite with ubiquitin]. The zeta potential values of the WS2 NS–Ag2O NP nanocomposite with ubiquitin are also commensurate with the value of WS2 NSs (Table S3, ESI†). The excitation spectrum (Fig. S24, ESI†) of WS2 NSs at an emission position of 450 nm shows two peaks at ca. 265 and 360 nm, referring to two electronic states available for the system. The WS2 NS–Ag2O NP nanocomposite, on the other hand, has only one major peak at around 310 nm. The disappearance of the longer wavelength peak is attributed to the effective energy transfer due to changes in surface states. However, the re-appearance of the longer wavelength excitation peak (∼360 nm) in the case of the WS2 NS–Ag2O NP nanocomposite with ubiquitin at 360 nm verifies the shutting off of FRET between WS2 NSs and Ag2O NPs in the presence of protein and the regeneration of the fluorescence intensity. The mass spectra of pure protein and protein in the presence of the WS2 NS–Ag2O NP composite are given in Fig. S25a and b, ESI.† The characteristic protein peaks are evident in the mass spectrum of pure protein. In contrast, these peaks are absent in the case of the analyte-containing sensor solution (ubiquitin + WS2 NS–Ag2O NP solution) (see Fig. S25a, ESI†). This implies that the protein was bound to the Ag2O NPs and did not appear in the mass spectrum.
Evaluation of the WS2 NS–Ag2O NP nanocomposite utility as a sensor for basic AAs in real sample detection was also performed using blood and urine samples. Detection and quantification of Lys are significant, as they give a straightforward diagnosis of various disorders and diseases with the presence of Lys in urine and plasma.50 We chose Lys as our analyte for real sample analysis. The samples were prepared by spiking known concentrations of Lys and the fluorescence spectra were recorded after addition to the WS2 NS–Ag2O NP nanocomposite at room temperature in buffer solutions. The results obtained are shown in Table 1. The recovery and relative standard deviation of the measurements (% RSD) were evaluated to check the reliability of the present sensor. For both the blood sample and the urine sample, excellent recovery within 2% RSD was obtained, which is superior to those of already reported methods.51–53 The result indicates the reliability of the sensor for analytical applications.
| Sample | Spiked (μM) | Observed (mean; μM) | Recovery % | % RSD |
|---|---|---|---|---|
| Blood sample | 1.5 | 1.5188 ± 0.044 | 101.25 | 1.67 |
| 2 | 2.0038 ± 0.027 | 100.19 | 0.81 | |
| 2.5 | 2.5037 ± 0.019 | 100.15 | 0.97 | |
| Urine sample | 1.75 | 1.7716 ± 0.072 | 101.24 | 1.75 |
| 2 | 2.0031 ± 0.093 | 100.16 | 2.03 | |
| 2.25 | 2.2772 ± 0.108 | 101.21 | 2.14 | |
| 2.5 | 2.5232 ± 0.092 | 100.93 | 1.66 |
4. Conclusion
In the present study, we demonstrated a luminescent 2D material, exfoliated WS2 nanosheets, for sensing biologically relevant molecules. Highly fluorescent and stable WS2 nanosheets were synthesized using a facile hydrothermal synthetic route. Interestingly, Ag2O NPs were formed on the WS2 nanosheets instantaneously by the addition of the AgNO3 solution. The formation of Ag2O NPs leads to the reduction of PL from WS2 NSs via FRET. We investigated the effect of positively charged basic nanoparticles on the PL of the WS2 NS–Ag2O NP composite and fabricated a turn-on sensor for three basic amino acids viz. His, Lys and Arg. The easy, straightforward synthetic method for the formation of stable and highly fluorescent WS2 NSs without using any hazardous solvents, the in situ formation of stable Ag2O NPs without an extra stabilizing agent, and devising a simple aggregation strategy to shut the FRET pathway, thereby ensuring a turn-on detection and screening of all the basic amino acids, are the highlights of the present sensor system. The practical application of the sensor was demonstrated for the detection of protein: ubiquitin and Lys from real samples – blood and urine.Live subject statement
All experiments were performed in accordance with the guidelines “National ethical guidelines for biomedical and health research involving human participants, Indian Council for Medical Research (ICMR), Government of India”, and approved by the ethics committee at “Institutional Ethics Committee (IEC), Indian Institute of Space Science and Technology, Thiruvananthapuram”. Informed consents were obtained from the human participants of this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge IIST Thiruvananthapuram for funding and the PSG Institute of Advanced Studies, Coimbatore and The National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram for analysis.References
- X. Gu, B. Li and R. Yang, J. Appl. Phys., 2016, 119, 085106 CrossRef.
- M.-W. Lin, I. I. Kravchenko, J. Fowlkes, X. Li, A. A. Puretzky, C. M. Rouleau, D. B. Geohegan and K. Xiao, Nanotechnology, 2016, 27, 165203 CrossRef PubMed.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- P. Neema, A. M. Tomy and J. Cyriac, TrAC, Trends Anal. Chem., 2020, 124, 115797 CrossRef CAS.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, ACS Nano, 2014, 8, 1102–1120 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- Y. Zhou and J. Yoon, Chem. Soc. Rev., 2012, 41, 52–67 RSC.
- S. S. Sharma and K.-J. Dietz, J. Exp. Bot., 2006, 57, 711–726 CrossRef CAS PubMed.
- H. Yoshida, Y. Nakano, K. Koiso, H. Nohta, J. Ishida and M. Yamaguchi, Anal. Sci., 2001, 17, 107–112 CrossRef CAS PubMed.
- D. Xiong, M. Chen and H. Li, Chem. Commun., 2008, 880–882 RSC.
- T. Liu, N. Li, J. X. Dong, Y. Zhang, Y. Z. Fan, S. M. Lin, H. Q. Luo and N. B. Li, Biosens. Bioelectron., 2017, 87, 772–778 CrossRef CAS PubMed.
- X. Lu, W. Wang, Q. Dong, X. Bao, X. Lin, W. Zhang, X. Dong and W. Zhao, Chem. Commun., 2015, 51, 1498–1501 RSC.
- S. T. R. Naqvi, T. Rasheed, M. N. Ashiq, M. N. ul Haq, S. Majeed, B. Fatima, R. Nawaz, D. Hussain and S. Shafi, Polyhedron, 2020, 114426 CrossRef.
- H. Liu, J. Shao, L. Shi, W. Ke, F. Zheng and Y. Zhao, Sens. Actuators, B, 2020, 304, 127333 CrossRef CAS.
- E. V. Suprun, E. V. Karpova, S. P. Radko and A. A. Karyakin, Electrochim. Acta, 2020, 331, 135289 CrossRef CAS.
- M. Hasanzadeh, A. Karimzadeh, N. Shadjou, A. Mokhtarzadeh, L. Bageri, S. Sadeghi and S. Mahboob, Mater. Sci. Eng., C, 2016, 68, 814–830 CrossRef CAS PubMed.
- J. L. da Silva, M. A. Beluomini, G. C. Sedenho and N. R. Stradiotto, Microchem. J., 2017, 134, 374–382 CrossRef.
- B. R. Kolanu, V. Boddula, S. Vadakedath and V. Kandi, Cureus, 2017, 9(3), e1091 Search PubMed.
- G. Patel and S. Menon, Chem. Commun., 2009, 3563–3565 RSC.
- Y. Sun, L. Zhang and H. Li, New J. Chem., 2012, 36, 1442–1444 RSC.
- T. Minami, N. A. Esipenko, B. Zhang, L. Isaacs and P. Anzenbacher, Chem. Commun., 2014, 50, 61–63 RSC.
- H. Zeng, G.-B. Liu, J. Dai, Y. Yan, B. Zhu, R. He, L. Xie, S. Xu, X. Chen and W. Yao, Sci. Rep., 2013, 3, 1–5 Search PubMed.
- X. Zhang, X.-F. Qiao, W. Shi, J.-B. Wu, D.-S. Jiang and P.-H. Tan, Chem. Soc. Rev., 2015, 44, 2757–2785 RSC.
- Y. Zhao, X. Luo, H. Li, J. Zhang, P. T. Araujo, C. K. Gan, J. Wu, H. Zhang, S. Y. Quek and M. S. Dresselhaus, Nano Lett., 2013, 13, 1007–1015 CrossRef CAS PubMed.
- H. Li, J. Wu, Z. Yin and H. Zhang, Acc. Chem. Res., 2014, 47, 1067–1075 CrossRef CAS PubMed.
- A. Molina-Sanchez and L. Wirtz, Phys. Rev. B, 2011, 84, 155413 CrossRef.
- H. Zeng, G.-B. Liu, J. Dai, Y. Yan, B. Zhu, R. He, L. Xie, S. Xu, X. Chen and W. Yao, Sci. Rep., 2013, 3, 1608 CrossRef PubMed.
- Y. Cheng, J. Peng, B. Xu, H. Yang, Z. Luo, H. Xu, Z. Cai and J. Weng, IEEE Photonics J., 2016, 8, 1 Search PubMed.
- X. Zhang, W. Lei, X. Ye, C. Wang, B. Lin, H. Tang and C. Li, Mater. Lett., 2015, 159, 399–402 CrossRef CAS.
- G.-Q. Han, Y.-R. Liu, W.-H. Hu, B. Dong, X. Li, Y.-M. Chai, Y.-Q. Liu and C.-G. Liu, Mater. Chem. Phys., 2015, 167, 271–277 CrossRef CAS.
- K. Zhang, L. Fu, W. Zhang, H. Pan, Y. Sun, C. Ge, Y. Du and N. Tang, ACS Omega, 2018, 3, 12188–12194 CrossRef CAS PubMed.
- X. Mao, Y. Xu, Q. Xue, W. Wang and D. Gao, Nanoscale Res. Lett., 2013, 8, 430 CrossRef PubMed.
- S. X. Leong, C. C. Mayorga-Martinez, X. Chia, J. Luxa, Z. Sofer and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 26350–26356 CrossRef CAS PubMed.
- A. Bayat and E. Saievar-Iranizad, J. Lumin., 2017, 185, 236–240 CrossRef CAS.
- A. K. Mishra, K. V. Lakshmi and L. Huang, Sci. Rep., 2015, 5, 15718 CrossRef CAS PubMed.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P.-H. Tan and G. Eda, ACS Nano, 2013, 7, 791–797 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- B. Zhu, X. Chen and X. Cui, Sci. Rep., 2015, 5, 9218 CrossRef PubMed.
- L. Yuan and L. Huang, Nanoscale, 2015, 7, 7402–7408 RSC.
- K. T. Sullivan, C. Wu, N. W. Piekiel, K. Gaskell and M. R. Zachariah, Combust. Flame, 2013, 160, 438–446 CrossRef CAS.
- M. Padilla Villavicencio, A. Escobedo Morales, M. Ruiz Peralta, M. Sánchez-Cantú, L. Rojas Blanco, E. Chigo Anota, J. H. Camacho Garcia and F. Tzompantzi, Catal. Lett., 2020, 150, 2385–2399 CrossRef CAS.
- Z. Lin, Y. Lu and J. Huang, Cellulose, 2019, 26, 6683–6700 CrossRef CAS.
- L. Xu, B. Wei, W. Liu, H. Zhang, C. Su and J. Che, Nanoscale Res. Lett., 2013, 8, 1–7 CrossRef PubMed.
- T. Tsuru, T. Shutou, S.-I. Nakao and S. Kimura, Sep. Sci. Technol., 1994, 29, 971–984 CrossRef CAS.
- Z. Liu, T. Fan, D. Zhang, X. Gong and J. Xu, Sens. Actuators, B, 2009, 136, 499–509 CrossRef CAS.
- W. Lin, L. Long, L. Yuan, Z. Cao and J. Feng, Anal. Chim. Acta, 2009, 634, 262–266 CrossRef CAS PubMed.
- V. Chau, J. W. Tobias, A. Bachmair, D. Marriott, D. J. Ecker, D. K. Gonda and A. Varshavsky, Science, 1989, 243, 1576–1583 CrossRef CAS PubMed.
- C. Wloka, V. Van Meervelt, D. van Gelder, N. Danda, N. Jager, C. P. Williams and G. Maglia, ACS Nano, 2017, 11, 4387–4394 CrossRef CAS PubMed.
- W. Song, W. Duan, Y. Liu, Z. Ye, Y. Chen, H. Chen, S. Qi, J. Wu, D. Liu and L. Xiao, Anal. Chem., 2017, 89, 13626–13633 CrossRef CAS PubMed.
- V. D. O. Torres, R. C. Piva, W. F. A. Junior and C. A. L. Cardoso, Orbital: Electron. J. Chem., 2018, 10, 1–8 CAS.
- V. I. Chalova, I. B. Zabala-Díaz, C. L. Woodward and S. C. Ricke, World J. Microbiol. Biotechnol., 2008, 24, 353–359 CrossRef CAS.
- A. Hayat, T. M. Jahangir, M. Y. Khuhawar, M. Alamgir, A. J. Siddiqui and S. G. Musharraf, J. Cereal Sci., 2014, 60, 356–360 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sd00009a |
| This journal is © The Royal Society of Chemistry 2022 |