
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical methods for on-site multidrug detection at festivals†
Robin
Van Echelpoel‡
 ab,
Jonas
Schram‡
ab,
Marc
Parrilla
ab,
Devin
Daems
ab,
Amorn
Slosse
c,
Filip
Van Durme
c and
Karolien
De Wael
*ab
ab,
Jonas
Schram‡
ab,
Marc
Parrilla
ab,
Devin
Daems
ab,
Amorn
Slosse
c,
Filip
Van Durme
c and
Karolien
De Wael
*ab
aA-Sense Lab, Department of Bioscience Engineering, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium. E-mail: karolien.dewael@uantwerpen.be
bNANOlab Center of Excellence, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
cDrugs and Toxicology Department, National Institute for Criminalistics and Criminology (NICC), Vilvoordsesteenweg 100, 1120, Brussels, Belgium
First published on 1st June 2022
Abstract
Music festivals have emerged as an important setting for the consumption of illicit drugs, harming both consumers and society. Therefore, law enforcement present at these events requires straightforward, robust and accurate screening tools to obtain a rapid indication of the presence of these drugs in suspicious samples encountered on-site. Electrochemical profile (EP)-based drug sensing has proven to offer the desired affordability, portability and high-performance for this purpose. However, previous studies have mainly focused on the detection of only one drug type, rather than the simultaneous detection of multiple drugs. In this work, two innovative electrochemical methods (i.e. the flowchart and dual-sensor) towards the rapid and accurate detection of the four main illicit drugs encountered at festivals (cocaine, 3,4-methylenedioxymethamphetamine -MDMA-, amphetamine and ketamine) are developed and assessed based on their practicality, performance and limitations. The flowchart method employs sequential measurements in different measuring conditions, following a flowchart, combining good practicality, affordability and performance. The dual-sensor method combines the EP recorded in parallel at two electrodes with different measuring conditions into a superprofile. As the combined electrochemical information of the recorded EPs provides an increased selectivity, this method obtains the highest accuracy (87.5% vs. 80.0% for the flowchart) when applied to a set of confiscated samples. Interestingly, both methods outperform a portable Raman device (60%) that analyzed the same set of confiscated samples. Overall, these electrochemical methods offer law enforcement a rapid, portable and accurate screening method for the analysis of the large variety of suspicious samples encountered at music festivals.
Introduction
Music festivals have become an important setting for the use of illicit drugs. Studies have shown the link between music, nightlife and substance abuse, as many people attending music festivals consider this setting as an ideal place to experiment with and/or consume illicit drugs.1–3 However, the consumption of illicit drugs has an adverse health, social and economic impact on the user, while also negatively affecting society and the environment.4,5 Health-related risks associated with substance use at festivals include harmful side effects such as hyperthermia, seizures and multi-organ failure, which could even result in death.6 Additionally, the presence of other substances (e.g. adulterants, diluents, other illicit drugs) in drug samples may cause harm, while strong variations in purity between different countries increase the risk of overdoses.6 Moreover, concomitant consumption of alcohol or other drugs (polydrug abuse) occurs frequently in these settings and often exponentially increases the health risks.7–9The monitoring of drug use patterns at festivals is performed in various ways, including through surveys, samples seized by law enforcement and the analysis of pooled urine or wastewater.10–13 Depending on the type of festival and location, a wide variety of substances can be in circulation, going from traditionally popular drugs such as cannabis and cocaine to more exotic drug types such as synthetic cathinones and psychedelics.14,15 Furthermore, studies have shown the link between the music genre of festivals and drug use patterns.16–18 Electronic music festivals in particular have been extensively linked with higher drug consumption and particularly increased use of amphetamine-type stimulants (ATS) such as 3,4-methylenedioxymethamphetamine (MDMA).16,18
Law enforcement agencies (LEAs) are tasked with preventing illicit drugs from reaching the festival site by performing searches at the entrance.19,20 When a suspicious sample is encountered, LEAs require a fast on-site indication of the presence of illicit substances to decide on further actions. Typically used laboratory techniques such as gas chromatography coupled with mass spectrometry (GC-MS), which are regarded as the gold standard in drug analysis, are not suitable for this purpose due to their low portability, time-consuming measurements and need for trained personnel.20,21 Therefore, LEAs present at music festivals need portable on-site screening methods for the rapid and accurate analysis of the expectedly large amount of samples encountered during these searches. Moreover, these methods need to be capable of analyzing different sample types such as tablets, capsules, powders, crystals and liquids which come in various shapes and colors.15,20
Presumptive color tests are generally used by LEAs in the field for the fast on-site screening of suspicious samples. Despite their low cost and simplicity in use, these tests lack specificity as they are prone to producing false positive and false negative results.22,23 Furthermore, different color tests are preferred for different drug classes (e.g. the cobalt thiocyanate test for cocaine or the Marquis reagent for opium alkaloids and several synthetic drugs).23 Therefore, to cover a range of different drugs and reduce the false positives and false negatives, unknown substances are subjected to sequences of different tests, which can be laborious and time-consuming.23,24 Spectroscopic methods such as Raman and Fourier transformed infrared (FTIR) are also available as portable or benchtop devices and are suitable alternatives for on-site drug analysis due to their broad spectral libraries.25,26 Drawbacks of spectroscopic methods include the relatively high cost of the instruments and the possible influence of pigments and binders present in samples on the analysis, as is the case with fluorescence interference in Raman measurements.27
Electrochemical sensors have great potential for use in forensic applications due to their high portability, rapid measurements and affordability.28–31 In addition, they offer strong analytical performances in complex matrices, as they are not affected by optically absorbing and fluorescent molecules, nor by the dyes in samples or their morphology.28,29 More specifically, sensing based on electrochemical profiles (EP), in which the characteristic electrochemical signal or profile of a compound in a given analytical context is used for its identification, is considered an inviting approach.32 It allows the simultaneous detection of multiple compounds in a sample, as is the case for drug samples containing adulterants, diluents and other electroactive compounds. Carbon-based screen-printed electrodes (SPEs) in particular are perfect candidates for the on-site analysis of suspicious samples: they are highly portable, cheap and disposable.29 Utilizing the characteristic EP for identification on SPEs (unmodified or including a short pre-treatment step or coating), previous studies have reported accurate detection strategies for numerous drugs in the presence of adulterants, including cocaine,33,34 MDMA,35–37 heroin38,39 and ketamine.40 However, these studies generally focus on the detection of one drug, while LEAs at music festivals and other nightlife settings require screening tools for a variety of drugs. The state-of-the-art approach used in electrochemical illicit drug sensors involves recording an EP at a single electrode and subsequently processing this EP with peak identification software (Fig. 1).41 This becomes cumbersome if multiple target compounds, some sharing a similar structure, have to be detected by the same sensor. More electrochemical information of the sample is thus required, and it is hypothesized that this additional information can be obtained through extending the measurement from a single electrode towards multiple electrodes. By diversifying the measuring conditions at each electrode, different EPs can be obtained from the same sample, which will greatly enhance the electrochemical information available to decide on the sample's identity.
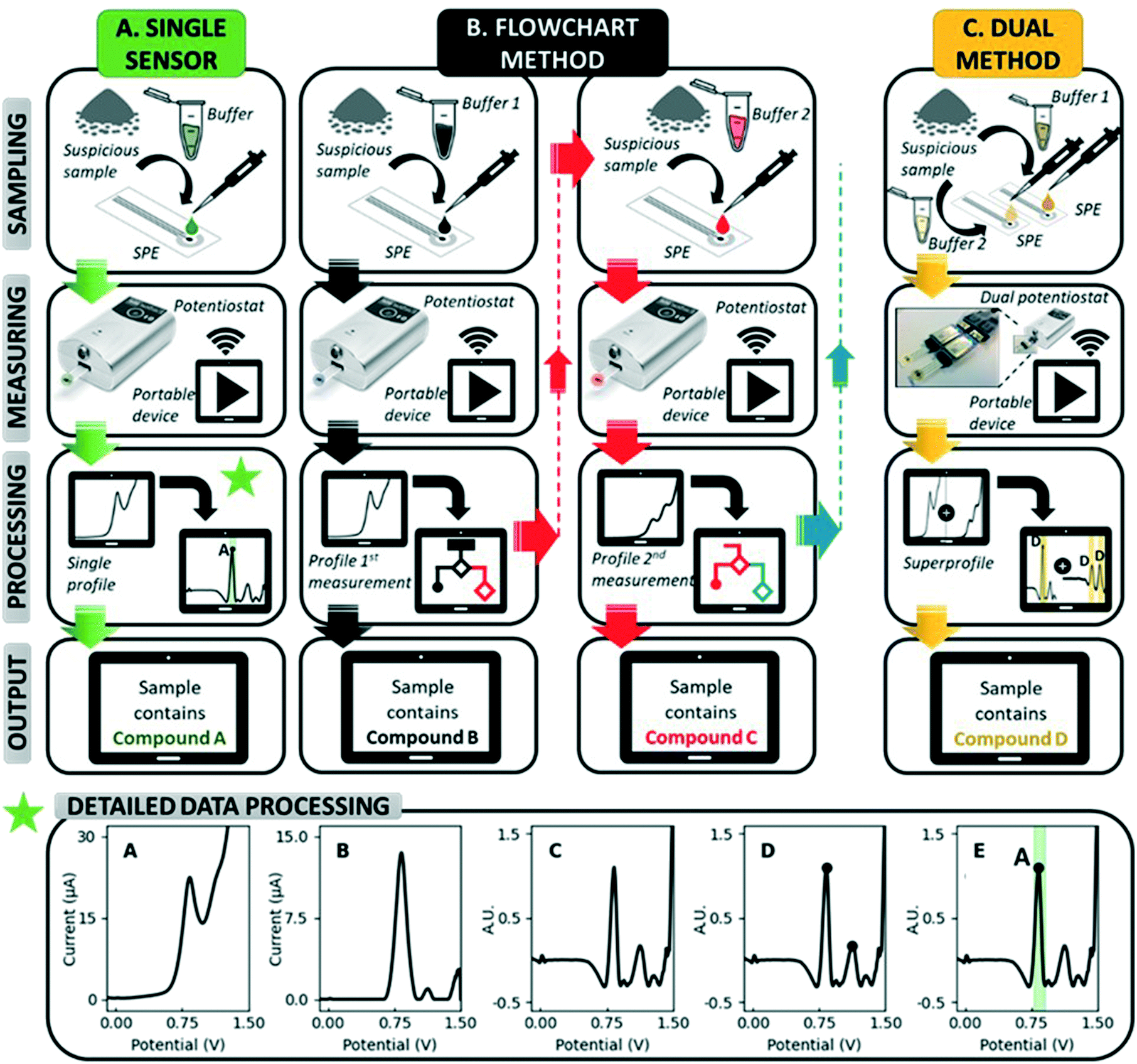 | ||
| Fig. 1 Schematic overview of the state-of-the-art approach used in electrochemical drug sensors (single sensor). In this work, two novel methods are developed: a flowchart method and a dual sensor method. In the flowchart method, the approach is similar to the single sensor, however, a statement on the presence/absence of the illicit drug is not directly given. Rather, depending on the outcome of the first measurement, subsequent measurements are proposed and required prior to the final verdict. The dual sensor method requires simultaneous sampling using two different buffers, followed by a simultaneous measurement at two SPEs. The resulting electrochemical profiles are data processed together (superprofile). A schematic display of the data processing of each method is displayed at the bottom of the figure. These data processing steps involve: pre-processing of the raw voltammogram (A) with a baseline correction (B) and digital filter (C), identification of the relevant peaks (D) and assignment of compounds to these peaks using an internal database (E). | ||
Herein we present innovative electrochemical methods for the simultaneous detection of the four illicit drugs most commonly encountered at festivals in Europe:12–14 cocaine, MDMA, amphetamine and ketamine (Fig. S1†). After demonstrating the shortcomings of single sensors for the detection of multiple drugs, two novel multidrug methods (Fig. 1) are assessed based on their practicality, performance and limitations: (i) a flowchart based on sequential measurements in different measuring conditions (pH, buffer composition, …) and (ii) a dual-sensor which simultaneously measures an EP at two SPEs using different measuring conditions. In the dual sensor, the simultaneously recorded EPs are combined into a so-called superprofile. This superprofile links the information from both individual EPs, thereby creating a wealth of information about the measured sample. Both novel methods are developed using a training set of samples containing pure drugs, pure adulterants and relevant binary mixtures between drugs and adulterants. Thereafter, the methods are validated with a test set containing 10 confiscated street samples for each of the four target drugs (40 in total) and their performance compared to that of a portable Raman spectroscopic device which is commercially available for the analysis of confiscated samples. Overall, these methods aim to offer LEAs tools for the rapid, affordable and high-performance on-site screening for multiple drugs at music festivals, while also providing them with the possibility to use the device for other duties such as border control and cargo analysis.
Experimental
Reagents and confiscated samples
Standards of D,L-MDMA·HCl, D,L-amphetamine·sulphate, ketamine·HCl, D-methamphetamine·HCl, butylone·HCl and codeine·HCl were purchased from Lipomed (Arlesheim, Switzerland). A standard of cocaine·HCl was purchased from Chiron AS (Trondheim, Norway). Standards of phenacetin, paracetamol, lidocaine, procaine and benzocaine were purchased from Sigma-Aldrich (Diegem, Belgium), a standard of levamisole was purchased from Acros Organics (Geel, Belgium), a standard of caffeine was purchased from VWR Chemicals (Leuven, Belgium) and a standard of creatine monohydrate was purchased from J&K Scientific (Lommel, Belgium).Confiscated samples containing cocaine, MDMA, amphetamine and ketamine were provided by the National Institute for Criminalistics and Criminology (NICC) in Belgium. Confiscated samples were provided in different physical forms (tablets, powders, crystals, pastes), colors (white, yellow, pink, orange), compositions (presence of adulterants and diluents) and purities (6.6 to 100%). Qualitative and quantitative analysis of the confiscated samples were performed by NICC using GC-MS and GC-flame ionization detection (GC-FID) respectively.
Analytical grade salts of KH2PO4 and KCl, as well as KOH and HCl for pH-corrections and the formalin solution (aqueous formaldehyde solution, 37% w/w), were all purchased from Sigma-Aldrich (Overijse, Belgium).
All solutions were prepared in 18.2 MΩ cm−1 doubly deionized water (Milli-Q water systems, Merck Millipore, Germany). Monitoring of the pH was performed using a 914 pH/conductometer from Metrohm (Herisau, Switzerland).
Instrumentation and methods
All square wave voltammetry (SWV) measurements were performed using a MultiPalmSens4 potentiostat (Palm-Sens, Houten, The Netherlands) with PSTrace/MultiTrace software. Disposable carbon ItalSens IS-C Screen Printed Electrodes (SPEs) containing a graphite working electrode (Ø = 3 mm), a carbon counter electrode, and a silver (pseudo) reference electrode were used for all measurements (single use) and were also provided by PalmSens. All experiments were performed by applying 50 μL of the solution onto the SPE. The SWV parameters that were used: potential range of −0.1 to 1.5 V, frequency 10 Hz, amplitude 25 mV and step potential 5 mV. All electrochemical measurements shown were performed three times.Phosphate buffer saline (PBS) pH12 used in the experiments contains 0.020 M K2HPO4 and 0.1 M KCl (further referred to as “pH12”). Another buffer is also employed: PBS pH 7F contains 0.1 M KH2PO4 and 0.1 M KCl, as well as 30% v/v formalin solution (11.1% v/v formaldehyde) (further referred to as “pH7F”). All measurements utilizing this buffer solution were started after a reaction time of 28 seconds (which adds to a total of 1 minute including the measurement time). Measurements of pure compounds and binary mixtures used a concentration of 0.5 mM per compound in the pH12 buffer and 1 mM in the pH7F buffer. Moreover, all street samples measured were diluted to a concentration of 0.3 mg mL−1 in the former buffer and to 2.0 mg mL−1 in the latter.
For the flowchart, a first sampling is performed in the proposed buffer solution for the first measurement. Depending on the result of the analysis, a second sampling could be proposed in a different buffer solution. In contrast, the dual-sensor requires two samplings before each measurement, one in each of the proposed buffer solutions.
The spectroscopic measurements were performed using a Bruker Bravo Handheld Raman spectrometer (Bruker Optik GmbH, Ettlingen, Germany). Further information on the used parameters and library is included in the ESI.†
Data processing
All raw voltammograms (Fig. 1A) were background corrected using the “moving average iterative background correction” (peak width = 1) tool in the PSTrace software and subsequently digitally filtered with a top hat filter (wt = 7) (Fig. 1B and C, respectively). This digital filtering and all further pre-processing steps are executed utilizing an in-house developed MATLAB script. The resulting pre-processed voltammograms are displayed throughout the manuscript. After the pre-processing of the voltammograms, the relevant peaks are selected based on a minimum peak prominence and minimum peak height threshold (Fig. 1D). This is to make sure only peaks are used that hold information about the sample. Subsequently, compounds are assigned to the selected peaks using an internal database (Fig. 1E). An exception module in the data processing software allows the incorporation of exceptions. A detailed description of this data processing approach can be found in the manuscript of Van Echelpoel et al.41Results and discussion
In the EP-based sensing of illicit drugs, identification is based on the peak potential associated with a particular redox signal of a target compound. First, the electrochemical behavior of the target is studied in specific measuring conditions to identify reliable signals. Second, a potential interval is defined around each peak to account for small variations in peak potential due to temperature, concentration or the effect of electroactive adulterants in drug samples. The collection of these intervals composes the internal database of the peak recognition approach in the data processing. The presence of a peak in a recorded voltammogram can then be linked to the presence of a specific compound in the sample if that peak lies within one of the predefined intervals of the internal database. Some compounds have multiple peaks, and thus an increased selectivity is obtained by requiring the presence of all those peaks. If these conditions are fulfilled for a specific drug, the analysis is positive for this compound.Shortcomings of single sensors for multidrug detection
When developing a sensor for the detection of a single drug (Fig. 1), the measuring conditions (buffer, pH, electrode material) are selected in such a way that the drug yields a clear EP that can be successfully distinguished from other electroactive compounds present in real samples. The optimization of measuring conditions has previously been reported for the detection of cocaine,34,42,43 MDMA35–37 and ketamine.44,45 In particular, the use of pH12 buffer on unmodified carbon SPEs is highly suitable for the detection of these three illicit drugs in the presence of their adulterants.33,37,40 Meanwhile, the detection of amphetamine is complicated by the high oxidation potentials of primary amines,46 which fall outside the accessible potential window (>1.5 V) in an aqueous environment on unmodified graphite screen-printed electrodes.To overcome this, derivatization approaches using various reagents have been reported.47–50 For example, the formaldehyde approach developed in our group successfully employs pH7F buffer (containing 30 v% formalin solution) to detect amphetamine and enrich the EP of other drugs with additional characteristic peaks.48
When multiple target compounds have to be detected by the same single sensor, this strategy becomes cumbersome due to potentially overlapping peaks. This is illustrated in Fig. 2, which contains the EP of the four target drugs, measured in the previously mentioned pH7F and pH12 measuring conditions.
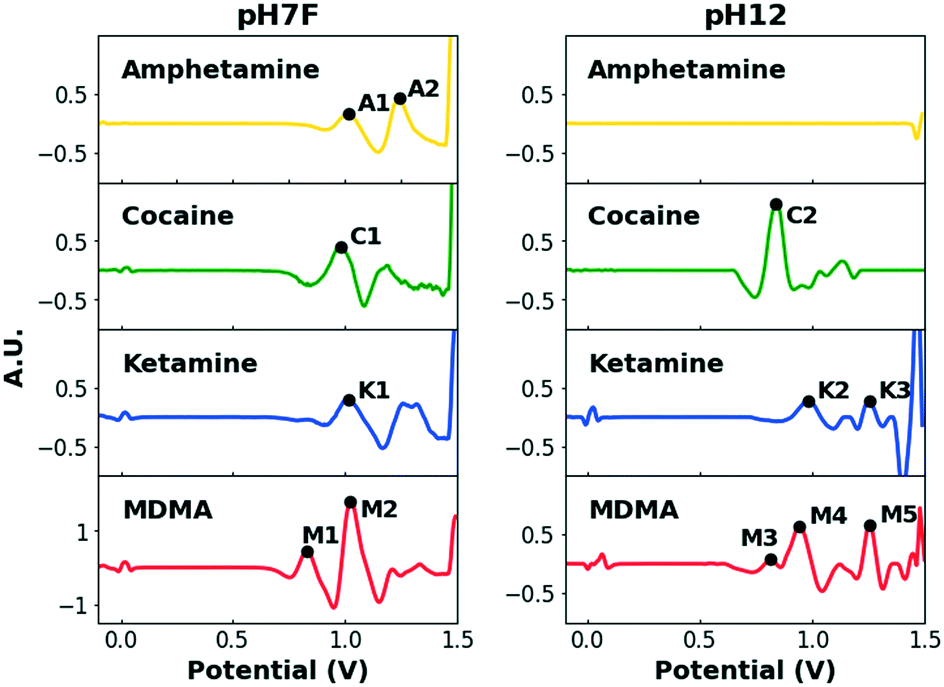 | ||
| Fig. 2 Overview of the pre-processed EPs of the four selected illicit drugs in pH7F (left) and pH12 (right). The relevant peaks are assigned a peak name. Concentrations in pH7F: 1 mM. Concentrations in pH12: 0.5 mM. | ||
Table 1 summarizes the oxidation potentials of the characteristic peaks of these compounds. In both buffers, significant overlap between the signals of the target drugs is observed, especially considering that potential intervals need to be defined around each signal to account for variations caused by various factors (i.e. temperature, concentration and composition). Although some drugs yield multiple signals to facilitate detection, the strong similarities between the EPs of cocaine and ketamine in pH7F and the non-detectable nature of amphetamine in pH12 are only two examples to demonstrate the shortcomings of single sensors when selective multidrug detection is desired.
| Drug | pH7F | pH12 | ||
|---|---|---|---|---|
| Peak name | Peak potential (V) | Peak name | Peak potential (V) | |
| Amphetamine | A1 | 1.01 | ||
| A2 | 1.23 | |||
| Cocaine | C1 | 0.98 | C2 | 0.83 |
| Ketamine | K1 | 0.99 | K2 | 0.98 |
| K3 | 1.26 | |||
| MDMA | M1 | 0.81 | M3 | 0.81 |
| M2 | 1.03 | M4 | 0.95 | |
| M5 | 1.24 | |||
As the use of multiple electrodes can provide the necessary additional electrochemical information, two multi-SPE methods are developed (Fig. 1): (i) the flowchart method starts with a first measurement and depending on the resulting EP, a second measurement is executed in different measuring conditions. Exactly which conditions are used for this second measurement is linked to a specific output in the first measurement. Additional measurements can be performed if necessary (Fig. 1B), (ii) the dual-sensor method measures an EP simultaneously at two SPEs using different measuring conditions at each electrode. Both EPs are then combined into a superprofile that links the information of both individual EPs with each other (Fig. 1C). Both multi-SPE methods thus make use of the same EPs, but use them in a different way to come to a result. The flowchart can be seen as a stepping stone to the dual sensor, since the former tries to obtain a result with as few SPEs as possible, while the latter always uses the information from two SPEs to arrive at a result. The combination of these two recorded EPs into a superprofile, results in more than double the information since signals in the first EP can be linked to the presence/absence of signals in the second EP, and vice versa.
Building the database through a selected training set
A set of samples, the training set, is selected to optimize the flowchart and dual-sensor methods. It may be clear that both methods thus use the same set of EPs, i.e. the training set, for their optimization. An overview of the selected samples is given in Table S1.† Apart from the four illicit drugs, this set contains eight adulterants and 21 relevant binary mixtures (drugs with adulterants). The adulterants are an important part of this training set as they are frequently added to drug samples and could influence the EP of the drug in the sample. These adulterants in particular are selected based on their common occurrence in formulations containing one of the drugs discussed in this work.51–53 Furthermore, they could also be encountered as the main ingredient in legal formulations, in which case differentiation with the target drugs is necessary as an overlap could lead to false positive results. Therefore, it is important to carefully choose the potential intervals defined for each of the characteristic peaks of the targeted drugs, based on their electrochemical behavior in the used measuring conditions.First, the adulterants in the training set were analyzed in pH7F and pH12 (Fig. S2†), with resulting peak potentials summarized in Table S2.† It can be observed that benzocaine, codeine, lidocaine and procaine produce signals located in a similar potential area to the target drugs (Table 1). This is mainly the case in pH7F, as these measuring conditions have an enriching effect on the EPs of not only drugs but all compounds containing primary or secondary amines. The proximity of all these signals again highlights the necessity of combining multiple measuring conditions to obtain differentiation.
The influence of these adulterants on the EPs of the drugs in the binary mixtures (Fig. S3†) is assessed based on the change in potential of the characteristic drug peaks (Table S3†) they cause. Importantly, these findings lead to the definition of potential intervals for each peak. For example, if the presence of the adulterants of a drug tends to cause a positive shift in the peak potential of that drug, then the interval is made larger on that side of the peak. Several observations made from Table S3† are discussed below.
Firstly, the shifts of the C2 and K2 peaks (+0.09 V) in the binary mixtures of cocaine and ketamine with benzocaine in pH12 stand out. Additionally, K3 is completely suppressed. In previous work, de Jong et al. found that, upon oxidation, benzocaine creates a local near-surface pH effect causing the redox peaks of other compounds (e.g. cocaine and ketamine) to shift or be suppressed.54 To avoid false negative results, this effect needs to be taken into account when defining intervals. However, simply broadening it to compensate for this strong shift is not desirable, as several other compounds produce peaks in this potential zone (0.80–1.00 V) and narrow intervals are, therefore, preferred. As this effect is, to our knowledge, specific to benzocaine, a more selective solution is proposed. In the peak recognition software, it is programmed that, when a signal is detected in the interval 0.45–0.55 V (where the characteristic peak of benzocaine is located) in pH12, the potential intervals of the C2 and K2 peaks are shifted to higher potentials and extended, while K3 is no longer a required peak (Fig. 3).
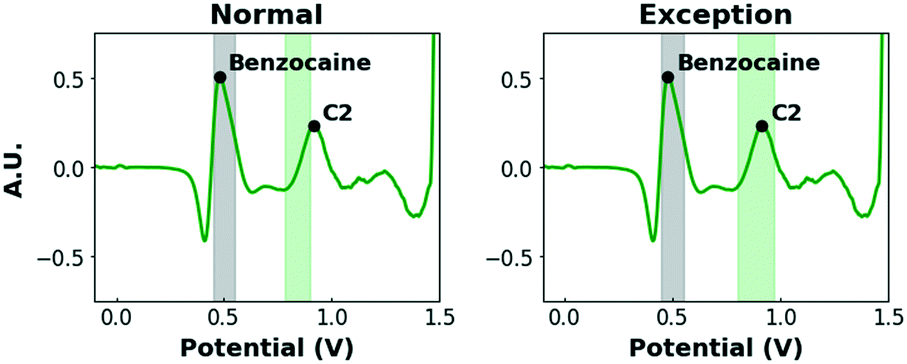 | ||
| Fig. 3 Left: A pre-processed voltammogram of an equimolar cocaine/benzocaine mixture (0.5 mM/0.5 mM) in pH12 with the normal potential interval visually shown (in green). Right: Same voltammogram but with a potential interval that is shifted/extended. If a peak is detected in the grey interval (0.45–0.55 V), it is positive for benzocaine and the exception is activated. | ||
Secondly, large shifts are also observed for the A2 (+0.10 V) and M5 (+0.06 V) peaks in their binary mixtures with caffeine (in pH7F and pH12 respectively). This is due to the overlapping of A2 and M5 with the caffeine signal, resulting in one broad peak. As fewer characteristic peaks of target drugs occur in this potential zone, a wide interval is proposed covering both the pure compounds and the binary mixtures (Table 2). Thirdly, the M3 peak (pH12) is not detected in the binary mixtures of MDMA with amphetamine and ketamine due to a peak prominence below the threshold. This low peak prominence is a common issue for signals which are present as a shoulder on a different peak in the raw voltammogram and are separated by the pre-processing step, as is the case for M3 (Fig. 2). To overcome this, the requirement of this peak for MDMA identification is set to optional as two characteristic signals in each pH7F and pH12 can provide the necessary selectivity (Table 2).
| pH7F | pH12 | |||||
|---|---|---|---|---|---|---|
| Peak name | Potential interval (V) | Required peaks | Peak name | Potential interval (V) | Required peaks | |
| a Detection of M3 is optional. | ||||||
| Amphetamine | A1 | 0.96–1.04 | 2/2 | 0/0 | ||
| A2 | 1.15–1.35 | |||||
| Cocaine | C1 | 0.95–1.06 | 1/1 | C2 | 0.78–0.90 | 1/1 |
| C2(b) | 0.80–0.97 | |||||
| Ketamine | K1 | 0.97–1.07 | 1/1 | K2 | 0.92–1.02 | 2/2 |
| K2(b) | 1.02–1.15 | 1/1(b) | ||||
| K3 | 1.23–1.40 | |||||
| MDMA | M1 | 0.78–0.86 | 2/2 | M3a | 0.80–0.86 | 2/2a |
| M2 | 0.98–1.08 | M4 | 0.92–0.97 | |||
| M5 | 1.24–1.32 | |||||
Flowchart method
A flowchart starts by performing a first test (on one SPE) to obtain an EP of the sample. Based on this, either a result is shown or a follow-up measurement, requiring a second sampling and measurement on a different SPE, is proposed by the software in different measuring conditions to improve differentiation. Thus, compounds that produce overlapping signals in the conditions used for the first test can be grouped in one joint interval. When a peak is detected in this interval, a follow-up measurement is proposed in conditions that allow differentiation between the drugs included. The overview of possible measurement sequences is subsequently summarized in a flowchart (Fig. 4).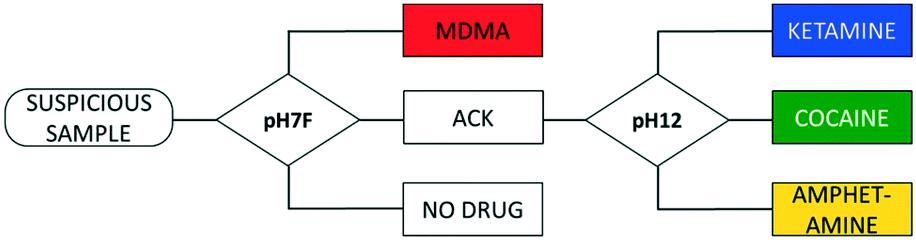 | ||
| Fig. 4 Representation of the flowchart method and its dedicated databases. First, a measurement is executed in pH7F, and depending on the outcome, a second measurement might be executed in pH12. ACK = joint interval for amphetamine, cocaine and ketamine in pH7F (0.92–1.07 V). | ||
Since LEAs at music festivals expectedly have a large number of suspicious samples to analyze, including numerous negative samples, it makes sense to minimize the measurement sequence in general and in particular for negative results. Therefore, the choice of measuring conditions for the first measurement needs to cover all the drugs targeted by the sensor, in this case by using the pH7F conditions.
As was demonstrated in Fig. 2, the oxidation potentials of the C1, A1, M2 and K1 peaks in pH7F are all located in a narrow potential zone (0.98–1.03 V). While MDMA can be selectively detected by utilizing the M1 peak, differentiation between the other three drugs is complicated.
Although the EP of amphetamine contains a second characteristic peak (A2), the presence of caffeine, considered to be one of the most prevalent adulterants in both cocaine and ketamine samples,51,55 in the analyzed sample could also trigger this interval. Consequently, amphetamine, cocaine and ketamine are combined in one interval, named ‘ACK’ (0.92–1.07 V), to avoid wrongful identifications (Fig. 4). For MDMA, both characteristic peaks are required for identification, and despite one of those occurring in the ACK interval, MDMA can be selectively identified based on the presence of the other. If neither the conditions for ACK detection are fulfilled nor those of MDMA, then the result of the measurement is negative. For this reason, a sample that does not contain any of the target drugs would only require one sampling and one measurement.
If a peak is detected in the ACK interval, a follow-up measurement (in this case in the pH12 condition) is proposed to differentiate between these three drugs. The EPs of cocaine and ketamine are sufficiently separated in these conditions, while amphetamine yields no peaks. It can be programmed in the peak recognition software that the latter is detected when neither the cocaine nor the ketamine intervals are activated in pH12. However, Table S2† shows that three pure adulterants also activate the ACK interval in pH7F: benzocaine, codeine and lidocaine. As none of them triggers the intervals of cocaine and ketamine in pH12, the analysis of these compounds would give a false positive result for amphetamine. Since they are unlikely to be encountered in real amphetamine samples (which mainly contain caffeine, creatine and non-electroactive diluents53,56), an exception is built into the software that utilizes the difference in electrochemical behavior between the mentioned adulterants and amphetamine in pH12. While the former all produce characteristic oxidation peaks between 0.47 and 0.80 V, the latter is non-electroactive. Thus, it is programmed that amphetamine is only identified by the sensor if: (a) the ACK interval is activated in pH7F, and (b) no peaks are identified in the potential range 0.47–0.80 V in pH12. If a peak is detected in the defined range, the compound is deemed to be an adulterant and will give a negative result.
The major advantage of the flowchart over the single sensor is that the results of a first measurement can be linked to an optional, second measurement. Next, a dual-sensor method is investigated to further explore the possibilities of linking two EPs to each other.
Dual sensor method
A second method, coined the dual-sensor method, involves the combined analysis of two electrochemical profiles recorded simultaneously at two parallel SPEs. This method thus always requires only one sampling step (comprising sampling in one or multiple different buffer solutions), as opposed to the flowchart method which might require more than one sampling step (in one buffer solution each time). The fusion of the two simultaneously recorded electrochemical profiles results in a so-called superprofile that holds much more information about the sample than either one of these two individual profiles separately. In fact, the information is more than doubled with the superprofile because the presence or absence of signals in both individual profiles can be linked to each other. The data interpretation approach of Van Echelpoel et al. has been extended for this dual-sensor method to maximally exploit this information contained in the superprofile.41 Both electrochemical profiles are still pre-processed and subsequently assigned compounds using their own, unique database. However, only one, unique exception module which comprises both profiles is employed, rather than two exception modules for each profile separately. As a result, the presence of signals in both conditions can be required prior to the assignment of a specific compound.For these experiments, the pH7F and pH12 measuring conditions are used in parallel (Fig. S4†). The combination of a set of measuring conditions capable of detecting all drugs of interest (pH7F) and another set of conditions to provide improved differentiation among those drugs (pH12), is highly suitable for this purpose. As each analysis combines the EPs obtained in pH7F and pH12, a joint ‘ACK’ interval is no longer required and the intervals of the individual drugs can be used. The issue of false positives for amphetamine caused by adulterants is then reduced to those that activate both amphetamine intervals in pH7F, which is the case for codeine and lidocaine (Fig. 5). Again, it is programmed that amphetamine can only be detected if: (i) both its intervals are triggered in pH7F and (ii) no peak is detected in the potential range 0.47–0.80 V in pH12.
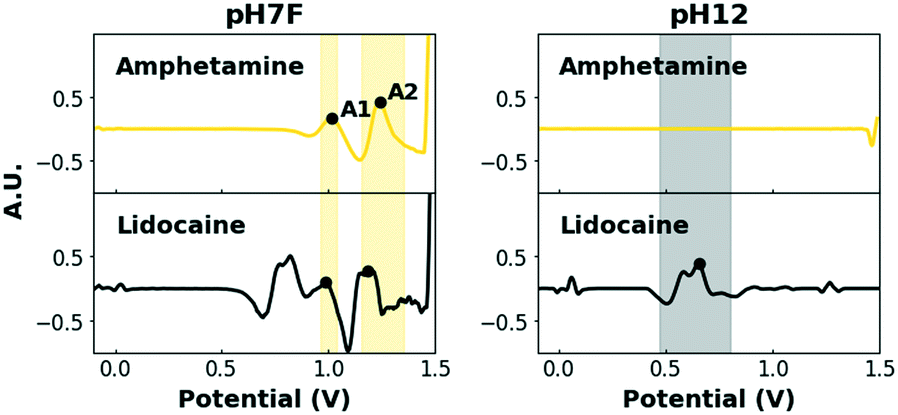 | ||
| Fig. 5 Pre-processed voltammograms of lidocaine and amphetamine in pH7F and in pH12. Amphetamine is identified if both diagnostic peaks in pH7F are present and no peak is present in the interval (0.47–0.80 V). If a peak is present in the latter interval, as is the case for lidocaine, amphetamine is not identified, thereby avoiding potential false positives. | ||
Validation using confiscated samples
Finally, the two multidrug sensing methods are validated by analyzing a set of confiscated samples (40 in total, Table S4†). These confiscated samples, previously analyzed at NICC by the standard methods of a forensic laboratory (i.e. GC-MS and GC-FID), provide valuable information about what is encountered on-site and are difficult to mimic in the lab due to their varying composition and the limited information available in the literature on these compositions. The performance of both methods is subsequently assessed using several metrics related to the accuracy of detection and the practicality of the method, summarized in Table 3. In addition, their performance is compared to that of a portable Raman spectroscopic device (Bruker Bravo) which is commercially available for the purpose of illicit drug detection.| Flowchart method | Dual-sensor method | Portable Raman | |
|---|---|---|---|
| Accuracy confiscated samples (40) | 80.0% | 87.5% | 60.0% |
| Time per analysis (seconds) | ∼90 | ∼60 | 20–60 |
| Average electrodes used | 1.54 | 2 | — |
| Different buffers required | 1.54 | 2 | — |
The flowchart method uses the additional electrochemical information provided by follow-up measurements to improve performance. Application of this method to the voltammograms obtained for the 40 confiscated samples in pH7F and pH12 (Fig. S5 and Table S5†) leads to a correct identification in 80.0% of the cases (Table 3). The main source of wrongful classifications is the series of ketamine samples, with 6 out of 10 classed as MDMA (Table S5†). It appears that a shoulder on the K1-peak of ketamine arises at high concentrations in pH7F, which is separated into a peak by the pre-processing step and which activates the M1 interval of MDMA (Fig. S5 and Table S5†). This feature is not included in the EP of ketamine due to its presence depending on the concentration, but could be encountered in on-site measurements and, therefore, needs to be taken into account. In this flowchart, no follow-up measurement was proposed in the case of MDMA detection in pH7F as it could be differentiated from the other drugs in these conditions. Moreover, this work aimed to make the flowchart as short as possible to showcase its balance between feasible practicality and performance. Indeed, the metrics displayed in Table 3 show that, of the two methods discussed, the proposed flowchart offers a good balance between being practical (limiting the number of samplings and SPEs used) and providing accurate results for this set of target drugs. At the cost of some practicality, an additional analysis could be introduced in the case of an MDMA positive in the first test to further improve the performance. The choice of measuring conditions for this additional measurement could be pH12, in which case this flowchart becomes the sequential version of the dual-sensor when a drug is detected, or another set of conditions to verify the presence of MDMA in the sample.
The dual-sensor method combines two parallel measurements (in two different measuring conditions) to achieve accurate detection. This method always requires the use of two SPEs and two samplings, but measurement time remains low due to the parallel character. Its key advantage is that, for each sample, the EP is recorded in two measuring conditions so each identification is well-founded. As expected, this method obtains the highest accuracy for the confiscated sample analysis in comparison to the standard methods (87.5%, Table 3). Two of the wrongfully classed samples are false negatives (1C and 9C, Table S5†), while the other three are classified as another illicit drugs. For all five, the expected characteristic peaks of the drug present in the sample were detected but fell outside the defined potential intervals (Fig. S5†).
Optimizing the choice of interval limits is a gradual process and the more samples are analyzed, the more accurate these become. Therefore, this outcome is promising as further optimization on relevant drug samples will filter out these few false identifications and thereby improve the performance.
Finally, the same set of confiscated samples was analyzed using the portable Raman device. The data evaluation was limited to finding the three main components as non-specialized personnel are unlikely to perform an extensive manual mixture analysis in the field. The results are summarized in Table S5† and the obtained accuracy is included in Table 3. The Raman device obtained a considerably lower accuracy (60.0%) than the electrochemical methods. Incorrect results include the identification of another (legal) substance present in the sample (e.g. caffeine in 3A and creatine in 8A) or of structurally related compounds (e.g. norephedrine in 1A and phenethylamine in 2A). In the field, LEAs often combine the use of a portable Raman device with other techniques to avoid false negatives.
It is important to emphasize that these results reflect the capabilities of the tested device for this specific purpose, rather than those of Raman spectroscopy in general.
Conclusions
In this work, two innovative electrochemical methods for the simultaneous detection of the four most commonly encountered illicit drugs at festivals in Europe (cocaine, MDMA, amphetamine and ketamine) were developed and validated. LEAs at music festivals require portable screening tests for the rapid on-site analysis of suspicious samples encountered during searches. Therefore, both methods were compared based on their practicality regarding on-site use and performance on a series of confiscated samples.The flowchart method introduces the possibility of performing follow-up measurements based on the result of an initial test. These then provide the electrochemical information necessary to achieve the desired differentiation. Furthermore, this method has the practical advantage of eliminating the need for further measurements in some cases, such as for negative samples (when the recorded EP does not resemble that of any of the target drugs). Already reaching an accuracy of 80.0% in the proposed form, this can be further increased by introducing additional measurements.
Expectedly, the dual-sensor is the most reliable method (accuracy 87.5%) as it bases all identifications on a double EP recorded in different measuring conditions. For each of the wrongfully classed confiscated samples, further optimization of the potential intervals used in the peak identification software through testing more samples could offer the solution. When further expanding the scope of the sensor to detect other drugs, this principle of parallel measurements (two or more) collecting sufficient electrochemical information for selective detection will be essential, as the measurement sequences used in flowcharts will then become too laborious and time-consuming.
Overall, these electrochemical multidrug methods proved to be viable options for the on-site screening of suspicious samples encountered during searches at music festivals, as evidenced by the performance comparison with the commercially available portable Raman device, which reached a considerably lower accuracy. They are portable, affordable, rapid, cover several different drugs simultaneously and reach high accuracies. Moreover, these findings demonstrated the robustness of this electrochemical sensor for the detection of the target drugs in different physical forms (tablets, powders, crystals, pastes), colors (e.g. white, yellow, pink, orange), compositions (presence of adulterants and diluents) and purities (ranging from 6.6 to 100%). This work will serve as a foundation for the development of new electrochemical tools to detect illicit drugs in decentralized settings, thereby empowering the LEAs capabilities towards a safe society.
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation programme under Grant Agreement No. 833787, BorderSens. The authors acknowledge financial support from the Fund for Scientific Research (FWO) Flanders, Projects, Infrastructure (screen printing) and Grant 1SB8122N and VLAIO IM [HBC.2019.2181], Brussels, Belgium.Notes and references
- T. F. M. ter Bogt, S. N. Gabhainn, B. G. Simons-Morton, M. Ferreira, A. Hublet, E. Godeau, E. Kuntsche and M. Richter, Subst. Use Misuse, 2012, 47, 130–142 CrossRef PubMed.
- P. Miller, N. Droste, F. Martino, D. Palmer, J. Tindall, K. Gillham and J. Wiggers, J. Subst. Use, 2015, 20, 274–281 CrossRef.
- E. Dilkes-Frayne, Int. J. Drug Policy, 2016, 33, 27–35 CrossRef PubMed.
- V. Pavluković, T. Armenski and J. M. Alcántara-Pilar, Tour. Manag., 2017, 63, 42–53 CrossRef.
- G. Maasz, E. Molnar, M. Mayer, M. Kuzma, P. Takács, Z. Zrinyi, Z. Pirger and T. Kiss, Environ. Toxicol. Chem., 2021, 40, 1491–1498 CrossRef CAS PubMed.
- N. Day, J. Criss, B. Griffiths, S. K. Gujral, F. John-Leader, J. Johnston and S. Pit, Harm Reduct. J., 2018, 15, 1 CrossRef PubMed.
- B. C. Kelly and J. T. Parsons, Am. J. Drug Alcohol Abuse, 2008, 34, 774–781 CrossRef.
- J. J. Palamar, P. Acosta, A. Le, C. M. Cleland and L. S. Nelson, Int. J. Drug Policy, 2019, 73, 81–87 CrossRef PubMed.
- P. Calle, N. Sundahl, K. Maudens, S. M. Wille, D. Van Sassenbroeck, K. De Graeve, S. Gogaert, P. De Paepe, D. Devriese, G. Arno and P. Blanckaert, Prehosp. Disaster Med., 2018, 33, 71–76 CrossRef PubMed.
- F. Y. Lai, P. K. Thai, J. O'Brien, C. Gartner, R. Bruno, B. Kele, C. Ort, J. Prichard, P. Kirkbride, W. Hall, S. Carter and J. F. Mueller, Drug Alcohol Abuse Rev., 2013, 32, 594–602 CrossRef PubMed.
- T. Mackuľak, P. Brandeburová, A. Grenčíková, I. Bodík, A. V. Staňová, O. Golovko, O. Koba, M. Mackuľaková, V. Špalková, M. Gál and R. Grabic, Sci. Total Environ., 2019, 659, 326–334 CrossRef PubMed.
- L. C. G. Hoegberg, C. Christiansen, J. Soe, R. Telving, M. F. Andreasen, D. Staerk, L. L. Christrup and K. T. Kongstad, Clin. Toxicol., 2018, 56, 245–255 CrossRef CAS PubMed.
- L. Bijlsma, A. Celma, S. Castiglioni, N. Salgueiro-González, L. Bou-Iserte, J. A. Baz-Lomba, M. J. Reid, M. J. Dias, A. Lopes, J. Matias, L. Pastor-Alcañiz, J. Radonić, M. Turk Sekulic, T. Shine, A. L. N. van Nuijs, F. Hernandez and E. Zuccato, Sci. Total Environ., 2020, 725, 138376 CrossRef CAS PubMed.
- E. Deconinck, C. Aït-Kaci, A. Raes, M. Canfyn, J. Bothy, C. Duchateau, C. Mees, K. De Braekeleer, L. Gremaux and P. Blanckaert, Drug Test. Anal., 2021, 13, 679–693 CrossRef CAS PubMed.
- L. Gremeaux and E. Plettinckx, Substance use at music festivals: What is burning up the dance floor?, 2017 Search PubMed.
- A. R. Winstock, P. Griffiths and D. Stewart, Drug Alcohol Depend., 2001, 64, 9–17 CrossRef CAS PubMed.
- T. Van Havere, W. Vanderplasschen, J. Lammertyn, E. Broekaert and M. Bellis, Subst. Abuse: Treat. Prev. Policy, 2011, 6, 18 Search PubMed.
- T. F. M. ter Bogt, S. N. Gabhainn, B. G. Simons-Morton, M. Ferreira, A. Hublet, E. Godeau, E. Kuntsche, M. Richter and The HBSC Risk Behavior and the HBSC, Subst. Use Misuse, 2012, 47, 130–142 CrossRef PubMed.
- C. E. Hughes, V. Moxham-Hall, A. Ritter, D. Weatherburn and R. MacCoun, Int. J. Drug Policy, 2017, 41, 91–100 CrossRef PubMed.
- P. Calle, K. Maudens, S. Lemoyne, S. Geerts, D. Van Sassenbroeck, P. Jensen, J. Van Overloop, E. Deconinck and P. Blanckaert, Forensic Sci. Int., 2019, 299, 174–179 CrossRef CAS PubMed.
- E. Deconinck, R. Van Campenhout, C. Aouadi, M. Canfyn, J. L. Bothy, L. Gremeaux, P. Blanckaert and P. Courselle, Talanta, 2019, 195, 142–151 CrossRef CAS PubMed.
- M. de Jong, A. Florea, J. Eliaerts, F. Van Durme, N. Samyn and K. De Wael, Anal. Chem., 2018, 90, 6811–6819 CrossRef CAS PubMed.
- M. Philp and S. Fu, Drug Test. Anal., 2018, 10, 95–108 CrossRef CAS PubMed.
- E. Cuypers, A.-J. Bonneure and J. Tytgat, Drug Test. Anal., 2016, 8, 136–140 CrossRef CAS PubMed.
- L. Harper, J. Powell and E. M. Pijl, Harm Reduct. J., 2017, 14, 52 CrossRef PubMed.
- W. R. de Araujo, T. M. G. Cardoso, R. G. da Rocha, M. H. P. Santana, R. A. A. Muñoz, E. M. Richter, T. R. L. C. Paixão and W. K. T. Coltro, Anal. Chim. Acta, 2018, 1034, 1–21 CrossRef CAS PubMed.
- E. Gerace, F. Seganti, C. Luciano, T. Lombardo, D. Di Corcia, H. Teifel, M. Vincenti and A. Salomone, Drug Alcohol Rev., 2019, 38, 50–56 CrossRef PubMed.
- A. Florea, M. de Jong and K. De Wael, Curr. Opin. Electrochem., 2018, 11, 34–40 CrossRef CAS.
- B. Zanfrognini, L. Pigani and C. Zanardi, J. Solid State Electrochem., 2020, 24, 2603–2616 CrossRef CAS.
- H. Teymourian, M. Parrilla, J. R. Sempionatto, N. F. Montiel, A. Barfidokht, R. Van Echelpoel, K. De Wael and J. Wang, ACS Sens., 2020, 5, 2679–2700 CrossRef CAS PubMed.
- E. De Rycke, C. Stove, P. Dubruel, S. De Saeger and N. Beloglazova, Biosens. Bioelectron., 2020, 169, 112579 CrossRef CAS PubMed.
- G. Moro, H. Barich, K. Driesen, N. Felipe Montiel, L. Neven, C. Domingues Mendonça, S. Thiruvottriyur Shanmugam, E. Daems and K. De Wael, Anal. Bioanal. Chem., 2020, 412, 5955–5968 CrossRef CAS PubMed.
- M. de Jong, A. Florea, A.-M. de Vries, A. L. N. van Nuijs, A. Covaci, F. Van Durme, J. C. Martins, N. Samyn and K. De Wael, Anal. Chem., 2018, 90, 5290–5297 CrossRef CAS PubMed.
- R. G. Rocha, J. S. Stefano, I. V. S. Arantes, M. M. A. C. Ribeiro, M. H. P. Santana, E. M. Richter and R. A. A. Munoz, Electroanalysis, 2019, 31, 153–159 CrossRef CAS.
- L. R. Cumba, J. P. Smith, K. Y. Zuway, O. B. Sutcliffe, D. R. do Carmo and C. E. Banks, Anal. Methods, 2016, 8, 142–152 RSC.
- G. Murilo Alves, A. Soares Castro, B. R. McCord and M. F. Oliveira, Electroanalysis, 2021, 33, 635–642 CrossRef CAS.
- S. Thiruvottriyur Shanmugam, R. Van Echelpoel, G. Boeye, J. Eliaerts, M. Samanipour, H. Y. V. Ching, A. Florea, S. Van Doorslaer, F. Van Durme, N. Samyn, M. Parrilla and K. De Wael, ChemElectroChem, 2021, 8, 4826–4834 CrossRef CAS.
- A. Florea, J. Schram, M. De Jong, J. Eliaerts, F. Van Durme, B. Kaur, N. Samyn and K. De Wael, Anal. Chem., 2019, 91, 7920–7928 CrossRef CAS PubMed.
- N. Felipe Montiel, M. Parrilla, V. Beltrán, G. Nuyts, F. Van Durme and K. De Wael, Talanta, 2021, 226, 122005 CrossRef CAS PubMed.
- J. Schram, M. Parrilla, N. Sleegers, N. Samyn, S. M. Bijvoets, M. W. J. Heerschop, A. L. N. van Nuijs and K. De Wael, Anal. Chem., 2020, 92, 13485–13492 CrossRef CAS PubMed.
- R. Van Echelpoel, M. de Jong, D. Daems, P. Van Espen and K. De Wael, Talanta, 2021, 233, 122605 CrossRef CAS PubMed.
- L. Asturias-Arribas, M. A. Alonso-Lomillo, O. Domínguez-Renedo and M. J. Arcos-Martínez, Anal. Chim. Acta, 2014, 834, 30–36 CrossRef CAS PubMed.
- M. F. Muzetti Ribeiro, J. W. da Cruz Júnior, E. R. Dockal, B. R. McCord and M. F. de Oliveira, Electroanalysis, 2016, 28, 320–326 CrossRef.
- B. Deiminiat and G. H. Rounaghi, Sens. Actuators, B, 2018, 259, 133–141 CrossRef CAS.
- K. Fu, R. Zhang, J. He, H. Bai and G. Zhang, Biosens. Bioelectron., 2019, 143, 111636 CrossRef CAS PubMed.
- A. Adenier, M. M. Chehimi, I. Gallardo, J. Pinson and N. Vilà, Langmuir, 2004, 20, 8243–8253 CrossRef CAS PubMed.
- F. M. Ivison, J. W. Kane, J. E. Pearson, J. Kenny and P. Vadgama, Electroanalysis, 2000, 12, 778–785 CrossRef CAS.
- J. Schram, S. T. Shanmugam, N. Sleegers, A. Florea, N. Samyn, A. L. N. van Nuijs and K. De Wael, Electrochim. Acta, 2021, 367, 137515 CrossRef CAS.
- M. Parrilla, N. F. Montiel, F. Van Durme and K. De Wael, Sens. Actuators, B, 2021, 337, 129819 CrossRef CAS.
- R. Van Echelpoel, R. F. Kranenburg, A. C. van Asten and K. De Wael, Forensic Chem., 2022, 27, 100383 CrossRef CAS.
- C. Cole, L. Jones, J. McVeigh, A. Kicman, Q. Syed and M. Bellis, Drug Test. Anal., 2011, 3, 89–96 CrossRef CAS PubMed.
- J. Broséus, N. Gentile and P. Esseiva, Forensic Sci. Int., 2016, 262, 73–83 CrossRef PubMed.
- M. F. Andreasen, C. Lindholst and E. Kaa, Open Forensic Sci. J., 2009, 2, 16–20 CrossRef CAS.
- M. de Jong, N. Sleegers, J. Schram, D. Daems, A. Florea and K. De Wael, Anal. Sens., 2021, 1, 54–62 Search PubMed.
- C. V. Giné, I. F. Espinosa and M. V. Vilamala, Drug Test. Anal., 2014, 6, 819–824 CrossRef PubMed.
- C. Cole, L. Jones, J. McVeigh, A. Kicman, Q. Syed and M. A. Bellis, Cut, a guide to adulterants, bulking agents and other contaminants found in illicit drugs, 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sd00043a |
| ‡ Robin Van Echelpoel and Jonas Schram contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |