
Coke-promoted Ni/CaO catal-sorbents in the production of cyclic CO and syngas†
Seongbin
Jo‡
 a,
Jong Heon
Lee‡
b,
Jin Hyeok
Woo
b,
Tae-Young
Kim
b,
Ho-Jung
Ryu
c,
Byungwook
Hwang
c,
Jae Chang
Kim
*b,
Soo Chool
Lee
*d and
Kandis Leslie
Gilliard-AbdulAziz
*ae
a,
Jong Heon
Lee‡
b,
Jin Hyeok
Woo
b,
Tae-Young
Kim
b,
Ho-Jung
Ryu
c,
Byungwook
Hwang
c,
Jae Chang
Kim
*b,
Soo Chool
Lee
*d and
Kandis Leslie
Gilliard-AbdulAziz
*ae
aDepartment of Chemical and Environmental Engineering, University of California–Riverside, Riverside, California 92521, USA. E-mail: klabdulaziz@engr.ucr.edu
bDepartment of Chemical Engineering, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: kjchang@knu.ac.kr
cKorea Institute of Energy Research, Daejeon 34129, Republic of Korea
dResearch Institute of Advanced Energy Technology, Kyungpook National University, Daegu, 41566, Republic of Korea. E-mail: soochool@knu.ac.kr
eDepartment of Material Science and Engineering, University of California–Riverside, Riverside, California 92521, USA
First published on 1st December 2021
Abstract
Compared with conventional CO2 capture and utilization (CCU), the integrated CCU (ICCU) process has attracted attention in reducing total thermal energy and ensuring a simplified process. CO2 capture and the subsequent dry reforming of methane (DRM) during the ICCU employing Ni/CaO catal-sorbents has been proposed for the conversion of waste CO2 with CH4 into syngas. Here, coke-promoted Ni/CaO catal-sorbents were fabricated via CH4 pretreatment, and a highly efficient ICCU process for producing CO and syngas was proposed. High CO2 conversion and high CO production were achieved in the CO2 conversion step employing the reverse Boudouard reaction in tandem with CO2 capture. In the following CH4 conversion step, the spent catal-sorbents were regenerated into syngas via a reaction with CH4, and the carbon sources for the reverse Boudouard reaction were supplied by CH4 decomposition. The C-Ni/CaO catal-sorbent exhibited excellent performances of CO2 capture capacity, and CO and H2 productivities in consecutive CO2 and CH4 conversions. However, the coke-promoted Ni/CaO catal-sorbents were not completely regenerated in the CH4 conversion step after the 5th cycle, which might be due to the sintering of Ni0/NiO and CaO/CaCO3 materials and the excess amount of coke deposited on the surface of C-Ni/CaO catal-sorbents.
Introduction
An increase in CO2 emissions from the excessive combustion of fossil fuels has rapidly increased its atmospheric concentration, and this is the main cause of the climate emergency.1–3 To achieve a low-carbon or carbon-neutral society, many researchers have extensively investigated innovative CO2 mitigation technologies, such as efficient electrical energy production and consumption; coal gasification; and CO2 capture, utilization, and storage (CCUS), in the past few decades.4 CO2 can be utilized as a carbon source to produce fuels and value-added chemicals in several CO2 green technologies.4–6 CO2 capture and utilization (CCU) is among the most effective climate mitigation technologies for separating CO2 from large-scale industrial, as well as other emission sources; transporting the separated CO2 to specific sites; and utilizing it for different chemical processes.7,8Calcium looping (CaL) technology is considered a potential inexpensive technology for separating CO2; it utilizes regenerable calcium oxide (CaO)-based solid sorbents with excellent CO2 sorption capacity (786 mgCO2 gsorbent−1 or 17.8 mmolCO2 gsorbent−1): CaO(s) + CO2(g) ↔ CaCO3(s).9–12 However, the application of CaL technology in the conventional CCU process requires high total energies for carbonation and subsequent decarbonation in a temperature swing system, followed by the transportation and conversion of CO2via several catalytic reactions.11,13–15 Additionally, the CaO particles undergo severe thermal sintering, which causes a gradual decrease in their CO2 capture performance (capacity and kinetics) under conventional CaL conditions (carbonation: 10–15% CO2 at 650 °C and decarbonation: concentrated CO2 at 950 °C).9–11
Dry reforming of methane (DRM), which converts two greenhouse gases (CO2 and CH4) into industrial feedstock syngas (H2 and CO), is widely investigated using various techniques such as thermal catalysis,16–21 electrocatalysis,22–24 and photocatalysis.25–29 Among these techniques, thermal catalysis is a way to convert CO2 with a fast reaction rate, high selectivity, and large throughput. Considering that H2 is industrially produced from methane, DRM is considered more economical for CO2 utilization.30–32 Ni-based catalysts are the most investigated for DRM because of their low cost and comparable activities to noble metal catalysts.17,19,21,24,33,34 Apart from the hydrogenation of CO2 (methanation and rWGS), the most significant factor that affected the stability of the severe process during DRM was carbon deposition.35 The formation of carbon during DRM was mainly ascribed to the decomposition of CH4 (CH4(s) ↔ C(s) + 2H2(g)) and the Boudouard reaction (2CO(g) ↔ C(s) + CO2(g)).35 To achieve high conversion and avoid catalytic coking, DRM should be conducted at a high temperature (870–1040 °C) with a CH4/CO2 ratio of 1 because carbon deposition proceeds from the decomposition of CH4 and the Boudouard reaction in the temperature range of 557–700 °C.35
Some researchers have recently attempted to combine effectively CaL technologies with utilization of CO2 using catal-sorbents or dual-functional materials (DFMs) to develop a novel integrated CCU (ICCU) process for reducing the total thermal energy, as well as achieving a simplified process compared with that of conventional CCU.15,36–45 The catal-sorbents or DFMs consist of CaO as a CO2 sorbent and catalysts such as Ru, Rh, or Ni metals.15,36,38,40–42,46–50 First, CO2 is captured by CaO to form CaCO3 in the CO2 capture step, then a reactive gas such as H2, CH4 or C2H6 is flowed to regenerate CaCO3 to CaO, activate catalysts, and convert captured CO2 to value added chemicals via methanation,15,36,38,40,41,49 reverse water gas shift (rWGS),42,50 or dry reforming of hydrocarbons.46–48
Presently, only a few studies have attempted to develop catal-sorbents for CaL combined with DRM in the ICCU process using Ni/CaO catal-sorbents.15,48 In the CO2 capture step, CaO in the Ni/CaO catal-sorbents reacts with CO2, forming CaCO3: NiO/CaO(s) + CO2(g) ↔ NiO/CaCO3(s). During the subsequent DRM step, the NiO phase was reduced to metallic Ni by CH4, and the captured CO2 in the spent catal-sorbents was completely desorbed into the CH4 stream, after which the released CO2 reacted with CH4 to produce syngas: NiO/CaCO3(s) + CH4(g) ↔ Ni0/CaO(s) + 2CO(g) + 2H2(g).51 Moreover, a higher thermodynamic equilibrium was observed in the conversion of CaCO3 by CH4 compared with the conversion with H2 at a high temperature (>600 °C). Kim et al. demonstrated the feasibility of coupled CO2 capture and conversion reactions performed at 720 °C using a mixture of CaO (CO2 sorbent) and Ni/MgO–Al2O3 (DRM catalyst).52 The released CO2 (regeneration of the CO2 sorbent) is converted into syngas with an approximately equimolar ratio of H2 to CO with minimal CO2 slip; however, the CO2 uptake decreases during 10 cycles from thermal sintering of CaO at 720 °C. Tian et al. coupled the CaL-based CO2 capture and DRM in ICCU employing a CaO/Ni bi-functional sorbent catalyst to exploit the possibility of producing syngas from waste CO2 in an energy-efficient manner.15 Compared with conventional CaL processes, the captured CO2 was desorbed with superior reaction kinetics, and the desorbed CO2 reacted with CH4 at 800 °C when Ni-catalyzed CH4 reforming was integrated with CaCO3 regeneration, following Le Chatelier's principle. However, the CO2 capture capacity of the material gradually decreased with the cycle number because of the thermal sintering of the CaO phase. This low amount of CO2 in the following DRM step marginally decreased the syngas yield and coke formation. Although the deposited carbon could be gasified via the reverse Boudouard reaction in the following CO2 capture step, the deposited carbon is accumulated in the DRM process.
The DRM process was conducted at a high temperature (>800 °C) to avoid carbon deposition and achieve high conversions of CO2 and CH4. Nevertheless, carbon was still deposited on the surface of the Ni phase of the Ni/CaO catal-sorbents during DRM. Moreover, the CO2 capture capacity decreased gradually because of the thermal sintering of CaO at a high temperature, and this decreased the production of syngas in the subsequent DRM process. Here, coke-promoted Ni/CaO (C-Ni/CaO) catal-sorbents were developed through CH4 pretreatment, after which a highly efficient process of producing CO and syngas at a relatively low temperature was proposed. The effect of the CH4 treatment temperature on coke, which was deposited on the coke-promoted Ni/CaO catal-sorbents, was investigated in detail. Moreover, the C-Ni/CaO catal-sorbents were applied to hybrid CO and syngas production via CO2 and CH4 conversions during cyclic ICCU.
Experimental
Materials
Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O), and citric acid were purchased from Sigma-Aldrich. Ca(NO3)2·4H2O and Ni(NO3)2·6H2O were utilized as the precursors of CaO and Ni, respectively. Citric acid was employed as a gelling agent.Synthesis of the catal-sorbent
Desired amounts of Ca(NO3)2·4H2O, Ni(NO3)2·6H2O, and citric acid were dispersed in deionized water, and the mixture was continuously stirred for 1 h. The mixed solution was evaporated for 12 h in a drying oven at 100 °C until a dried gel was formed. The dried gel was calcined at 800 °C in a muffle furnace for 5 h in air with the temperature ramp rate of 10°C min−1. The resulting sample was denoted as a NiO/CaO catal-sorbent. Based on the ICP analysis, the Ni metal content in the NiO/CaO catal-sorbents was found to be 9.3 wt% (Table S1†).Pretreatment of the NiO/CaO catal-sorbent
Before the tests, the as-prepared NiO/CaO catal-sorbents were pretreated in H2 or CH4 to reduce NiO to Ni0. In the presence of 10% H2, the NiO/CaO catal-sorbents were treated at 500 °C for 2 h, which were denoted as Ni/CaO catal-sorbents. In the presence of 10% CH4, the NiO/CaO catal-sorbents were treated at 600, 650, and 700 °C for 2 h to deposit carbon on the surface of the Ni, which were denoted as C-Ni/CaO catal-sorbents.Characterization
The temperature-programmed reduction (TPR) profiles of the NiO/CaO catal-sorbents were obtained by 10% H2 and 10% CH4 treatments from 100 °C to 800 °C, respectively, at a temperature ramping rate of 10 °C min−1. The outlet gases were recorded via a gas chromatograph (GC) equipped with thermal conductivity detectors (TCD). The crystal structure of the catal-sorbents was analyzed with a Cu-Kα radiation source on a Phillips X'PERT X-ray diffractometer at the Korea Basic Science Institute, Daegu. The crystallite size of the metal phase was calculated by using the Scherrer equation. Raman spectroscopy was performed to determine the degree of graphitization in the C-Ni/CaO catal-sorbents. Derivative thermogravimetry-temperature-programmed oxidation (DTG-TPO) was conducted from 100 °C to 800 °C, with a temperature ramping rate of 10 °C min−1 to determine the type of coke species that was deposited in the C-Ni/CaO catal-sorbents. The crystalline phase was determined by field-emission transmission electron microscopy (FE-TEM, JEOL, JEM-2100F), which was performed at the Korea Basic Science Institute, Daegu.CO production and syngas production tests
The production of CO, and the subsequent production of syngas in the ICCU process, were determined by monitoring their concentrations via GC. The prepared catal-sorbent (0.5 g) was packed in a fixed-bed reactor with a diameter of ½-inch, and the reactor was placed in an electric furnace at atmospheric pressure. Prior to the tests, the Ni/CaO catal-sorbents were pretreated with CH4 as mentioned above. During each CO2 conversion and subsequent CH4 conversion process, the reaction temperature was maintained at 600, 650, and 700 °C. In the CO2 conversion step, 10 vol% CO2 and balance N2 were passed through the bed for 2 h at 40 mL min−1. When the CO2 concentration of the outlet gas matched that of the inlet CO2 feed gas, the gas composition was changed to pure N2 to purge CO2 from the reactor for 2 min. Afterwards, the gas composition was changed to 10 vol% CH4. During the subsequent conversion of CH4, the reaction temperature was maintained at 600 °C, 650 °C, and 700 °C. Moreover, N2 gas was employed as the standard and balance gas. To prevent the condensation of water vapor, the inlet and outlet lines of the reactor were maintained at temperatures greater than 100 °C employing a heating tape before the injection of gas into the reactor and GC column. The outlet gases were analyzed on a gas chromatograph (Agilent 6890), which was equipped with two TCDs. A Carboxen 1000 packed column was connected to one TCD to analyze the N2, CO2, and H2O gases; a Porapak Q packed column was connected to the other TCD to analyze the H2, N2, CO, CH4, and CO2 gases. The amount of coke deposited at each cycle was calculated using eqn (1)–(4)| Coke deposited in the CH4 treatment step (mmol g−1) = CH4 consumed − CO produced | (1) |
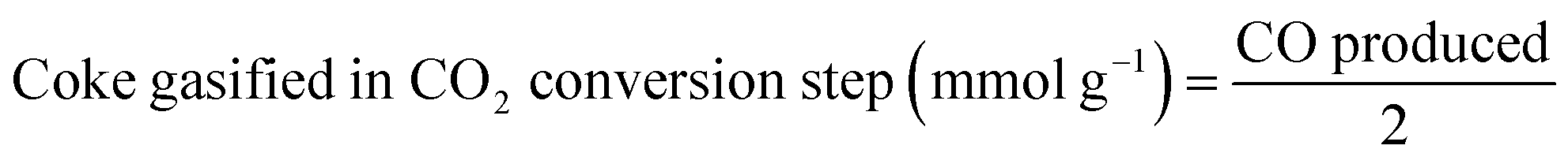 | (2) |
 | (3) |
| Coke accumulated (mmol g−1) = eqn (1) − eqn (2) + eqn (3) | (4) |
Results and discussion
Process description
We propose a cyclic production of CO and syngas in the ICCU process using the coke-promoted Ni/CaO (C-Ni/CaO) catal-sorbent as illustrated in Fig. 1; the main reactions that occurred in this process are summarized in Table 1. Regarding the CO2 conversion step, the reverse Boudouard reaction, CO2 splitting, and CO2 capture processes proceeded simultaneously with C-Ni/CaO to produce CO gas in a high yield while also capturing CO2: C-Ni/CaO(s) + CO2(g) ↔ NiO/CaCO3(s) + CO(g). Regarding the CH4 conversion step, the decomposition of CH4, partial oxidation of CH4, and DRM proceeded simultaneously in the presence of NiO/CaCO3 to produce syngas in a high yield while achieving the complete regeneration of CaCO3: NiO/CaCO3(s) + CH4(g) ↔ C-Ni/CaO(s) + CO(g) + H2(g). Given that the process proposed in this study outweighs a high CO and syngas production from CO2 and CH4 conversion as well as multicycle stability by suppressing the thermal sintering of Ni or CaO, the optimum CH4 pretreatment and reaction temperatures are further examined in this paper.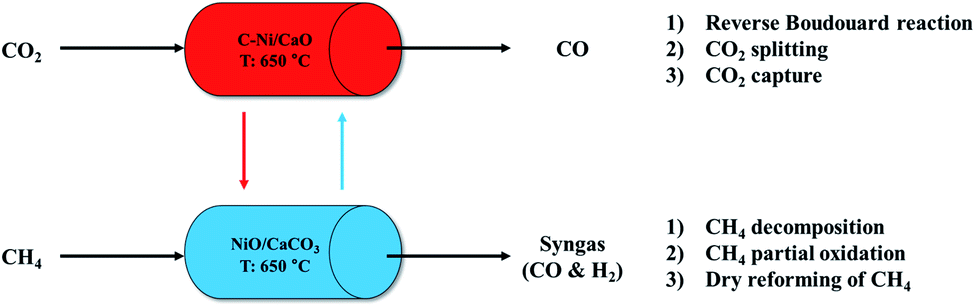 | ||
| Fig. 1 Schematics of CO and syngas production in the ICCU process utilizing C-Ni/CaO. | ||
| CO2 conversion | CH4 conversion |
|---|---|
| C(s) + CO2(g) ↔ 2CO(g) | NiO(s) + CH4(g) ↔ Ni0(s) + CO(g) + 2H2(g) |
| Ni0(s) + CO2(g) ↔ NiO(s) + CO(g) | CaCO3(s) + CH4(g) ↔ CaO(s) + 2CO(g) + 2H2(g) |
| CaO(s) + CO2(g) ↔ CaCO3(s) | CH4(g) ↔ C(s) + 2H2(g) |
Fig. 2 shows the TPR profiles of the NiO/CaO catal-sorbents under (a) 10% H2 and (b) 10% CH4 treatment conditions from 100 °C to 800 °C, with a temperature ramping rate of 10 °C min−1 after the pretreatment with N2 for 1 h at 200 °C to desorb the adsorbed gases. The H2 consumption curves of the NiO/CaO catal-sorbents exhibited two reduction peaks, which corresponded to the reducible NiO species in the NiO/CaO catal-sorbents: NiO(s) + H2(g) ↔ Ni0(s) + H2O(g).15,45 The lower and higher temperature peaks at 500 °C and 600 °C were attributed to the reductions of the free NiO and NiO interacting with the CaO support, respectively. During the reduction by CH4, CO was detected between 500 °C and 600 °C, which corresponded to the same reduction peaks observed under the H2 condition. CO and H2 were produced via the partial oxidation of CH4 employing NiO as an oxygen source: NiO(s) + CH4(g) ↔ Ni0(s) + CO(g) + 2H2(g).53 Notably, the H2/CO ratio was higher than the value (∼2) obtained from the partial oxidation of CH4, and the CH4 consumption and H2/CO ratio increased as the reaction temperature increased. The improved production of H2 during the reduction of CH4 could be attributed to the decomposition of CH4 at temperatures above 450 °C: CH4(g) ↔ C(s) + 2H2(g).51,54
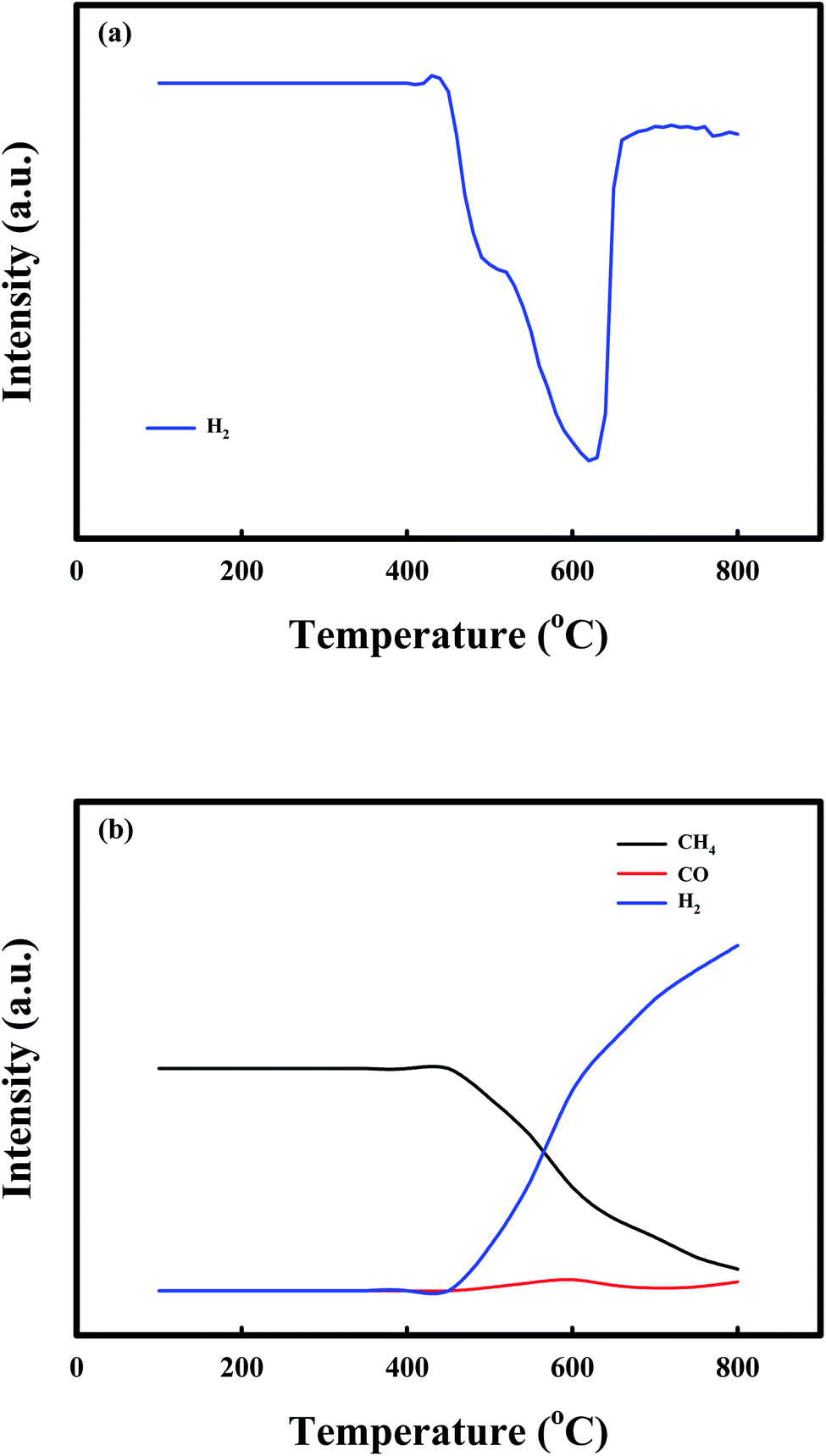 | ||
| Fig. 2 TPR profiles of the NiO/CaO catal-sorbents under the (a) 10% H2 and (b) 10% CH4 conditions from 100 to 800 °C at a temperature ramping rate of 10 °C min−1. | ||
Fig. 3 shows the XRD patterns of the catal-sorbents in the calcined (NiO/CaO) and reduced states employing H2 at 800 °C (Ni/CaO) or CH4 at 650 °C (C-Ni/CaO). The catal-sorbents exhibited sharp peaks corresponding to the CaO phase (JCPDS no. 48-1467) in all the states. In addition to the CaO phase, the XRD patterns of the catal-sorbents in the calcined state (NiO/CaO) contained the NiO phase (JCPDS no. 43-1003) with small peaks of the Ca(OH)2 phase (JCPDS no. 84-1263). The XRD patterns of the catal-sorbents after they were reduced by H2 at 800 °C (Ni/CaO) exhibited the metallic Ni phase (JCPDS no. 70-1849) without the NiO phase, indicating the complete reduction of NiO to Ni0. After they were reduced by CH4, deposited carbon phases and the Ni0 phase are observed. This result corresponds to the TPR results in Fig. 1. Almost no phase change was observed in the XRD patterns of the C-Ni/CaO samples that were reduced by CH4 at 600, 650, and 700 °C (Fig. S1†).
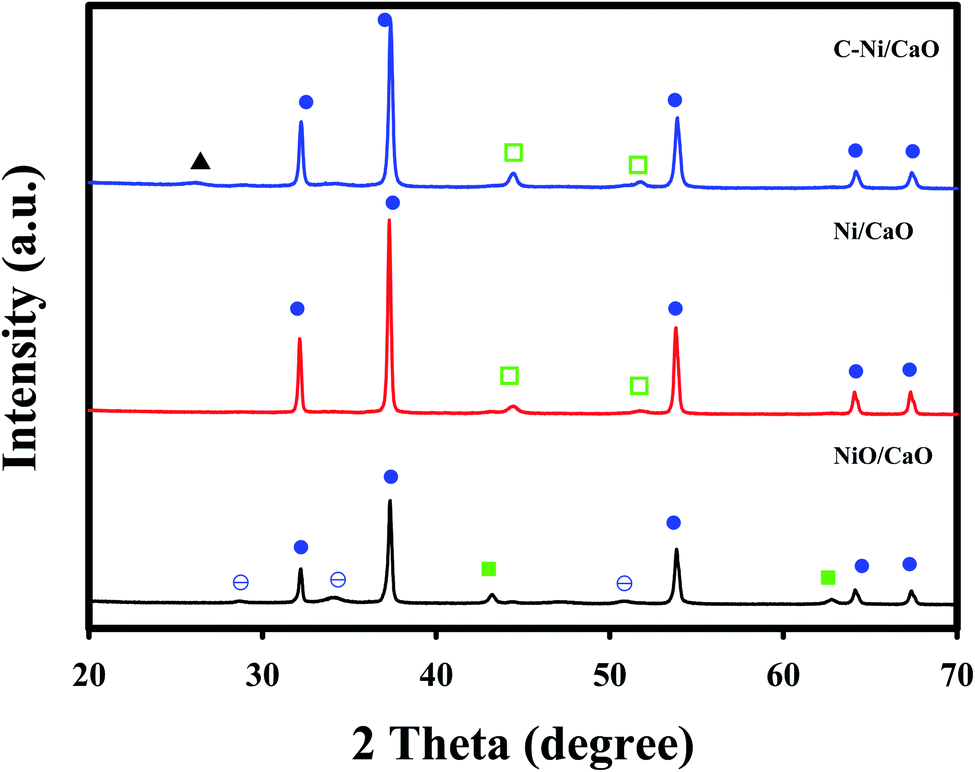 | ||
Fig. 3 XRD patterns of the Ni/CaO catal-sorbents in the calcined (NiO/CaO) and reduced states employing H2 at 800 °C (Ni/CaO) or CH4 at 650 °C (C-Ni/CaO): ( ) NiO, ( ) NiO, ( ) Ni0, ( ) Ni0, ( ) CaO, ( ) CaO, ( ) Ca(OH)2 and (▲) coke. ) Ca(OH)2 and (▲) coke. | ||
Raman spectroscopy, DTG-TPO, and TEM analyses afforded additional information on the deposition of coke during the reduction by CH4. The phase chemistry of the coke, which was deposited on the C-Ni/CaO, as revealed by Raman spectroscopy (Fig. 4a), exhibited two sharp characteristic bands in all the samples. The D band at 1340 cm−1 was ascribed to the presence of disordered or amorphous carbons, and the G band at 1580 cm−1 corresponded to the well-crystallized graphite phase.55–57 The ratios of the intensities of the D to G bands (ID/IG) of coke, which was deposited on the CH4-reduced Ni/CaO catal-sorbents at 600, 650, and 700 °C, were 1.01, 0.79, and 0.77, respectively. This indicated that the graphitization degrees of the carbon increased with increasing CH4 reduction temperatures. The DTG-TPO analyses of C-Ni/CaO at different CH4 reduction temperatures were performed to determine the type of coke that was deposited under air conditions (Fig. 4b). Two types of oxidation peaks were observed: a small one at ∼380 °C and a large one at 600–700 °C. Following the literature, the small peaks at a lower combustion temperature were attributable to the combustion of encapsulating coke with an amorphous nature, whereas the latter peaks at a higher temperature corresponded to the filamentous carbon.33,58 The DTG-TPO results indicated that the amorphous coke deposit decreased while the filamentous carbon increased with increasing CH4 reduction temperature; this is consistent with the Raman spectra. Fig. S2a† shows that the Ni particles with a crystallite size of 15–20 nm in the Ni/CaO sample were well dispersed in the NiO/CaO sample (Fig. S3†), and this corresponds to the crystallite size obtained by XRD (Fig. 3). After the CH4 treatment at 650 °C (Fig. S2b†), no change was observed in the crystallite size of the Ni particles, whereas the amorphous and filamentous coke deposits exhibited changes (Fig. S4†); these results correspond to the Raman and TPO results.
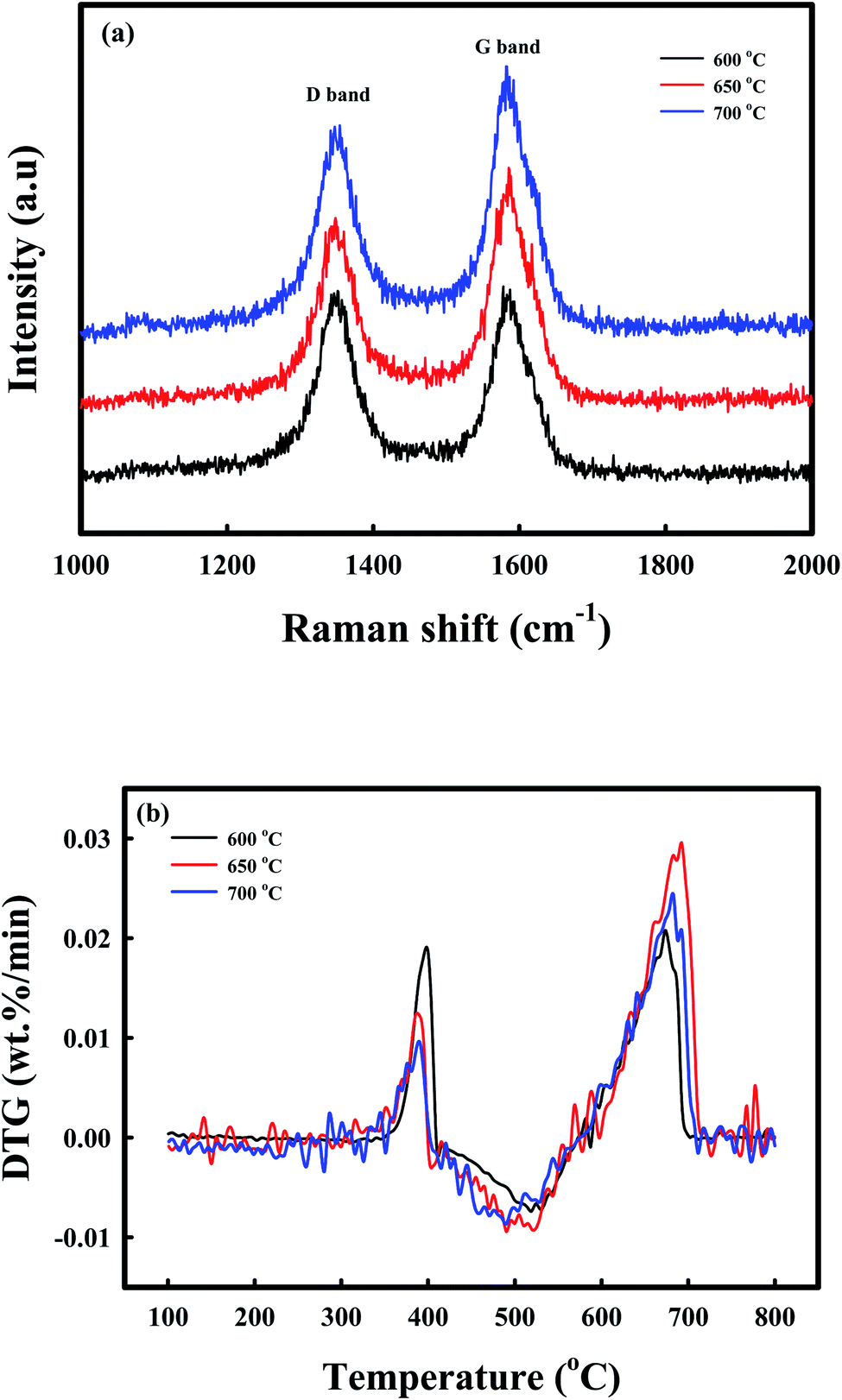 | ||
| Fig. 4 Coke characterization: (a) Raman spectra, (b) DTG-TPO of the C-Ni/CaO catal-sorbents that were reduced at different temperatures (600 °C, 650 °C, and 700 °C). | ||
Fig. 5 shows the CO2 consumption and CO production performances of the Ni/CaO and C-Ni/CaO catal-sorbents in the CO2 conversion step under the 10 vol% CO2 condition at 650 °C. The Ni/CaO and C-Ni/CaO catal-sorbents were prepared by pretreating the as-prepared NiO/CaO samples with H2 and CH4 at 800 °C and 650 °C, respectively. The total CO2 consumption capacity and CO productivity of the Ni/CaO catal-sorbent were 12.2 mmolCO2/g−1 and 1.05 mmolCO g−1, respectively (Fig. 5a). The metallic Ni in Ni/CaO reacted with CO2 to yield CO via CO2 splitting, as follows: Ni0(s) + CO2(g) ↔ NiO(s) + CO(g) (Fig. S5a†). However, most CO2 reacted with CaO and was stored as the CaCO3 phase (11.6 mmolCO2 g−1): CaO(s) + CO2(g) ↔ CaCO3(s). The conversion of CO2 decreased rapidly to zero after achieving complete CO2 capture and splitting (Fig. 5b and c). Conversely, the total CO2 consumption capacity and CO productivity of C-Ni/CaO were 30.2 mmolCO2 g−1 and 36.9 mmolCO g−1, respectively. Notably, the total CO2 consumption capacity and CO productivity of C-Ni/CaO were significantly improved via the reverse Boudouard reaction: C(s) + CO2(g) ↔ 2CO(g). Nevertheless, the carbon deposited in C-Ni/CaO are observed in XRD analysis after the CO2 conversion step (Fig. S5b†). The amount of CO produced from CO2 splitting employing Ni0 as a reductant was assumed to be insignificant. Additionally, the CO2 gas was also stored as CaCO3 (11.7 mmolCO2 g−1), which is the same as the amount of CO2 that was captured by the Ni/CaO catal-sorbent. Therefore, the simultaneous CO2 capture and reverse Boudouard reaction ensured that the C-Ni/CaO catal-sorbent maintained a high CO2 conversion, as well as CO mole fraction, at the early stage of the reaction (∼40 min). After saturation of CO2 capture, the amount of CO that was produced via the reverse Boudouard reaction was maintained, thus decreasing the CO2 conversion and mole fraction of CO (Fig. 5b and c). Although the reverse Boudouard reaction with increasing CH4 reduction temperatures increased the production of CO, C-Ni/CaO catal-sorbents exhibited the highest CO2 conversion and CO mole fraction at the early stage of the reaction at 650 °C (Fig. S6†). This is because CaO sorbents exhibited the highest CO2 capture performance at 650 °C. At 700 °C, however, the catal-sorbents did not show enhancement of CO2 conversion at the early stage of the reaction because the excess carbon deposition in the CH4 treatment step blocks the access of the CO2 to CaO phase. Owing to the unfavorable conditions, the thermodynamic equilibrium has revealed that CO2 splitting at a low temperature (<900 °C) could not achieve high CO production and complete CO2 conversion.53 However, C-Ni/CaO could achieve high CO2 conversion and the production of a large amount of CO at relatively low temperature via the reverse Boudouard reaction with CO2 capture: C-Ni0/CaO(s) + CO2(g) ↔ NiO/CaCO3(s) + CO(g).
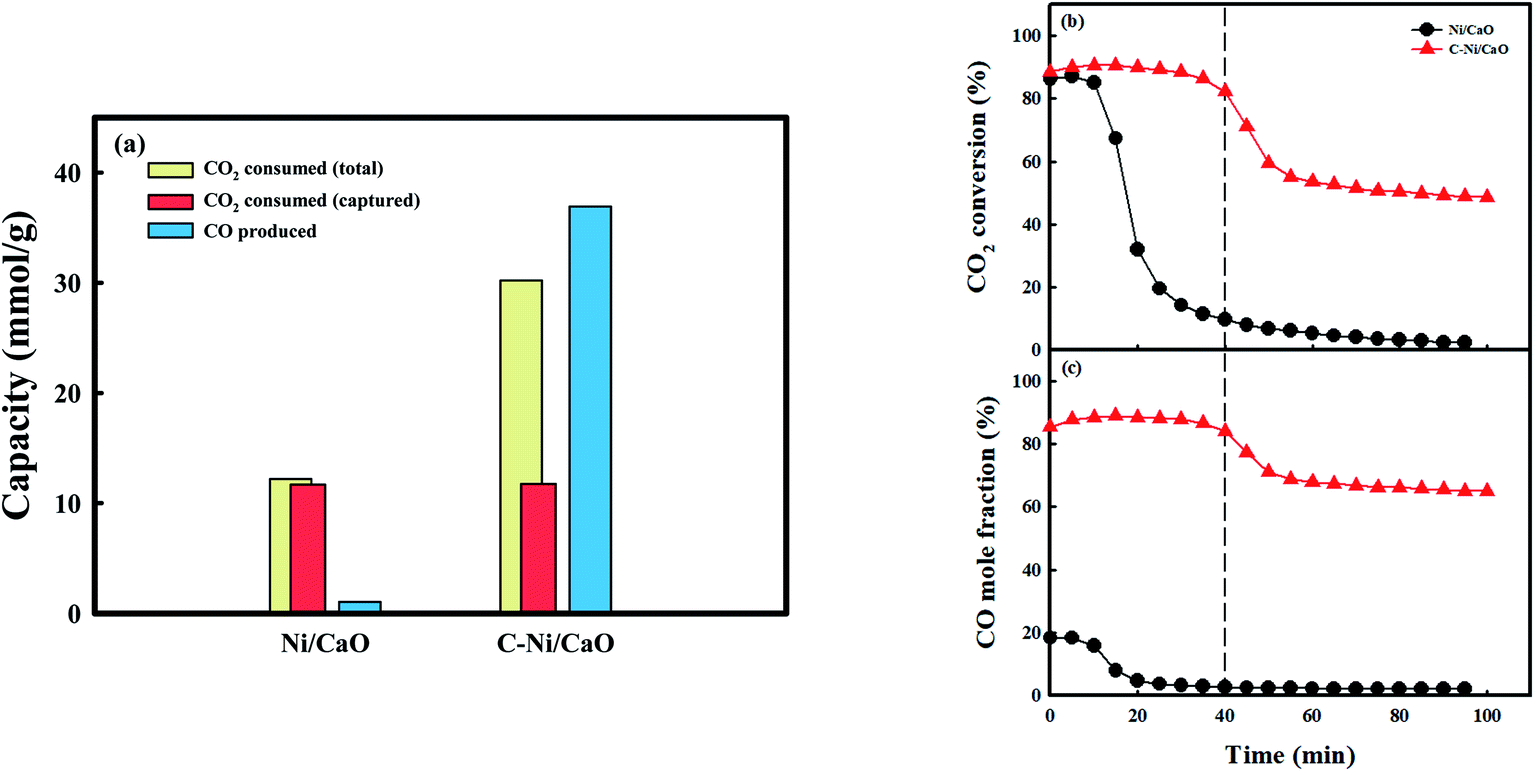 | ||
| Fig. 5 CO2 consumption and CO production performances in the CO2 conversion step: (a) CO2 consumption and CO production capacity, (b) CO2 conversion and (c) CO mole fraction obtained with Ni/CaO and C-Ni/CaO under the 10 vol% CO2 condition at 650 °C. | ||
Fig. 6 exhibits syngas productivity of the CH4 conversion step employing the as-prepared NiO/CaO and NiO/CaCO3 catal-sorbents under the 10 vol% CH4 condition at 650 °C. NiO/CaCO3 was obtained from C-Ni/CaO after the conversion of CO2 at 650 °C. The syngas productivities of NiO/CaO were 48.4 mmolH2 g−1 and 0.94 mmolCO g−1, respectively. A low amount of CO is produced via the partial oxidation of CH4 utilizing NiO as the oxygen source: NiO(s) + CH4(g) ↔ Ni(s) + CO(g) + 2H2(g) (Fig. S7a†). Contrarily, H2 was produced via the partial oxidation, as well as the decomposition, of CH4. The CH4 conversion performance decreased dramatically at the early stage of the reaction (∼40 min) after the partial oxidation of CH4 (reduction of nickel). Coke, which was deposited on the surface of nickel might contribute significantly to the gradual deactivation of CH4 conversion as the reaction proceeded, by blocking the access of the reactant gases to the catalytic sites.52,57 Conversely, the NiO/CaCO3 catal-sorbent shows higher syngas production (110.6 mmolH2 g−1 and 18.5 mmolCO g−1) and initial CH4 conversion compared with NiO/CaO. The improved CH4 conversion by NiO/CaCO3 at the early stage of the reaction (∼120 min) was due to DRM with CO2 that was stored as CaCO3, which produced syngas (CO and H2), as follows: CaCO3(s) + CH4(g) ↔ CaO(s) + 2CO(g) + 2H2(g) (Fig. S7b†). However, the NiO/CaCO3 catal-sorbent shows rapid deactivation at the early stage compared to NiO/CaO catal-sorbents because the high CO concentration from enhanced CH4 conversion promoted the Boudouard reaction: 2CO(g) ↔ C(s) + CO2(g). The amount of CO, which was produced by the partial oxidation of CH4 employing metallic NiO as an oxidant, was assumed to be negligible. Moreover, the conversion of CH4 decreased dramatically, and there was no observed production of CO after DRM; the production of H2via the decomposition of CH4 proceeded. As the reaction temperature increased, the conversion of CH4, as well as the production of H2, increased because of its favorable thermodynamics regarding the decomposition of CH4 (Fig. S8†). At 700 °C, the NiO/CaCO3 catal-sorbent shows a relatively stable CH4 conversion because the Boudouard reaction is unfavorable at temperatures >700 °C.15 However, the amount of CO produced from DRM at 700 °C was low because the amount of CO2 that was captured in the CO2 conversion step was the lowest. Therefore, improved CH4 conversion and syngas production were achieved via DRM and CH4 decomposition at 600–700 °C: C-Ni0/CaO(s) + CO2(g) ↔ NiO/CaCO3(s) + CO(g). Additionally, almost all the captured CO2 was utilized in the production of syngas, and the carbon source was supplied for the production of CO in the CO2 conversion step from the decomposition of CH4 at relatively low temperatures.
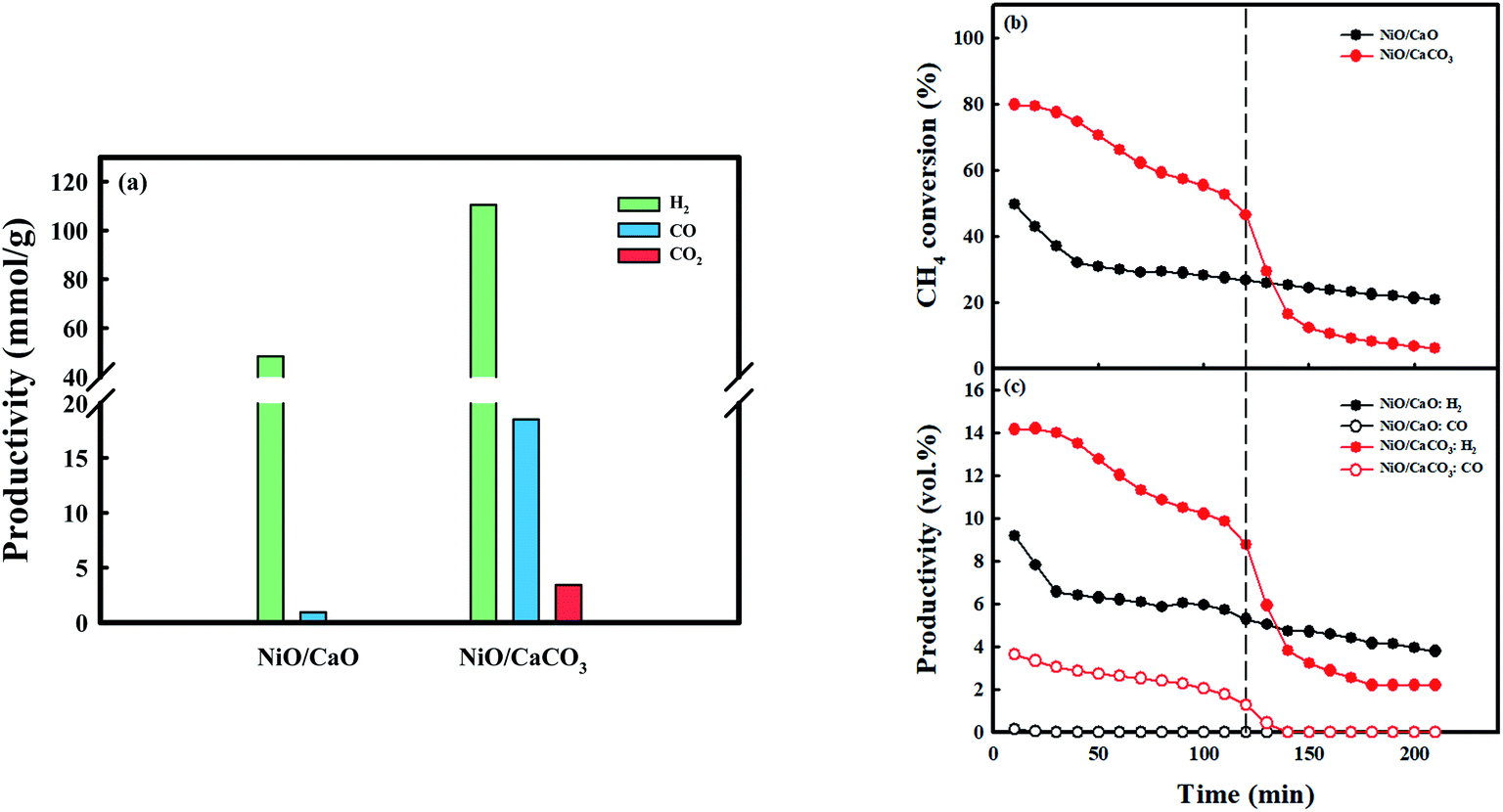 | ||
| Fig. 6 Syngas production performances in the CH4 conversion step: (a) syngas (H2 and CO) productivity and capacity for desorbing CO2, (b) CH4 conversion, and (c) syngas mole fraction obtained by utilizing NiO/CaO and NiO/CaCO3 under the 10 vol% CH4 condition at 650 °C. | ||
The long-term stability of C-Ni/CaO was also investigated in ten consecutive cycles of CO2 and CH4 conversions (CO2 conversion: 10 vol% CO2 for 2 h, and CH4 conversion: 10 vol% CH4 for 4 h at 650 °C) and these results are illustrated in Fig. 7a and summarized in Table S2.† As shown in Fig. 7a, CO production via coke gasification in the CO2 conversion step remained unchanged, whereas that via DRM in the CH4 conversion step decreased slightly. This is because the captured CO2 was not desorbed completely in the subsequent CH4 conversion step after the 5th cycle (Fig. 7b), leading to a decrease in CO2 capture capacity in the following cycles of CO2 conversion. This phenomenon might be due to the sintering of Ni0 and CaO/CaCO3 materials (Table S3†). Moreover, H2 productivity decreased (∼48.3 mmolH2 g−1) gradually and the amount of coke accumulated reached a maximum value (∼85.8 mmolC g−1) after 7 cycles of CH4 conversions and remained unchanged. During the multicycle stability tests, the C-Ni/CaO catal-sorbents exhibited excellent amount of CO and H2 productivity in cyclic CO2 and CH4 conversions after saturation of coke deposition. However, the Ni and CaO/CaO materials are sintered during the cycles, as observed by XRD (Fig. 7b and Table S3†), leading to decrease in performances of CO2 capture capacity and CO and H2 productivity. The combined H2/CO molar ratio that was obtained from the CO2 and CH4 conversion steps was constant (∼1.0). Additionally, the desired H2/CO ratios for producing value-added chemicals, e.g., liquid fuels, light olefins, and higher alcohol, could be controlled by changing the reaction time in the CH4 conversion step.
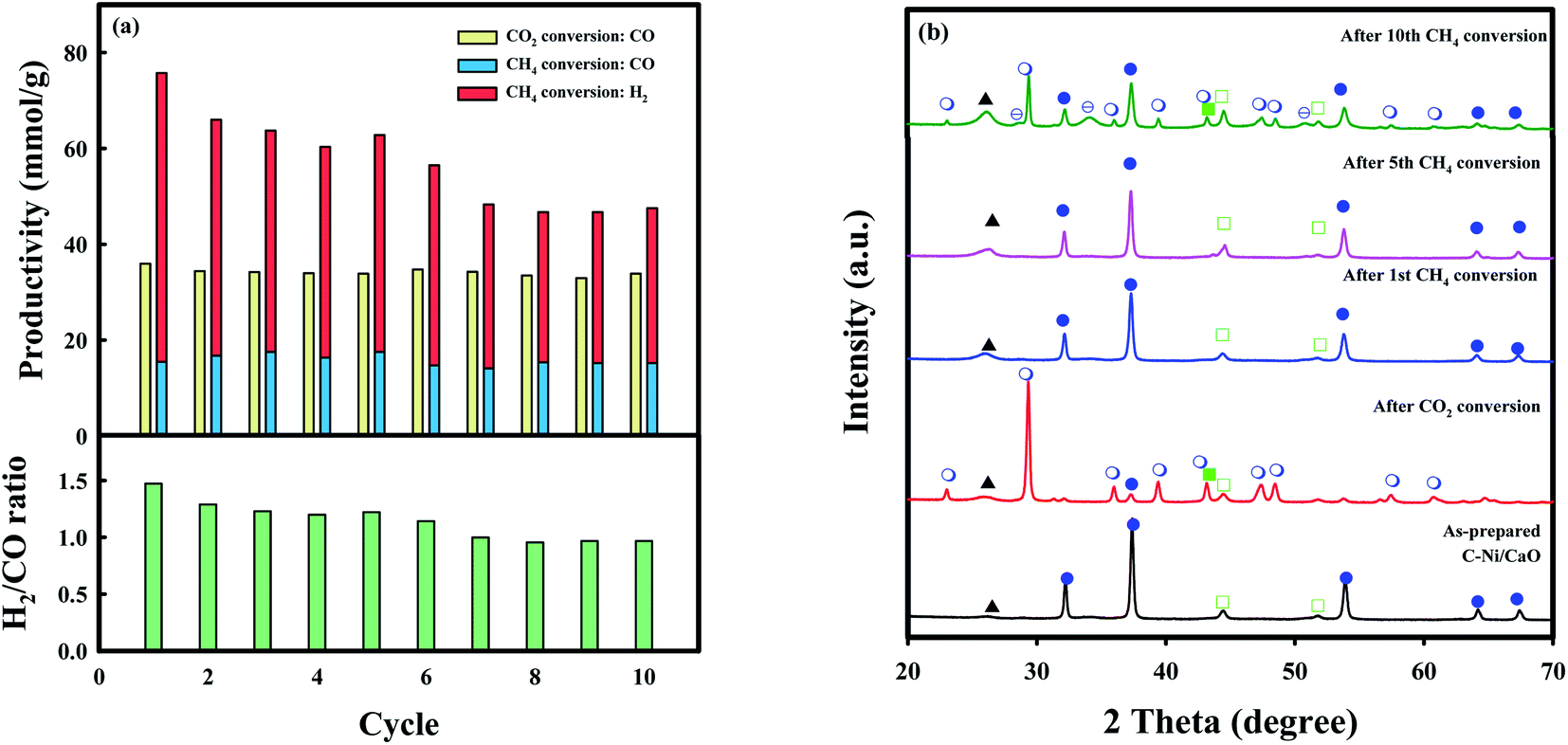 | ||
Fig. 7 Long-term stabilities of C-Ni/CaO in ten consecutive cycles of CO2 and CH4 conversions: (a) CO and syngas (H2 and CO) productivities in the CO2 and CH4 conversion steps, as well as the combined values of the H2/CO ratios of CO2 and CH4 conversions, respectively; (b) XRD patterns of C-Ni/CaO in ten consecutive cycles of CO2 and CH4 conversions (( ) NiO, ( ) NiO, ( ) Ni0, ( ) Ni0, ( ) CaO, ( ) CaO, ( ) CaCO3, ( ) CaCO3, ( ) Ca(OH)2 and (▲) coke). ) Ca(OH)2 and (▲) coke). | ||
Conclusions
Here, we proposed a highly efficient strategy for producing CO and syngas in a cyclic ICCU process employing C-Ni/CaO to reduce the total thermal energy and achieve a simplified process compared with that of conventional CCU. The C-Ni/CaO catal-sorbents were fabricated by the CH4 pretreatment of NiO/CaO. Further, they were applied to the production of CO and syngas in a cyclic ICCU process. In the CO2 conversion step, coke, which was deposited on the surface of the Ni catalysts, was gasified by CO2via the reverse Boudouard reaction, and the residual CO2 was simultaneously captured via CaO, thus enabling the high consumption of CO2, as well as the high production of CO. In the following CH4 conversion step, CaCO3 was regenerated, and the desorbed CO2 was converted into syngas by CH4. Moreover, the carbon source for the production of CO in the CO2 conversion step was the decomposition of CH4. The C-Ni/CaO catal-sorbent exhibited excellent performances of CO2 capture capacity, and CO and H2 productivities in consecutive CO2 and CH4 conversions. However, while CO productivity in the CO2 conversion step was constant, the coke-promoted Ni/CaO catal-sorbents were not completely regenerated in the CH4 conversion step after the 5th cycle. This might be due to the sintering of Ni0/NiO and CaO/CaCO3 materials and the excess amount of coke deposited on the surface of C-Ni/CaO catal-sorbents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea (No. 20182010600530).References
- D. Aaron and C. Tsouris, Sep. Sci. Technol., 2005, 40, 321–348 CrossRef CAS.
- D. J. Hofmann, J. H. Butler and P. P. Tans, Atmos. Environ., 2009, 43, 2084–2086 CrossRef CAS.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- Y. Zheng, W. Q. Zhang, Y. F. Li, J. Chen, B. Yu, J. C. Wang, L. Zhang and J. J. Zhang, Nano Energy, 2017, 40, 512–539 CrossRef CAS.
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- W. Li, H. Wang, X. Jiang, J. Zhu, Z. Liu, X. Guo and C. Song, RSC Adv., 2018, 8, 7651–7669 RSC.
- K. Arning, J. Offermann-van Heek, A. Sternberg, A. Bardow and M. Ziefle, Environ. Innov. Soc. Tr., 2020, 35, 292–308 CrossRef.
- H. J. Ho, A. Iizuka and E. Shibata, Ind. Eng. Chem. Res., 2019, 58, 8941–8954 CrossRef CAS.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- I. Martinez, G. Grasa, R. Murillo, B. Arias and J. C. Abanades, Energy Fuels, 2012, 26, 1432–1440 CrossRef CAS.
- A. Perejon, L. M. Romeo, Y. Lara, P. Lisbona, A. Martinez and J. M. Valverde, Appl. Energy, 2016, 162, 787–807 CrossRef CAS.
- H. J. Yoon and K. B. Lee, Chem. Eng. J., 2019, 355, 850–857 CrossRef CAS.
- A. Martinez, Y. Lara, P. Lisbona and L. M. Romeo, Environ. Sci. Technol., 2013, 47, 11335–11341 CrossRef CAS PubMed.
- I. Martinez, G. Grasa, R. Murillo, B. Arias and J. C. Abanades, Chem. Eng. J., 2013, 215, 174–181 CrossRef.
- S. Tian, F. Yan, Z. Zhang and J. Jiang, Sci. Adv., 2019, 5, eaav5077 CrossRef CAS PubMed.
- F. G. Wang, K. H. Han, W. S. Yu, L. Zhao, Y. Wang, X. J. Wang, H. Yu and W. D. Shi, ACS Appl. Mater. Interfaces, 2020, 12, 35022–35034 CrossRef CAS PubMed.
- K. H. Han, W. S. Yu, L. L. Xu, Z. Y. Deng, H. Yu and F. G. Wang, Fuel, 2021, 291, 120182 CrossRef CAS.
- T. G. de Araujo Moreira, J. F. S. de Carvalho Filho, Y. Carvalho, J. M. A. R. de Almeida, P. N. Romano and E. F. Sousa-Aguiar, Fuel, 2021, 287, 119536 CrossRef CAS.
- K. H. Han, S. Y. Xu, Y. Wang, S. Wang, L. Zhao, J. Kambonde, H. Yu, W. D. Shi and F. G. Wang, J. Power Sources, 2021, 506, 230232 CrossRef CAS.
- C. Anil, J. M. Modak and G. Madras, Mol. Catal., 2020, 484, 110805 CrossRef CAS.
- B. L. Han, L. Zhao, F. G. Wang, L. L. Xu, H. Yu, Y. Cui, J. M. Zhang and W. D. Shi, Ind. Eng. Chem. Res., 2020, 59, 13370–13379 CrossRef CAS.
- X. Tu, H. J. Gallon, M. V. Twigg, P. A. Gorry and J. C. Whitehead, J. Phys. D: Appl. Phys., 2011, 44, 274007 CrossRef.
- T. Yabe, K. Yamada, K. Murakami, K. Toko, K. Ito, T. Higo, S. Ogo and Y. Sekine, ACS Sustain. Chem. Eng., 2019, 7, 5690–5697 CrossRef CAS.
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, Fuel Process. Technol., 2017, 158, 96–103 CrossRef CAS.
- S. W. Wu, Y. Z. Li, Q. Q. Hu, J. C. Wu and Q. Zhang, ACS Sustain. Chem. Eng., 2021, 9, 11635–11651 CrossRef CAS.
- Z. C. Du, F. P. Pan, E. Sarnello, X. H. Feng, Y. Gang, T. Li and Y. Li, J. Phys. Chem. C, 2021, 125, 18684–18692 CrossRef CAS.
- A. A. Khan and M. Tahir, Appl. Catal. B: Environ., 2021, 285, 119777 CrossRef CAS.
- K. Shimura and H. Yoshida, Catal. Surv. Asia, 2014, 18, 24–33 CrossRef CAS.
- K. H. Han, Y. Wang, S. Wang, Q. Y. Liu, Z. Y. Deng and F. G. Wang, Chem. Eng. J., 2021, 421, 129989 CrossRef CAS.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- A. M. Ranjekar and G. D. Yadav, J. Indian Chem. Soc., 2021, 100002 CrossRef.
- I. V. Yentekakis, P. Panagiotopoulou and G. Artemakis, Appl. Catal. B: Environ., 2021, 120210 CrossRef CAS.
- Z. Alipour, M. Rezaei and F. Meshkani, J. Energy Chem., 2014, 23, 633–638 CrossRef.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. S. Hamill, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- W. J. Jang, J. O. Shim, H. M. Kim, S. Y. Yoo and H. S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- M. A. Arellano-Trevino, Z. Y. He, M. C. Libby and R. J. Farrauto, J. CO2 Util., 2019, 31, 143–151 CrossRef CAS.
- M. A. Arellano-Trevino, N. Kanani, C. W. Jeong-Potter and R. J. Farrauto, Chem. Eng. J., 2019, 375, 121953 CrossRef CAS.
- A. Bermejo-López, B. Pereda-Ayo, J. González-Marcos and J. González-Velasco, J. CO2 Util., 2019, 34, 576–587 CrossRef.
- L. F. Bobadilla, J. M. Riesco-Garcia, G. Penelas-Perez and A. Urakawa, J. CO2 Util., 2016, 14, 106–111 CrossRef CAS.
- M. Arellano, M. Duyar and R. Farrauto, Abstr. Pap., Jt. Conf. - Chem. Inst. Can. Am. Chem. Soc., 2015, 250, 370–376 Search PubMed.
- S. M. Kim, P. M. Abdala, M. Broda, D. Hosseini, C. Coperet and C. Muller, ACS Catal., 2018, 8, 2815–2823 CrossRef CAS.
- H. M. Sun, J. Q. Wang, J. H. Zhao, B. X. Shen, J. Shi, J. Huang and C. F. Wu, Appl. Catal. B: Environ., 2019, 244, 63–75 CrossRef CAS.
- S. X. Wang, E. T. Schrunk, H. Mahajan and R. J. Farrauto, Catalysts, 2017, 7, 88 CrossRef.
- Q. H. Zheng, R. Farrauto and A. C. Nguyen, Ind. Eng. Chem. Res., 2016, 55, 6768–6776 CrossRef CAS.
- S. B. Jo, J. H. Woo, J. H. Lee, T. Y. Kim, H. I. Kang, S. C. Lee and J. C. Kim, Sustainable Energy Fuels, 2020, 4, 4679–4687 RSC.
- J. Hu, P. Hongmanorom, P. Chirawatkul and S. Kawi, Chem. Eng. J., 2021, 130864 CrossRef CAS.
- A. Al-Mamoori, A. A. Rownaghi and F. Rezaei, ACS Sustain. Chem. Eng., 2018, 6, 13551–13561 CrossRef CAS.
- J. W. Hu, P. Hongmanorom, V. V. Galvita, Z. Li and S. Kawi, Appl. Catal. B: Environ., 2021, 284, 119734 CrossRef CAS.
- Q. Zheng, R. Farrauto and A. C. Nguyen, Ind. Eng. Chem. Res., 2016, 55, 6768–6776 CrossRef CAS.
- B. Shao, G. Hu, K. A. Alkebsi, G. Ye, X. Lin, W. Du, J. Hu, M. Wang, H. Liu and F. Qian, Energy Environ. Sci., 2021, 14, 2291–2301 RSC.
- S. B. Jo, J. H. Woo, J. H. Lee, T. Y. Kim, H. I. Kang, S. C. Lee and J. C. Kim, Sustainable Energy Fuels, 2020, 4, 5543–5549 RSC.
- K. Y. Koo, S.-h. Lee, U. H. Jung, H.-S. Roh and W. L. Yoon, Fuel Process. Technol., 2014, 119, 151–157 CrossRef CAS.
- J. S. Zhang, V. Haribal and F. X. Li, Sci. Adv., 2017, 3, e1701184 CrossRef PubMed.
- J. Ashok, M. Subrahmanyam and A. Venugopal, Int. J. Hydrogen Energy, 2008, 33, 2704–2713 CrossRef CAS.
- S. Mahamulkar, K. H. Yin, R. J. Davis, H. Shibata, A. Malek, C. W. Jones and P. K. Agrawal, Ind. Eng. Chem. Res., 2016, 55, 5271–5278 CrossRef CAS.
- K. Yin, R. J. Davis, S. Mahamulkar, C. W. Jones, P. Agrawal, H. Shibata and A. Malek, AIChE J., 2017, 63, 725–738 CrossRef CAS.
- X. Y. Zhao, Y. Cao, H. R. Li, J. P. Zhang, L. Y. Shi and D. S. Zhang, RSC Adv., 2017, 7, 4735–4745 RSC.
- C. T. Zhang, X. Hu, Z. M. Zhang, L. J. Zhang, D. H. Dong, G. G. Gao, R. Westerhof and S. S. A. Syed-Hassan, Fuel, 2018, 227, 307–324 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01136g |
| ‡ Seongbin Jo and Jong Heon Lee contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |