
Understanding the constant-voltage fast-charging process using a high-rate Ni-rich cathode material for lithium-ion batteries†
Kyojin
Ku
 *ab,
Seoung-Bum
Son
c,
Jihyeon
Gim
c,
Jehee
Park
c,
Yujia
Liang
a,
Anthony
Stark
a,
Eungje
Lee
c and
Joseph
Libera
*a
*ab,
Seoung-Bum
Son
c,
Jihyeon
Gim
c,
Jehee
Park
c,
Yujia
Liang
a,
Anthony
Stark
a,
Eungje
Lee
c and
Joseph
Libera
*a
aMaterials Engineering Research Facility, Applied Materials Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: kjku@hanbat.ac.kr; jlibera@anl.gov
bDepartment of Materials Science and Engineering, Hanbat National University, 125 Dongseodaero, Yuseong-Gu, Daejeon, 34158, Republic of Korea
cElectrochemical Energy Storage Department, Chemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 13th December 2021
Abstract
Fast-charging of lithium-ion batteries is a critical requirement for wider adoption of electric vehicles. However, it is subject to several difficulties, such as inhomogeneous delithiation, local heating, and lithium plating. Various charging protocols have been developed to resolve these problems, most of which comprise the combinations of constant-current (CC) and constant-voltage (CV) charging steps. Although it is one of the two main components in conventional charging protocols, CV charging is usually applied in a restrictive manner only to supplement CC charging, and its potential implications as a fast-charging protocol have not yet been fully explored. Herein, we study the effects of a CV-only charging protocol on the fast-charging efficiency of high-rate LiNi0.8Co0.1Mn0.1O2 cathode particles prepared by ultrasonic spray pyrolysis. A 15 minute full-charging is achieved by a single CV charging step without a significant capacity loss in the early cycles. However, gradual degradation of specific capacity and charging efficiency was observed at later cycles. Based on electro- and physico-chemical analysis data, we propose a two-step CV charging protocol that can improve the cycle stability alleviating the damage to the cathode particles. By revisiting CV charging protocols, this work provides a better understanding of the CV charging process that can contribute to the design of new charging protocols with advanced fast chargeability.
1. Introduction
Lithium-ion batteries (LIBs), the main energy storage systems for electric vehicles (EVs), are key technologies that will change the market share of the automotive industry in the near future.1,2 A critical factor that retards the growth of the EV market is the sluggish charging of LIBs, which takes much longer than refueling an internal combustion engine vehicle.3–5 Although fast charging of LIBs would provide more opportunities in the EV market, there are still questions to resolve in order to achieve fast charging. Lithium plating has been recognized as a major problem in fast charging: lithium metal formation on the surface of the anode can result in dendrites or even internal short circuits, which may cause serious safety issues.5–8 Although less attention has been paid to the effects of fast charging on cathode materials, their vulnerability has been noted as well.4,9,10 Promising cathode candidates for EVs, such as high-nickel LiNixCoyMnzO2 (NCMs) or lithium-rich layered oxides that aim to deliver high energy density, have problems including gas evolution, crack generation, and voltage decay that are directly related to cycle stability and safety of LIBs.11–15 Fast charging, furthermore, causes inhomogeneity, over-charge, and heat generation in electrode materials, which aggravates problems originating from cathode materials. Therefore, a fast-charging protocol should be carefully devised considering the material properties and the battery cell characteristics.Conventional LIB charging process is conducted by CCCV type protocols where the constant current (CC) charging step is supplemented by the constant voltage (CV) step at the end of the charging process. During the CV step in CCCV charging, as the applied current density and associated cell impedance gradually decrease, the extra capacity that was not accessible during the CC step is recovered, and any concentration gradient of lithium ions in active cathode particles is relaxed. Nevertheless, such a conventional CCCV charging protocol is not effective enough for fast charging and thus various modifications have been made to improve the fast charging capability.3,5,16 Anseán et al. proposed a multistage charging protocol consisting of two high- and low-current CC charges and one CV charge, which allows a LIB to charge in about 20 minutes.17 Notten et al. suggested boost charging, the combination of two high and normal CCCV protocols, which reduces total charging time as a large amount of capacities are delivered during the initial part of CCCV charging.18 Pulse charging, which is based on the repeated cycles of a short period of high-current CC charge and followed rest step, is another way to achieve fast charging while it suppresses a build-up of stresses and polarizations in electrode materials.19,20 Recent works also use machine learning or modeling to efficiently discover new charging protocols.16,21 Despite such wide variations, most of the modified fast-charging protocols use CC charge as the main charging method and CV charge only supplementary because of concerns about the initial surge of current density, which could be deleterious to the cell performances.18 CV charging, thus, is usually applied coupled with CC charging by which the current density changes in a much controllable manner, leading to the lack of understanding of the CV charging process.
In this work, we study the CV-based fast-chargeability of high-rate LiNi0.8Co0.1Mn0.1O2 (NCM811) synthesized by the mass scalable ultrasonic spray pyrolysis (USP) method. Less than 15 minutes were required to fully charge the LIB using a single CV-only charge step, and the following discharge profile was identical to that of normal CCCV-charged cells. However, gradual degradation of specific capacity and charging efficiency was observed at later cycles. Based on electro- and physico-chemical analyses data, we propose a two-step CV-only charging protocol that can better preserve fast chargeability with improved cycle stability. This study on CV-only charging without CC charging shows how CV charging affects cathode materials and electrochemical behaviors, which provides insights for designing an advanced fast-charging protocol.
2. Results and discussion
The precursor for NCM811 was obtained using the USP method. As it was reported previously as “reactive spray technology” from Cabot,22 the same instrument, which we call USP herein, provides several advantages for synthesizing cathode materials. In addition to mass scalability (Fig. S1†), USP is cost-effective and solvent waste-free. Unlike the conventional cathode synthesis process that requires a separate high-temperature reaction of lithium and transition metal (TM) precursors, the USP precursors already contain homogenously mixed lithium and transition metals, providing additional advantages for materials design and production. Using USP, we produced NCM811 precursor powder having cation disordered rock-salt structure, which indicates that lithium and all TMs are finely mixed (Fig. 1a). It exhibited a spherical morphology with some hollow structures, and particle size ranged from 1 to 3 μm (Fig. 1b, S2a, and S2b†). Layer structured NMC811 cathode materials (USP-NMC811) were obtained by annealing the NMC811 precursor at 775 °C (Fig. 1a). The morphology of annealed particles shows random agglomerations of ∼200 nm sintered primary particles (Fig. 1c, S2c, and S2d†).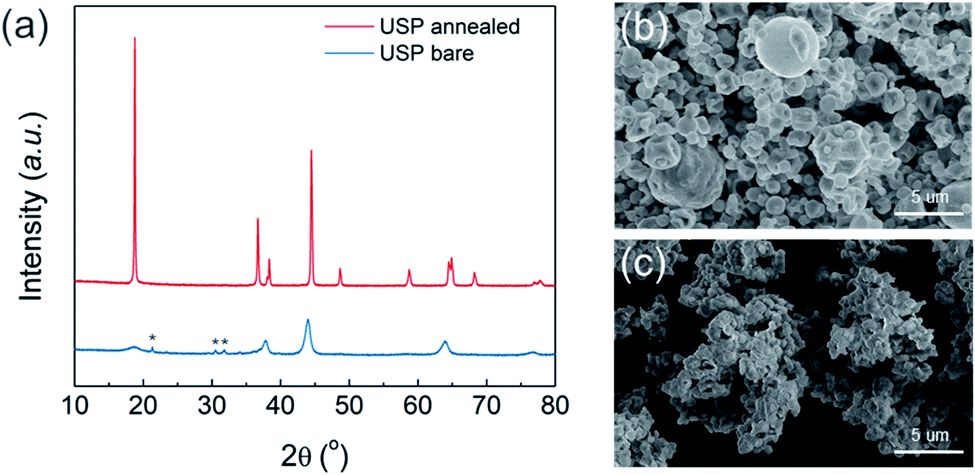 | ||
| Fig. 1 (a) X-ray diffraction (XRD) of NCM811 precursor (blue line) and annealed powders (red line) using USP. Asterisks present lithium carbonate impurities. Scanning electron microscopy (SEM) images of (b) USP precursor and (c) annealed powders. | ||
The USP-NCM811 cathode delivered a high capacity with an impressive rate capability. Fig. 2 shows the half cell performance conducted by a CCCV charing and CC discharging cycle protocol. During the two CCCV charge and CC discharge cycles at C/10, it exhibited an initial discharge capacity of 208 mA h g−1 (Fig. 2a). Moreover, it provided superior rate capability, where 154 mA h g−1 of capacity was obtained at 10C, almost 75% of 0.1C capacity (Fig. 2b). A Ragone plot in Fig. 2c illustrates the high rate property of NCM811 from USP. It delivers a high power density, ∼7151 W kg−1, at an energy density of 558 W h kg−1, which indicates that the rate capability of USP-NCM811 is comparable to or even better than that of NCM811 synthesized from the optimized co-precipitation method.23,24 The high rate capability of USP-NCM811 originates from a small primary particle size with a porous particle morphology, where a short lithium diffusion path and sufficient contact between active materials and electrolytes are provided.22
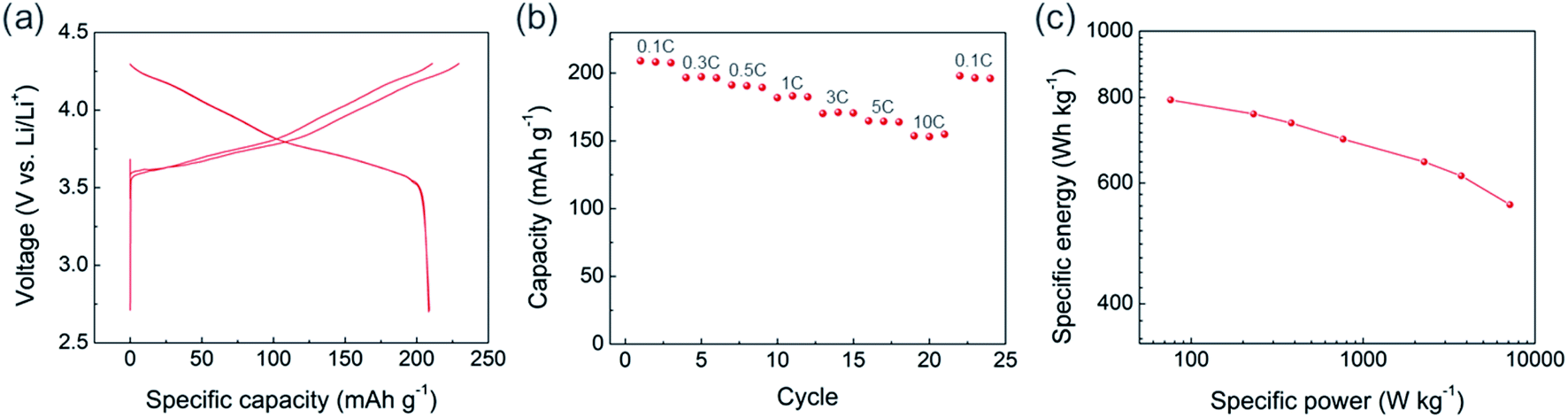 | ||
| Fig. 2 (a) Voltage profile of USP-NCM811 for initial 2 cycles. The cell was CCCV charged and CC discharged at 0.1C with a voltage range 2.7 to 4.3 V and a current cutoff of CV at 0.05C. (b) Rate performance and (c) Ragone plot of USP-NCM811. | ||
Using the high-rate USP-NCM811 cathode, we evaluated the CV charging capability and its effect on electrochemical behavior. CV charging was performed at 4.3 V, while the charging was set to end at a current of 0.05C. In Fig. 3a, the initial CV charging process is completed only in 14 minutes with a charge capacity of 228 mA h g−1, which is similar to 231 mA h g−1 from the CCCV charging case. Interestingly, in spite of the fast charging, the discharge profile after CV charging is almost the same as that after CCCV charging case. Fig. 3b shows that an extremely high current density is applied during the early period of CV charging and the current density rapidly diminishes (see blue dotted line). About 95% of the total charge capacity is obtained only in 5 minutes of the initial CV charging step. CC-only fast charging of USP-NCM811 was also conducted at 2C for comparison (Fig. S3†). In contrast to the CV-charging data, CC-charging results in 206 mA h g−1 initial charge capacity, which is 22 mA h g−1 less than that for CV-charging, as the charge profile encounters a cut-off voltage at an earlier SOC. This highlights the effectiveness of CV-charging as a fast-charging protocol and demonstrates that the high-current region of CV charging does not significantly alter the electrochemistry, at least during the initial cycles.
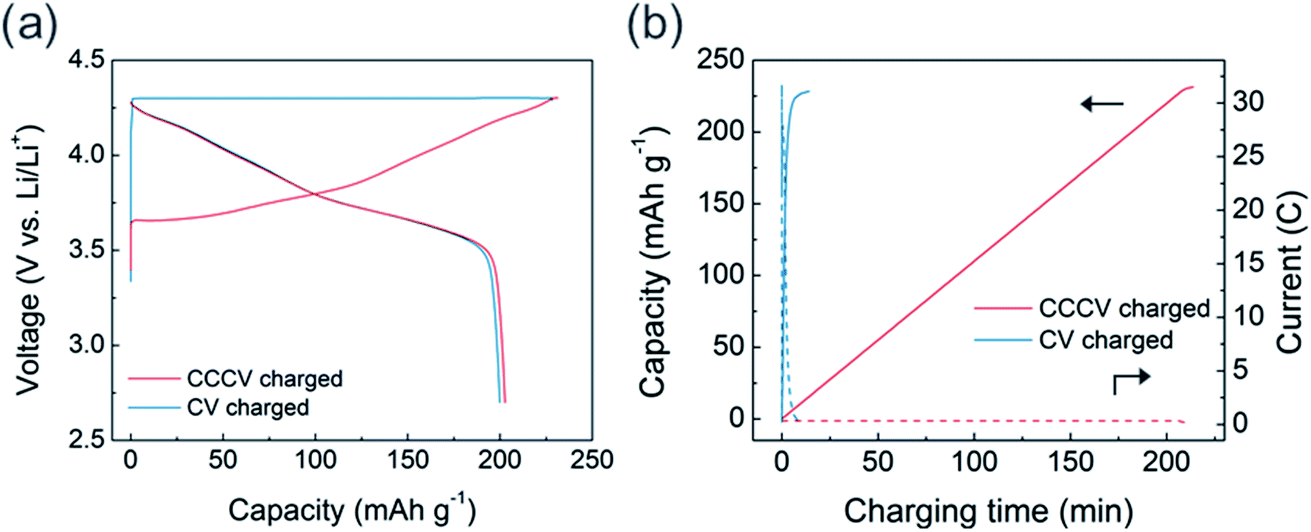 | ||
| Fig. 3 (a) Voltage profile of USP-NCM811 using CCCV (red line) and CV (blue line) charging protocol. (b) Capacity (solid line) and current (dotted line) profile with charging time. | ||
Although fast charging with sufficient charge–discharge capacity can be achieved in the initial cycles, the CV-only charging protocol appears to aggravate the capacity decay in the extended cycles. Fig. S4† illustrates the cycle stabilities of CV and CCCV charged cells in which all charged cells were discharged at 0.33C. After 50 cycles, the CCCV charged cell maintains 88% of its initial capacity, whereas the CV charged cell shows 74% capacity retention. We also observed that the fast chargeability of USP-NMC811 continuously decreases as the CV-charging cycle progresses. Fig. 4a clearly shows the cycle-to-cycle increase in charging time (gray arrow) and decrease in charge capacity (red arrow). In Fig. S5,† charging time gradually increases from 14 minutes to about 100 minutes throughout 40 cycles. To understand the relation between charging time and capacity, the current response (in C-rate) is plotted as a function of CV charging time for every 10 cycles in Fig. 4b and S6.† For the second cycle, the applied current density sharply rises up to 42C (=8.4 A g−1) at the start of the CV charging and continuously drops within several minutes. Note that such a high current density is not usually achievable by CC-only charging because the responding cell voltage will instantaneously shoot up higher than the cut-off voltage resulting in premature termination of the charging step. The peak C-rate gradually decreases on progressive cycling, and drops to 12C (=2.4 A g−1) at the 50th cycle. The reduction in the peak C-rate means more amounts of charge capacity are to be delivered at the later part of a CV charging where a lower C-rate is applied, extending the overall charging time. Fig. 4c illustrates the changes in the portion of charging capacity generated at different C-rates. While the capacities from the high C-rate region (>15C) gradually decreases to almost 0% at the 40th cycle, the capacities from the low (<5C) C-rate regions kept increasing. The capacity from the middle C-rate region constantly increases until 40th cycle and starts to diminish at 50th cycles. Hence, the charging time increased progressively as a larger portion of the charging reaction occurred at the low C-rate region with cycling.
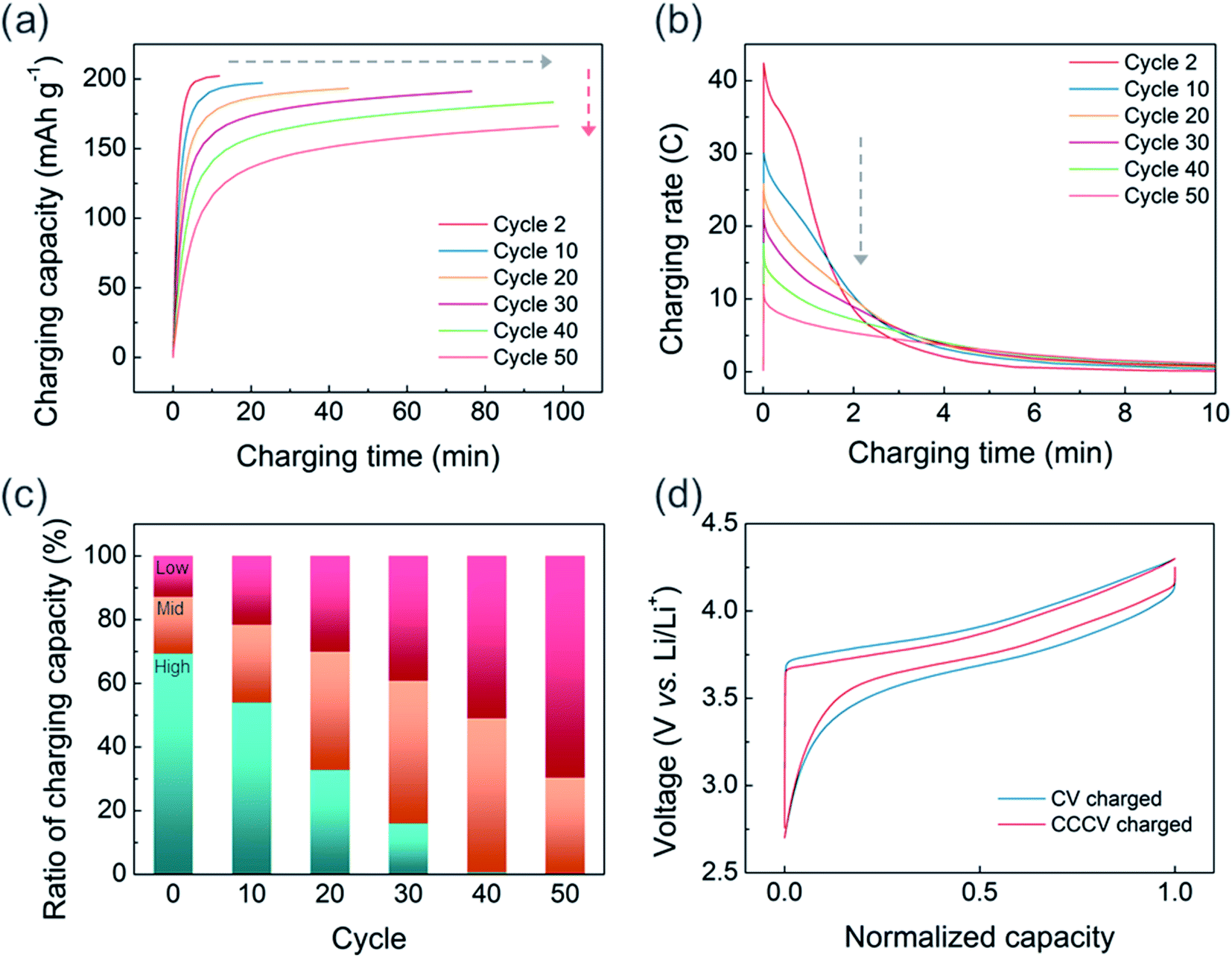 | ||
| Fig. 4 (a) Charging capacity profile and (b) charging rate profile of CV charging every 10 cycles. (c) Ratio of charging capacity delivered from high (>15C, blue), middle (orange) and low (<5C, red) C-rate. (d) Voltage profile with normalized capacity after 50 cycles of CV charging (blue line) and CCCV charging (red line). | ||
The origin of extended CV-charging time with cycles, accompanied by reduced peak C-rate, is the degradation of the NCM811 electrode with CV-charging cycles. Fig. 4d compares the normalized voltage profiles (CC charge/discharge at C/10) recorded after 50 CV- vs. CCCV-charging cycles. The charge–discharge curve for the cell experiencing CV charging exhibits a larger voltage gap between the charge and discharge profiles indicating a larger cell impedance. To eliminate the effects of the cycled lithium metal, we disassembled the CV-cycled NCM811 electrode and reassembled it with fresh lithium metal and a fresh electrolyte (Fig. S7†). However, the increased voltage hysteresis was still retained at the reassembled coin-cell; this demonstrates that the increase in voltage hysteresis is due to the deterioration of the NCM811 electrodes.
We found more prominent degradation from the surface region compared to the bulk of the particles. Fig. S8a† shows no noticeable differences in the XRD patterns for the cycled electrodes collected after 50 cycles of CV- vs. CCCV-charging suggesting the effect of CV-charging on the bulk structure is not particularly detrimental. On the other hand, X-ray photoelectron spectroscopy (XPS) reveals severe surface deterioration for the electrode cycled with CV-charging protocol. Fig. 5 illustrates the XPS spectra of F 1s, C 1s and O 1s measured from pristine (green), CCCV-charging (red), and CV-charging cycled (blue) electrodes. In Fig. 5a and S9a,† besides the sharp peak at 688 eV from the C–F bonding of polyvinylidene fluoride (PVDF) binder at the pristine electrode, the Li–F peak at 685.5 eV and the LixPFyOz peak at 686.7 eV arise from the cycled electrodes. The higher intensity ratio of [Li–F + LixPFyOz] to [C–F] for the CV-cycled electrode than that for the CCCV-cycled electrode implies that a larger amount of electrolyte decomposition products are covering the CV-cycled electrode.25,26 In Fig. 5b, the pristine electrode shows three sharp C1s XPS peaks at 284.7 eV (C–C from conductive carbon), 286.5 eV (C–H from PVDF), and 290.9 eV (C–F from PVDF). As in Fig. 5a, the overall peak intensities from PVDF and conductive carbons are reduced at the cycled electrodes. The peaks from various electrolyte decomposition products overlap in the region marked with orange box, and the deconvolution of the region (Fig. S9b†) indicates that C–O (285.7 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.0 eV) functionalities coexist where the intensities of the region are also higher and broader in CV-cycled electrodes than CCCV-cycled electrodes. The same trend is observed in Fig. 5c for O 1s spectra. The pristine electrode shows two main peaks from lattice oxygen, O–TM (529.4 eV), and surface oxygen, CO3 (531.9 eV) but they disappear in the cycled electrodes. Instead, new peaks from C–O (534.4 eV) and CO (532.8 eV) appear broadly with higher intensity at the CV-cycled electrode. XPS results indicate that new chemical substances, composed of LiF and various organic compounds, accumulated on the cycled electrode. This substance seems to be much thicker in CV-cycled electrodes. The observed surface chemical compounds originate from the oxidation or decomposition of electrolyte salt and solvents, which demonstrates that the extremely high C-rate in CV charging leads to the inhomogeneous charging reaction or oxidation of electrolytes, facilitating the formation of the thick and insulating surface layer.25,26
O (288.0 eV) functionalities coexist where the intensities of the region are also higher and broader in CV-cycled electrodes than CCCV-cycled electrodes. The same trend is observed in Fig. 5c for O 1s spectra. The pristine electrode shows two main peaks from lattice oxygen, O–TM (529.4 eV), and surface oxygen, CO3 (531.9 eV) but they disappear in the cycled electrodes. Instead, new peaks from C–O (534.4 eV) and CO (532.8 eV) appear broadly with higher intensity at the CV-cycled electrode. XPS results indicate that new chemical substances, composed of LiF and various organic compounds, accumulated on the cycled electrode. This substance seems to be much thicker in CV-cycled electrodes. The observed surface chemical compounds originate from the oxidation or decomposition of electrolyte salt and solvents, which demonstrates that the extremely high C-rate in CV charging leads to the inhomogeneous charging reaction or oxidation of electrolytes, facilitating the formation of the thick and insulating surface layer.25,26
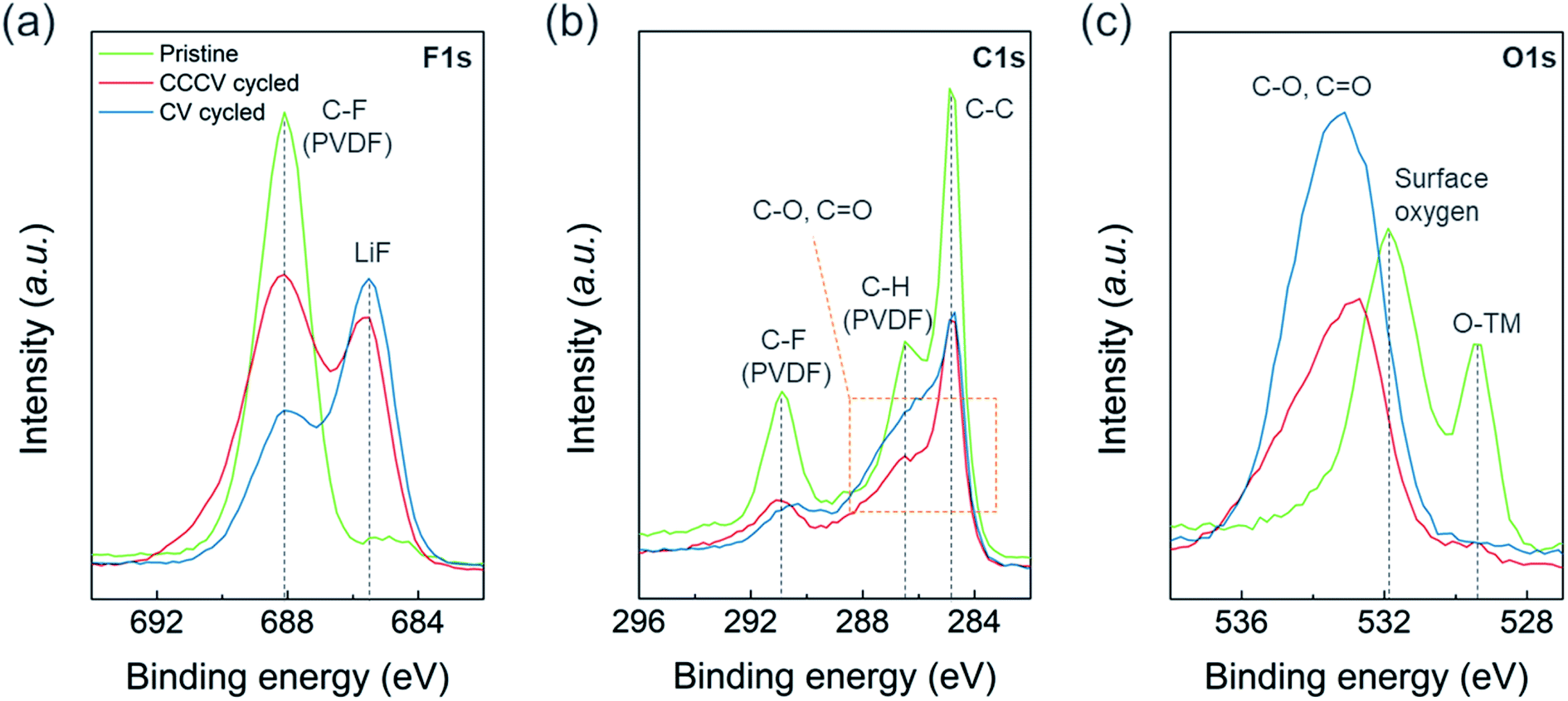 | ||
| Fig. 5 XPS spectra of (a) F 1s, (b) C 1s, and (c) O 1s regions measured from pristine (green lines), CCCV-cycled (red lines), and CV-cycled (blue lines) electrodes. | ||
Effect of surface degradation onto the electrochemical performance was also evaluated by electrochemical impedance spectroscopy (EIS) analyses after 50 cycles (Fig. S8b†). The equivalent circuit, shown in the inset of Fig. S8b,† is used for the analyses, where each term represents ohmic resistance (Rs), solid electrolyte interface (SEI) film resistance (Rf), charge transfer resistance (Rct), and Warburg impedance (W).26–28 A CV-cycled cell delivers 4.5 times and 2.4 times higher values of Rs and Rf, respectively, compared to a CCCV-cycled cell. As observed in the XPS analysis, significant surface reactions, such as the formation of an insulating SEI layer, during the CV charging cycle contribute to greater increases in ohmic and SEI film resistance. It also causes higher charge transfer resistance via forming a more resistive SEI layer and a deteriorated surface structure on the particle. Therefore, in order to minimize critical surface side reactions, the extremely high C-rate region should be avoided during fast-charging.
Based on these findings, a two-step CV charging protocol that includes a lower voltage CV pre-charging step is proposed (Fig. 6). Because the main purpose of pre-CV charging is to avoid extremely high current during the initial stages of 4.3 V CV charging while maintaining fast chargeability, we applied pre-CV charging for only 5 minutes at 4 V (Fig. 6a). About 58% of charging capacity was delivered from the 4 V charging region during the first cycle, and the following discharge profile matched well with those of the 4.3 V one-step CV-charged cell and the CCCV-charged cell (Fig. 3a). After pre-charging, the peak C-rate at 4.3 V was reduced to 15C, which is about one-third of the peak C-rate from one-step CV charging (Fig. 6b). Moreover, compared to the one-step CV charged cell where only 28% of the initial peak C-rate was retained after 50 cycles, the degradation rate was mitigated in the two-step CV-charged cell to that of 50% of CV charged cell.
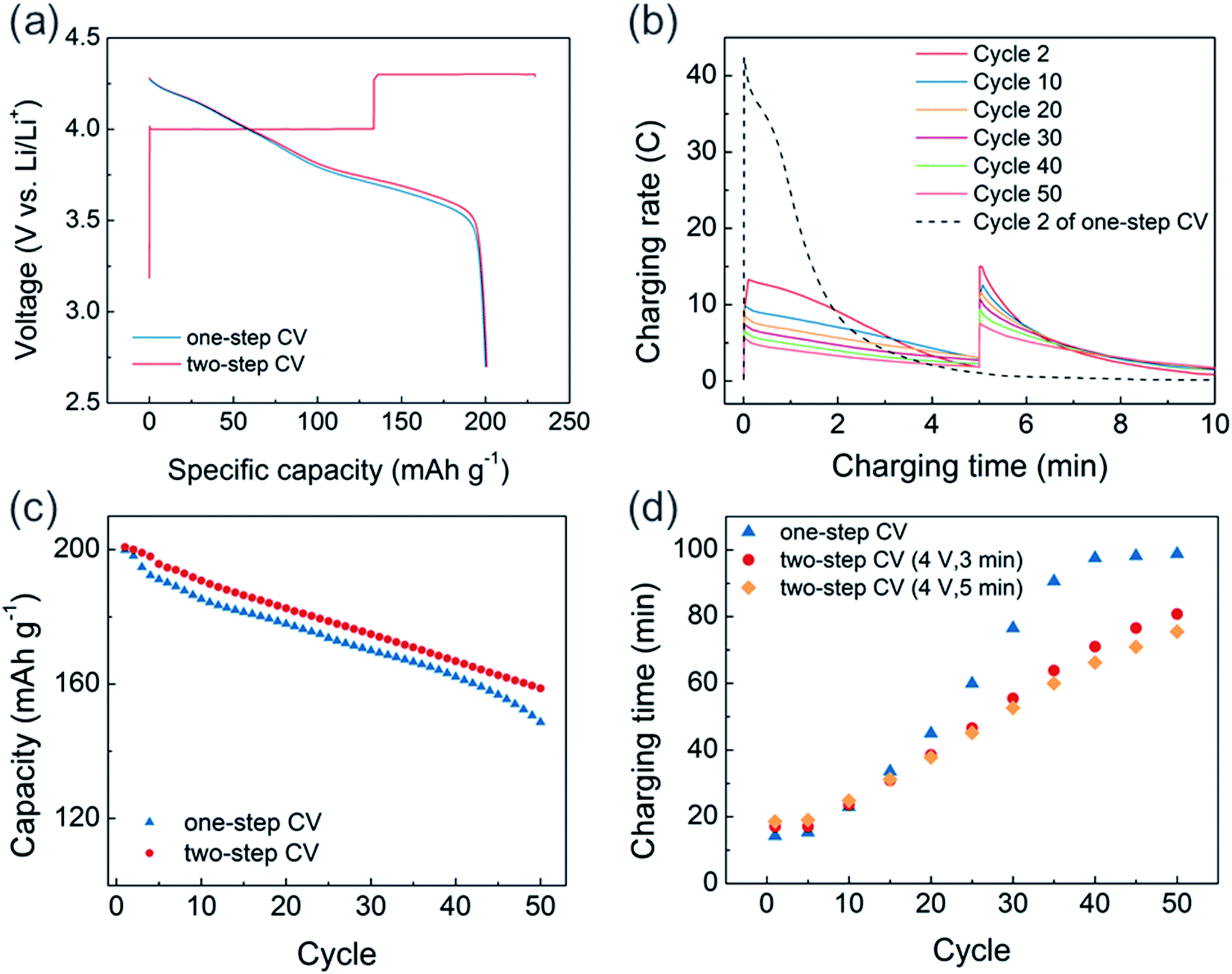 | ||
| Fig. 6 (a) Voltage profile of two-step CV charging (red lines) and one-step CV charging (blue lines, discharge only). (b) Charging rate profile of two-step CV charging every 10 cycles. Dotted line: charging rate profile from the initial cycle of one-step CV charging. (c) Cycle stability of one-step CV-charged (blue dots) and two-step CV-charged cells (red dots). (d) Charging time every 5 cycles: one-step CV-charged (blue dots), 3 minutes (red dots), and 5 minutes of two-step CV-charged cells (orange dots). | ||
As a result of minimizing extremely high-current region, the two-step CV-charged cell exhibited enhanced cycle stability and preserved the charging time better than the one-step CV-charged cell. As shown in Fig. 6c, we observed about 5% of the enhanced capacity retention at the 50th cycle in the two-step CV-charged cell, compared to the one-step CV-charged cell. Also, the capacity of one-step CV-charged cell was normalized by that of two-step CV-charged cell to directly compare cycle properties as shown in Fig. S10.† During initial 5 cycles, there exists a faster capacity decay in one-step CV-charged cell, while the acceleration of capacity decay starts to reappear from the 40th cycles. Based on our observation, faster capacity reduction at initial cycles would be related with the extremely high C-rate, which leads to the decomposition of electrolyte and the formation of thick surface layer. On the other hand, the two-step CV-charged cell was able to avoid the extremely high C-rate as we confirmed in Fig. 6b. Therefore, the two-step CV-charged cell delivered an improved capacity retention during the initial cycles and also better preserved the retention at later cycles. Moreover, the increase in charging time was also mitigated from 4 V pre-charged cells with cycles (Fig. 6d). Due to the fixed pre-charging time, about 3 minutes of additional charging time is required at earlier cycles in the 4 V pre-charged cells. However, it soon starts to take longer to charge the one-step CV-charged cell, since the rate of increasing charging time is slower in the pre-charged cells. In addition, the increasing rate is even lower in the 5 minute pre-charged cell (orange dots) than that of the 3 minute pre-charged cell (red dots), which demonstrates the effectiveness of minimizing extremely high-current region onto preserving charging time via pre-charging.
For the generality of the CV charging and the effectiveness of the two-step CV-charging protocols, we performed identical sets of experiments using commercial-grade NCM811 synthesized by co-precipitation method (Fig. S11a†). As we observed with the USP-NCM811 materials, all of the discharge profiles of CV and CCCV charged cells overlapped each other, indicating that CV-charging protocols are feasible as a fast-charging protocol during earlier cycles (Fig. S11b†). Although the CCCV-charged cell exhibited outstanding cycle performance, maintaining 98% of its initial capacity after 50 cycles, the CV-charged cell started to deteriorate rapidly after the 25th cycle (Fig. S11c†). In addition to the particle surface problem we showed in Fig. 5 and S8,† crack generation of NCM811 particles with spherical morphology would also affect the capacity degradation of the cells, which may accelerate under extremely high C-rate and inhomogeneous delithiation during CV charging.29 Therefore, the effect of pre-charging appears more clearly from co-precipitation NCM811 particles, possibly due to the reduction of extremely high C-rate that leads to more homogeneous and stable charging reaction. It also demonstrates the potential of multistep CV charging as a fast-charging protocol. Since the steps of CV charging can be finely tuned, it can further consider dynamic reaction kinetics of active materials, which is dependent on the lithium-ion concentration.30,31 Combined with machine learning, we expect some promising multistep CV charging protocol would be developed with reduced charging time and improved stability.21
3. Conclusion
In summary, we report here that CV charging is feasible for use as a fast-charging protocol, and demonstrate electrochemical behaviors during CV-charging cycles. Using a high-rate NCM811 cathode material synthesized using a novel USP method, we achieved fast charging that required just 15 minutes to fully charge the material using a CV-only charging protocol. Although discharge profiles precisely overlapped with normal CCCV charged cells, CV-charged cells exhibited faster capacity degradation with elongation of charging time as cycle proceeds. Moreover, the deterioration of NCM811 particle surface occurred at extremely high C-rate, as a result, the reduction of high C-rate region at later cycles leads to the extension of charging time. As we observed from XPS, an extremely high C-rate induces oxidation and decomposition of electrolytes at the surface region, causing cell impedance and capacity degradation to increase. Based on these findings, we proposed a two-step CV-charging protocol that can reduce charging at extremely high C-rate. Our suggested protocol delivered improved capacity retention and mitigated the increase of charging time, which demonstrates the importance of minimizing extremely high C-rate charging. Through our advanced understanding of CV charging, this work indicates that multistep CV charging has potential as a fast and stable charging protocol. It also provides the rationale for the currently used high-current CCCV, such as 4C or 6C CCCV,16 which can avoid extremely high C-rate regions with stable cycle properties.4. Experimental
4.1 Synthesis of materials
NCM811 was synthesized using the USP method. Stoichiometric amounts of LiNO3 (6% excess to compensate for lithium loss), Ni(NO3)2·6H2O, Co(NO3)2·6H2O, and Mn(NO3)2·4H2O were dissolved in deionized water with overall solution solid loading of 5 wt%. The solution was atomized through a 1.65 MHz submerged ultrasonic spray generator, and the resultant droplets went through a 700 °C reactor carried by ambient air. The precursor powder from USP was sintered at 775 °C for 12 hours with O2 flow.The precursor of NCM811 was also produced using co-precipitation. A 2 mol L−1 stoichiometric solution of NiSO4·6H2O, CoSO4·7H2O, and MnSO4·H2O (all precursors from Sigma-Aldrich) was prepared and continuously pumped into a 4 L continuously stirred tank reactor that was filled with 1.2 L of distilled water, purged with nitrogen. The appropriate amount of aqueous ammonia solution (5 mol L−1) as a chelating agent was separately dripped into the reactor, accurately controlling the concentration ratio between transition metal and ammonia. The pH was maintained at 11.0 with an aqueous solution of 4 mol L−1 sodium hydroxide, using a pH controller connected to the diaphragm metering pump. The temperature inside of reactor vessel was kept at 55 °C and the overhead stirrer was carefully controlled. The co-precipitated solution product was collected after a 24 hour reaction, and the precursor powder was obtained by washing and filtering. The cake powder from the filtration was transferred to the convection oven and dried overnight. The obtained NMC811 precursor was mixed with 3% excess LiOH·H2O and sintered at 770 °C for 12 hours under an O2 flow using a tube furnace with a wide, stable hot zone to avoid unwanted concentrations of heat.
4.2 Electrochemical tests
A slurry of 80 wt% active materials, 10 wt% carbon black (Super P, Timcal), and 10 wt% polyvinylidene fluoride (PVDF, Solvay), dissolved in N-methyl-2-pyrrolidone (NMP), was cast onto aluminum foil. The NMP was dried in an 80 °C oven overnight. The electrode was calendared before use. Coin cells (CR2032, Hohsen) were assembled with the working electrode, lithium metal disc, Celgard 2325 separator, and a 1.2 M solution of LiPF6 in ethylene carbonate/ethyl methyl carbonate (EC/EMC, 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 v/v) in an argon-filled glove box. The coin cells were tested using a MACCOR cycler (series 4000) between 2.7 and 4.3 V at 30 °C in a temperature-controlled chamber. For normal cycles, CCCV charging and CC discharging were applied at 0.1C (1C = 200 mA g−1) during the first cycles and 0.3C afterward with a current cutoff of CV at 0.05C. During the CV-only charging at 4.3 V, the current cutoff was 0.05C and the time cutoff was 2 hours. For two-step (or multistep) CV charging, only a time cutoff was applied for 3 or 5 minutes at the first CV charging, while a current cutoff at 0.05C and a time cutoff at 2 hours were maintained at the last 4.3 V CV-charging step. EIS measurements were performed on a BioLogic VSP-300. The EIS data was observed at a perturbation amplitude of 10 mV in the frequency range of 100 kHz to 10 mHz at the open circuit voltage of 3.6 V.
:7 v/v) in an argon-filled glove box. The coin cells were tested using a MACCOR cycler (series 4000) between 2.7 and 4.3 V at 30 °C in a temperature-controlled chamber. For normal cycles, CCCV charging and CC discharging were applied at 0.1C (1C = 200 mA g−1) during the first cycles and 0.3C afterward with a current cutoff of CV at 0.05C. During the CV-only charging at 4.3 V, the current cutoff was 0.05C and the time cutoff was 2 hours. For two-step (or multistep) CV charging, only a time cutoff was applied for 3 or 5 minutes at the first CV charging, while a current cutoff at 0.05C and a time cutoff at 2 hours were maintained at the last 4.3 V CV-charging step. EIS measurements were performed on a BioLogic VSP-300. The EIS data was observed at a perturbation amplitude of 10 mV in the frequency range of 100 kHz to 10 mHz at the open circuit voltage of 3.6 V.
4.3 Characterization of materials
The coin cells were disassembled in an argon-filled glove box for the ex situ analyses. The structure of the powders and electrodes were characterized using an XRD (D8 ADVANCE, Bruker), equipped with a Cu Kα radiation source (λ = 1.54178 Å). We analyzed the morphology of the particles using scanning electron microscopy (SEM) (Hitachi S-4700), operated at an accelerating voltage of 10 kV. XPS analysis was performed by the PHI 5000 VersaProbe II System (Physical Electronics). The spectra were obtained using an Al Kα radiation (hν = 1486.6 eV) beam (100 μm, 25 W), Ar+ and electron beam sample neutralization, in fixed analyzer transmission mode. Peak fitting was performed using Shirley background correction and the Gaussian–Lorentzian curve synthesis available in MultiPack software. XPS spectra were aligned to the carbon black component in the C 1s spectra at 284.8 eV.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Office of Vehicle Technologies of the U.S. Department of Energy, particularly from Peter Faguy and Dave Howell, is gratefully acknowledged. This work was carried out at the Materials Engineering Research Facility (MERF), which is supported within the core funding of the Applied Battery Research (ABR) for Transportation Program. The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up, nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.References
- M. Armand and J. M. Tarascon, Building better batteries, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- M. S. Whittingham, Lithium batteries and cathode materials, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS PubMed.
- P. Keil and A. Jossen, Charging protocols for lithium-ion batteries and their impact on cycle life—An experimental study with different 18650 high-power cells, J. Energy Storage, 2016, 6, 125–141 CrossRef.
- S. Ahmed, et al., Enabling fast charging – A battery technology gap assessment, J. Power Sources, 2017, 367, 250–262 CrossRef CAS.
- A. Tomaszewska, et al., Lithium-ion battery fast charging: A review, eTransportation, 2019, 1, 100011 CrossRef.
- J. Vetter, et al., Ageing mechanisms in lithium-ion batteries, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nanoscale Nucleation and Growth of Electrodeposited Lithium Metal, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- G. Yoon, S. Moon, G. Ceder and K. Kang, Deposition and Stripping Behavior of Lithium Metal in Electrochemical System: Continuum Mechanics Study, Chem. Mater., 2018, 30, 6769–6776 CrossRef CAS.
- T. R. Tanim, et al., Extreme Fast Charge Challenges for Lithium-Ion Battery: Variability and Positive Electrode Issues, J. Electrochem. Soc., 2019, 166, A1926–A1938 CrossRef CAS.
- T. Liu, et al., Bulk and surface structural changes in high nickel cathodes subjected to fast charging conditions, Chem. Commun., 2020, 56, 6973–6976 RSC.
- L. Wen, et al., Nickel-Rich Layered Lithium Transition-Metal Oxide for High-Energy Lithium-Ion Batteries, Angew. Chem., Int. Ed., 2015, 54, 4440–4457 CrossRef PubMed.
- J. Hong, H. Gwon, S.-K. Jung, K. Ku and K. Kang, Review—Lithium-Excess Layered Cathodes for Lithium Rechargeable Batteries, J. Electrochem. Soc., 2015, 162, A2447–A2467 CrossRef CAS.
- K. Ku, et al., Suppression of Voltage Decay through Manganese Deactivation and Nickel Redox Buffering in High-Energy Layered Lithium-Rich Electrodes, Adv. Energy Mater., 2018, 8, 1800606 CrossRef.
- P. Yan, et al., Intragranular cracking as a critical barrier for high-voltage usage of layer-structured cathode for lithium-ion batteries, Nat. Commun., 2017, 8, 14101 CrossRef CAS PubMed.
- K. Ku, et al., A new lithium diffusion model in layered oxides based on asymmetric but reversible transition metal migration, Energy Environ. Sci., 2020, 13, 1269–1278 RSC.
- W. Mai, A. M. Colclasure and K. Smith, Model-Instructed Design of Novel Charging Protocols for the Extreme Fast Charging of Lithium-Ion Batteries Without Lithium Plating, J. Electrochem. Soc., 2020, 167, 080517 CrossRef CAS.
- D. Anseán, et al., Fast charging technique for high power lithium iron phosphate batteries: A cycle life analysis, J. Power Sources, 2013, 239, 9–15 CrossRef.
- P. H. L. Notten, J. H. G. O. h. Veld and J. R. G. v. Beek, Boostcharging Li-ion batteries: A challenging new charging concept, J. Power Sources, 2005, 145, 89–94 CrossRef CAS.
- J. Li, E. Murphy, J. Winnick and P. A. Kohl, The effects of pulse charging on cycling characteristics of commercial lithium-ion batteries, J. Power Sources, 2001, 102, 302–309 CrossRef CAS.
- F. Savoye, P. Venet, M. Millet and J. Groot, Impact of Periodic Current Pulses on Li-Ion Battery Performance, IEEE Trans. Ind. Electron., 2012, 59, 3481–3488 Search PubMed.
- P. M. Attia, et al., Closed-loop optimization of fast-charging protocols for batteries with machine learning, Nature, 2020, 578, 397–402 CrossRef CAS PubMed.
- M. Oljaca, et al., Novel Li(Ni1/3Co1/3Mn1/3)O2 cathode morphologies for high power Li-ion batteries, J. Power Sources, 2014, 248, 729–738 CrossRef CAS.
- H. Ji, et al., Ultrahigh power and energy density in partially ordered lithium-ion cathode materials, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- J. Liang, et al., Well-ordered layered LiNi0.8Co0.1Mn0.1O2 submicron sphere with fast electrochemical kinetics for cathodic lithium storage, J. Energy Chem., 2020, 47, 188–195 CrossRef.
- Y. Li, M. Bettge, J. Bareño, S. E. Trask and D. P. Abraham, Exploring Electrochemistry and Interface Characteristics of Lithium-Ion Cells with Li1.2Ni0.15Mn0.55Co0.1O2 Positive and Li4Ti5O12 Negative Electrodes, J. Electrochem. Soc., 2015, 162, A7049–A7059 CrossRef CAS.
- R. Tatara, et al., The Effect of Electrode-Electrolyte Interface on the Electrochemical Impedance Spectra for Positive Electrode in Li-Ion Battery, J. Electrochem. Soc., 2018, 166, A5090–A5098 CrossRef.
- S. J. An, et al., Electrolyte Volume Effects on Electrochemical Performance and Solid Electrolyte Interphase in Si-Graphite/NMC Lithium-Ion Pouch Cells, ACS Appl. Mater. Interfaces, 2017, 9, 18799–18808 CrossRef CAS PubMed.
- J. Sung-Kyun, et al., Understanding the Degradation Mechanisms of LiNi0.5Co0.2Mn0.3O2 Cathode Material in Lithium Ion Batteries, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- Z. Xu, M. M. Rahman, L. Mu, Y. Liu and F. Lin, Chemomechanical behaviors of layered cathode materials in alkali metal ion batteries, J. Mater. Chem. A, 2018, 6, 21859–21884 RSC.
- J. Park, et al., Fictitious phase separation in Li layered oxides driven by electro-autocatalysis, Nat. Mater., 2021, 20, 991–999 CrossRef CAS PubMed.
- J. Lim, et al., Origin and hysteresis of lithium compositional spatiodynamics within battery primary particles, Science, 2016, 353, 566–571 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05767g |
| This journal is © The Royal Society of Chemistry 2022 |