
Understanding the high performance of PdSn–TaN(tantalum nitride)/C electrocatalysts for the methanol oxidation reaction: coupling nitrides and oxophilic elements†
Na
Ye‡
a,
Pengcheng
Zhao‡
b,
Xiaoying
Qi
a,
Wenchao
Sheng
b,
Zhao
Jiang
 *a and
Tao
Fang
a
*a and
Tao
Fang
a
aDepartment of Chemical Engineering, Shaanxi Key Laboratory of Energy Chemical Process Intensification, Engineering Research Center of New Energy System Engineering and Equipment, Xi'an Jiaotong University, Xi'an, 710049, China. E-mail: jiangzhao@mail.xjtu.edu.cn
bCollege of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai, 200092, China
First published on 24th November 2021
Abstract
Core–shell like PdSn–TaN(tantalum nitride)/C catalysts with tunable oxophilicity were prepared through a surfactant-free solvothermal method. Optimized Pd1Sn3–TaN/C exhibited good activity (3293.46 A gPd−1, 15.6 times that of commercial Pd/C), stability and tolerance to COad-like species towards the methanol oxidation reaction (MOR) in alkaline media, which can be ascribed to the interfacial structures and bi-functional effects. Briefly, the core–shell like structure can provide an effective Pd–TaN/C interface related to the large ECSA, and the introduction of Sn can further synergistically modulate the interfacial electronic structures, thereby significantly promoting the MOR performances, as revealed by HRTEM, XPS and electrochemical results. Furthermore, in situ ATR-SEIRAS (attenuated total reflection-surface enhanced infrared absorption spectroscopy) measurements displayed that PdSn–TaN/C went through the dual-channel pathways including a large amount of HCOOad and a small amount of COad towards MOR in alkaline medium, which is different from the single COad pathway on commercial Pd/C. Density functional theory (DFT) calculations indicated that the coadsorption intensity and amount of OH provided a chance for CO conversion into CO2 on Pd4–SnO2/TaN(001). Compared to Pd(111), the CO binding energy was evidently reduced on Pd4–SnO2/TaN(001). Meanwhile, Pd1Sn3–TaN/C also showed high mass activity towards the formic acid oxidation reaction (FAOR) in acidic media, which further confirmed that Pd1Sn3–TaN/C may be a promising bi-functional electrocatalyst with high efficiency.
1. Introduction
The rapidly growing demand for energy, global warming and pollution have stimulated worldwide interest for exploiting new energy sources to substitute for traditional fossil fuels.1 It is crucial to develop renewable and sustainable energy sources. Direct methanol fuel cells (DMFCs) and direct formic acid fuel cells (DFAFCs) have attracted remarkable attention owing to their advantages, such as renewability, safety, high energy density, and ease of handling, storage and transport.2–4 Besides, CO2 in the exhaust gas from methanol fuel cells operating on carbon-containing fuels is not diluted by nitrogen, as it is in the exhaust gas from the normal combustion engine, which is easily compressed.5 It means that carbon-neutral power generation is significantly easier to achieve by using fuel cells than by conventional combustion of fossil fuels.5 Also, CCS (carbon capture and storage) or CCU (carbon capture and utilization) technologies will play a major role in energy conversion by a decarbonizing process.6,7 As we all know, noble metals (i.e. Pt and Pd) are effective catalysts to catalyze small organic molecules, such as the methanol oxidation reaction (MOR) in alkaline media and the formic acid oxidation reaction (FAOR) in acidic media.1,8 However, Pd-based catalysts always suffer from sluggish kinetics and poisoning of COad intermediates during the MOR, which inhibit their commercial applications.1 Therefore, the rational design of high-efficiency and low-cost Pd-based catalysts for the MOR in alkaline media is crucial to meet the commercial requirements.Transition metal nitrides (TMNs), such as TaN (tantalum nitride),9 NbN (niobium nitrides),10,11 TiN (titanium nitride),12,13 MoN (molybdenum nitride),14 VN (vanadium nitride),15–17 and WN (tungsten nitride),18–20 have received growing attention in recent years owing to the fact that their electronic properties are similar to those of noble metal Pt. Besides, their low cost, large abundance and good electrochemical stability also allow transition metal nitrides to be promising catalytic materials for fuel cell applications.12,13,21,22 In particular, the combination of metals and transition metal nitrides is considered as an effective method to enhance the catalytic performances of new materials by interaction between them.23 For example, Jing et al.24 deposited Pt nanoparticles on tungsten nitride, which exhibited good oxygen reduction reaction (ORR) performances in acidic media. The enhancement can be explained by the synergistic effect between Pt and WN. Tian et al.25 reported that TiNiN doped with atomic layers of Pt presented high catalytic activity and durability for the ORR in acidic media, which may be because of the synergistic effects from Ni as well as the unique interaction between Pt and TiN. As reported by Roca-Ayats and coworkers,26 compared with pure Pd/C, the Pt/TiN catalyst displayed higher performance for the CO oxidation reaction. The improved tolerance of CO species could be due to the electronic effect between them. However, to our knowledge, there are few studies on TaN-doped Pd-based catalysts for their application in direct fuel cells (i.e. MOR and FAOR). The intrinsic reaction mechanism and structure–activity relationship are even more challenging. Interestingly, it is feasible that Pd–M/TaN–C catalysts are alternative catalysts to solve the slow reaction kinetics and low anti-CO poisoning ability issues for the MOR and FAOR from an economic viewpoint, because TaN is not only rather cheaper than noble metals but also has a special Pt-like electronic structure. Therefore, low-content Pd components supported on TaN/C are expected to be efficient and low-cost materials towards direct fuel cells.
As for Pd-based catalysts, it is well recognized that the second metal plays a vital role in improving the catalytic performance by different synergistic effects, such as the electronic effect and strain effect.27 According to previous studies, it has been indicated that the oxophilicity of the second metal also had an evident effect on organic molecule activation by the facilitated adsorption of oxygen-containing species, such as OHad, which could help the subsequent oxidation of CO species and improve the catalytic performance.28–32 Du et al.30 found that platinum-tin oxide nanoparticles with an optimized concentration and distribution of Sn could efficiently improve the selectivity of CO2 through breaking the C–C bond of ethanol molecules for the ethanol oxidation reaction. Wu et al.33 proposed that the oxophilic atoms on the surface were responsible for the enhancement of the OH adsorption sites, while the internal oxophilic atoms were related to the changed electronic structures for the host metals and not responsible for the OH adsorption sites. In addition, the metallic oxophilicity could also affect the pathway or selectivity for the corresponding reactions. Liang et al.34 found that Au@PtIr preferred following a direct C1-12e oxidation pathway, whereas PtIr/C would rather follow the CO-poisoned pathway. Similarly, Qi et al.35 indicated that a “non-CO” pathway occurred on PtZn/MWNT, while a CO-poisoned pathway occurred on the pure Pt catalyst, which might be due to the fact that the oxophilic Zn stabilized the adsorption of OH on the catalyst surface. Therefore, it could be proposed that introducing oxophilic metals may be an effective strategy to activate small organic molecule fuels. In our previous work,36 the Pd–Cu/TaN catalyst exhibited good catalytic performance for the methanol oxidation reaction in alkaline medium (1326.95 A gPd−1), indicating that the introduction of Cu and TaN enhanced the oxidation efficiency of methanol molecules evidently. Considering that Sn shows more oxophilic nature than Pd,30,33,37 it is expected to adjust the oxophilicity of PdxSny–TaN/C and increase the adsorption intensity of oxygen-containing species, which can facilitate catalytic activity and anti-CO poisoning ability for the MOR. Furthermore, it is necessary to clarify the corresponding MOR mechanism on Pd-based/TaN catalysts by combining in situ analysis with theoretical calculations. For one thing, in situ ATR-SEIRAS (attenuated total reflection-surface enhanced infrared absorption spectroscopy), as a spectrochemical technique, is a good tool to obtain the interfacial information of intermediates and products during the MOR process at the molecular level.38,39 For example, the mentioned promoting effects from the second oxophilic metal can be evidenced by qualitative and/or semi-quantitative analysis, that is, potential-dependent intermediates and/or products.38 For another, density functional theory (DFT) calculations are also accessible to provide the energy information for each adsorption/desorption or reaction step of the MOR on catalysts. Meanwhile, to better understand the underlying enhancement mechanism, the identification of intermediates and the clarification of the structure–activity relationship are also necessary.
Based on the above analysis, the work is devoted to studying the effects of the oxophilicity of the synthesized PdSn–TaN/C with a special core–shell like structure on the catalytic performance and reaction mechanism. Valence-dependent catalytic activity accompanied by the identification of reaction intermediates is present in the work. Specifically, a series of PdxSny–TaN/C catalysts with a core–shell like structure and tunable oxophilicity are prepared via a mild one-pot solvothermal method with no surfactant added, in which the synthesis parameters and Sn contents are elaborately regulated to tailor the oxophilic sites. The optimized Pd1Sn3–TaN/C catalyst is confirmed to be effective for the MOR in alkaline media and the FAOR in acidic media. The special structures provide more interfacial interaction and more actives sites are accordingly exposed, as demonstrated by high-resolution transmission electron microscopy (HRTEM), X-ray photoelectron spectroscopy (XPS) and electrochemical results. It is also found that the MOR catalytic performances are strongly dependent on the valence states related to the oxophilic nature. Meanwhile, the lower d-band center after the addition of Sn also accounts for the enhanced tolerance to COad by weakening the adsorption of COad species. In situ ATR-SEIRAS measurements show that the PdSn–TaN/C and Pd–TaN/C catalysts proceed via dual pathways with formate as an important intermediate for the MOR in alkaline media, which may result from the enhanced adsorption of the dangling OH species related to non-hydrogen-bonded adsorbed interfacial H2O with the assistance of TaN and Sn, as evidenced by in situ ATR-SEIRAS results. This result is consistent with the DFT calculations. Our work aims at obtaining a fundamental understanding of the MOR on Pd-based/TaN catalysts from the perspective of the interfacial structure and reaction mechanism, which can provide a strategy to develop efficient and low-cost catalysts for direct fuel cells.
2. Experimental
2.1. Reagents and materials
Carbon black (VXC-72R) was obtained from King-Chemical Corporation. Na2PdCl4 (≥99.9%), SnCl2·2H2O (≥99%) and TaN (≥99.5%) were purchased from Aladdin. KOH (≥85%) was purchased from Sigma Aldrich. The commercial Pd/C catalyst (10% on carbon) was bought from Alfa Aesar. Sodium citrate dihydrate, ethylene glycol, ethanol, methanol, formic acid and H2SO4 (≥98%) were of analytical grade and obtained from Sinopharm Chemical Reagent Co. Ltd. (China). Argon and CO (20%) gas were bought from Xi'an Tianze Gas Company. Ultrapure water (18.2 MΩ cm) used was obtained by using an ultra-pure purification system. All chemicals were used as received without further treatments.2.2. Preparation of PdxSny–TaN/C catalysts
Similar to our previous reports,36 for the preparation of Pd1Sn1–TaN/C-m-alkaline, 270 mg of TaN and 135 mg of VXC-72R carbon were ultrasonically dispersed in 67.5 mL of ethylene glycol for 3 hours, and 60 mg of sodium citrate dihydrate, 1 mL of KOH solution (1 M), 260 µL of Na2PdCl4 (19 mg mL−1) and 635 µL of SnCl2·2H2O (5 mg mL−1, dissolved in the solvent of ethylene glycol) were then added to 8 mL of the above mixture under vigorous stirring (the molar ratio of Pd to Sn was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and the Pd loading was 3.33 wt%). Subsequently, the resulting mixture was heated to 393 K and held for 5 hours. Afterwards, after cooling to room temperature, the final Pd1Sn3/C-m-alkaline catalyst was collected through the subsequent centrifugation (washed with water and ethanol 3 times) and drying in a vacuum oven at 333 K overnight.
:1 and the Pd loading was 3.33 wt%). Subsequently, the resulting mixture was heated to 393 K and held for 5 hours. Afterwards, after cooling to room temperature, the final Pd1Sn3/C-m-alkaline catalyst was collected through the subsequent centrifugation (washed with water and ethanol 3 times) and drying in a vacuum oven at 333 K overnight.
As control experiments, Pd–TaN/C-m-alkaline was also prepared by a similar method, except that Sn was not added. Pd1Sn3–TaN/C-m-alkaline was also prepared by a similar method, except that the corresponding molar ratios of Sn and TaN were changed. Pd1Sn3/C-m-alkaline was prepared by a similar method, except that TaN was not added. Pd1Sn3–TaN/C-w-alkaline was prepared by a similar method, except that KOH was not added. Pd1Sn3–TaN/C-s-alkaline was prepared by a similar method, except that 2 mL of KOH was added. The actual loading amounts of PdxSny–TaN/C and Pd1Sn3/C-m-alkaline catalysts were measured by ICP-MS and are shown in Table S1 (ESI†).
2.3. Characterization
XRD patterns of the PdxSny–TaN/C catalysts were acquired on an X-ray diffractometer (XRD, Bruker D8 Advance) with Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 40 mA. To further investigate the surface/interface structures of the PdxSny–TaN/C catalysts, XPS spectra were recorded on an X-ray photoelectron spectrometer (XPS, Thermo Fisher ESCALAB Xi+) with Al Kα radiation (400 W, 1486.6 eV). To further characterize the morphology of the PdxSny–TaN/C catalysts, a high-resolution transmission electron microscope (HRTEM, Talos F200X) was used with an accelerating voltage of 200 kV. To ensure the actual compositions of the PdxSny–TaN/C catalysts, inductively coupled plasma-mass spectroscopy (ICP-MS) was conducted using a NexION 350D equipment.2.4. In situ ATR-SEIRAS measurements
Attenuated total reflection-surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) was employed in this study to uncover the reaction mechanism. The working electrode was prepared by adding dropwise the catalyst suspension onto an Au film coated Si hemispherical prism. The detailed preparation procedures of the Au thin film (∼60 nm) were based on previous studies.40–42 A Pt wire and a saturated calomel electrode were used as the counter electrode and reference electrode, respectively. The Pd and Pd-based suspensions were sonicated for 10 min and then 10 µL of suspensions were added dropwise onto an Au/Si surface and dried in air. Before the in situ ATR-SEIRAS study, the catalyst should be cycled in 1 M KOH between 0 and 1.4 V vs. RHE until the curve was stable. Then, real-time ATR-IR spectra were recorded from 0 to 1.4 V vs. RHE in Ar-saturated 1 M CH3OH and KOH solution in the Series mode of OMNIC software. And each spectrum was coded with 16 single spectra with a resolution of 8 cm−1. The spectra were named according to the end potential of each potential window. All spectra were presented in absorbance, A = −log(R/R0), where R is the reflectance of the sample spectrum and R0 is the background spectrum. The ATR-SEIRAS measurements employed a Nicolet iS50 FTIR spectrometer equipped with a mercury cadmium telluride (MCT) detector cooled with liquid nitrogen. And the incident angle of the IR beam was ∼65° on the reflection plane of the working electrode. The IR background was collected between 1.2 and 1.15 V.2.5. Electrochemical measurements
A three-electrode cell was utilized for all the electrochemical tests on a CHI 650B electrochemical workstation at room temperature, in which a Pt wire, a modified glassy carbon electrode and an Ag/AgCl electrode were used as the counter electrode, the working electrode and the reference electrode, respectively. In the preparation of the working electrode, 4 mg of the electrocatalyst, 30 µL of Nafion solution (Dupont) and 1 mL of solution (ultrapure water:isopropanol = 1:1) were ultrasonically mixed for 3 hours. Subsequently, 10 µL of the resulting homogeneous ink was added dropwise onto a clean glassy carbon electrode and dried, which formed the working electrode.
For all the electrochemical tests, the solutions were needed to be bubbled with an inert gas such as Ar to be deoxygenated before the measurements. Besides, to remove the contaminants on the surface, the catalysts were needed to be activated by potential scanning from 0 to 1.2 V (vs. RHE) in a 0.5 M H2SO4 or 1 M KOH solution till obtaining a stable cyclic voltammogram (CV) curve. CO stripping voltammograms were recorded in an Ar-saturated 0.5 M H2SO4 solution, followed by purging with CO gas for one hour to ensure the saturated adsorption of CO. Subsequently, the electrode was shortly transferred to a fresh 0.5 M Ar-purged H2SO4 solution, and the CO stripping voltammograms were obtained by cycling the potential between 0 and 1.2 V vs. RHE at 20 mV s−1. The electrochemical surface area (ECSA) can be defined based on the CO oxidation peak, as shown in eqn (1):
| ECSA = Q/(mPd × 420) = (SCO/V)/(mPd × 420) | (1) |
2.6. DFT calculations
The Vienna Ab initio Simulation Package (VASP) code was used for DFT calculations. The general gradient approximation (GGA)-Perdew–Burke–Ernzerhof (PBE) method was used to address the electronic exchange along with related effects.43 The setting of 400 eV for kinetic wave cutoff energy was applied. A 3 × 3 × 1 k-point mesh was set for the Brillouin-zone integration along with a Gaussian smearing of 0.1 eV.44 The energy convergence criterion was 1 × 10−5 eV. The p(3 × 3) supercells with four-layers were employed to model Pd(111) surface, together with the selected adsorbates of 1/9 monolayer. Pd4/TaN(001) was built using the Pd4 cluster deposited on the TaN(001) surface, which was consistent with our previous study.36 The Pd4–SnO2/TaN(001) model was constructed by the co-adsorbed Pd4 and SnO2 clusters on the TaN(001) surface, which represented Pd3Sn1/TaN(001). For Pd4/TaN(001) and Pd4–SnO2/TaN(001), a 2 × 2 super-cell with four layers was employed, which are shown in Fig. S1 of the ESI.† A vacuum layer (20 Å) was selected in the vertical direction from its periodic images. Furthermore, the climbing image nudged elastic band (CI-NEB) method was employed to uncover the transition states associated with the corresponding reactions.The binding energies (BEs) of the related intermediates were defined (eqn (2)):
| BE = Eadsorbate/surface − Eadsorbate − Esurface | (2) |
3. Results and discussion
3.1. Characterization of the PdxSny–TaN/C catalysts
In the work, a series of PdxSny–TaN/C catalysts are prepared by a facile one-pot solvothermal synthesis employing ethylene glycol as the solvent, in which the Pd–TaN/C-m-alkaline, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts acted as the reference. Notably, VXC-72R carbon with a large BET surface area (254 m2 g−1) is used as a support for all the catalysts, which is beneficial to provide more adsorption sites. Similar to previous reports from our group,36 Pd1Sn3–TaN/C-m-alkaline also exhibits a BET surface area of 66 m2 g−1 and a pore structure of 6.7 nm, which imply mesoporous properties.Firstly, the XRD technique is performed to explore the crystalline structures of the PdxSny–TaN/C catalysts. For Pd1Sn3–TaN/C-w-alkaline in Fig. 1(a), most of the sharp diffraction peaks (marked with *) are completely consistent with those of pure TaN (JCPDS 39-1485). Some small and sharp diffraction peaks can also be observed and assigned to other tantalum nitride/oxides. However, almost no Pd(PdO) or Sn(SnOx) peaks are observed, which may be due to the fact that the TaN peaks are strong as well as the PdSn content is low. Similar to the Pd1Sn3–TaN/C-w-alkaline sample, as for Pd–TaN/C-m-alkaline, Pd1Sn3–TaN/C-s-alkaline, Pd1Sn3–TaN/C-m-alkaline and Pd1Sn1–TaN/C-m-alkaline in Fig. 1(a), there are also lots of sharp and strong diffraction peaks (marked with *) corresponding to TaN (JCPDS 39-1485). The difference from Pd1Sn3–TaN/C-w-alkaline is that no small and sharp diffraction peaks in Fig. 1(b) (as highlighted by the red rectangular frame) corresponding to other tantalum nitrides/oxides were observed for the Pd–TaN/C-m-alkaline, Pd1Sn3–TaN/C-s-alkaline, Pd1Sn3–TaN/C-m-alkaline and Pd1Sn1–TaN/C-m-alkaline catalysts. At the same time, the diffraction peak of other PdxSny/TaN catalysts at 47.7° corresponding to TaN0.8 (JCPDS card no. 25-1279) becomes stronger than that of Pd1Sn3–TaN/C-w-alkaline. It may be in consequence of the altered coordination condition of sodium citrate in alkaline medium.45,46 For the synthesized Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts in Fig. 1(a), as a catalytic reference, a broad peak at around 25° can be assigned to the carbon (002) plane.47 The peak at around 40° is characteristic of Pd (111) (JCPDS 46-1043).
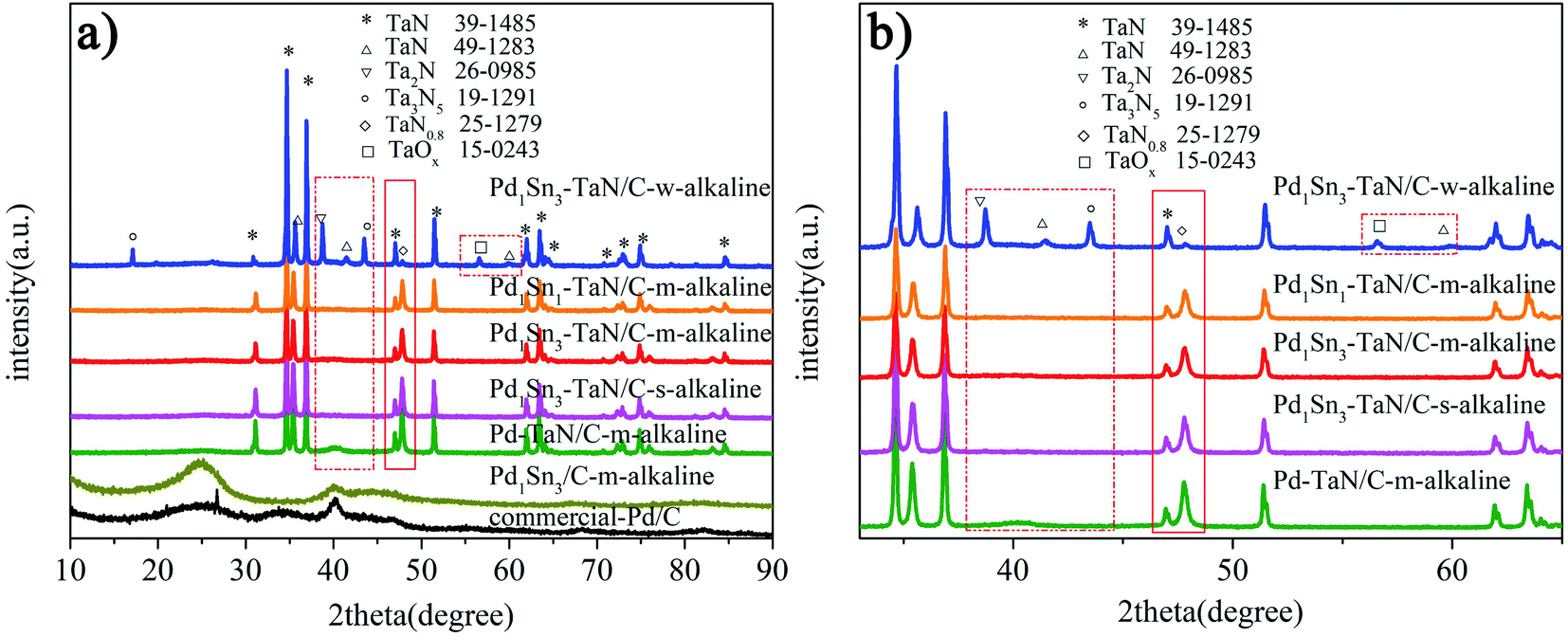 | ||
| Fig. 1 (a) XRD patterns and (b) the enlarged XRD patterns of the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts. | ||
In order to further characterize the morphology of Pd1Sn3–TaN/C-m-alkaline, TEM and HRTEM are conducted. As shown in Fig. 2(a), the well-dispersed Pd1Sn3–TaN/C-m-alkaline catalyst presents an average size of 1.91 nm. TEM images of other samples have been examined and shown in Fig. S2.† The small and dispersed morphology may result from the role of sodium citrate as a stabilizer in the ethylene glycol solvents.48Fig. 2(b) shows Pd particles with a clear lattice spacing of 0.227 nm, indicating that the crystallinity is good. The lattice spacing of 0.226 nm and 0.149 nm in Fig. 2(b)–(d) can be ascribed to the Pd(111) and TaN(300) planes. The slight increase in the lattice spacing of Pd may result from the lattice stretching of Sn atoms.49 EDX elemental mapping is carried out to further study the distribution information of different elements over the whole catalyst, as shown in Fig. 2(e)–(m). On the one hand, the EDX elemental mapping in Fig. 2(i) shows the structure of Pd1Sn3–TaN/C-m-alkaline with a TaN-rich shell and C-rich core, similar to our previous study.36 On the other hand, Pd and Sn atoms are uniformly dispersed at the same position in Fig. 2(l) and (m). Furthermore, in view of the large BET specific area of carbon, the core–shell like mesoporous TaN/C structure (verified by BET results) is beneficial to provide an effective interface to load PdSn, which has been verified by previous HRTEM results.36 Specifically, the interconnections between Pd and TaN are clearly observed in Fig. 2(d), where the lattice spacing of around 0.226 nm and 0.149 nm can be ascribed to the Pd(111) and TaN(300) planes. Interestingly, it is found that Pd and TaN are cross-distributed, which just looks like a sliced pizza, implying that a Pd–TaN interface structure is obviously formed.
 | ||
| Fig. 2 (a) TEM and (b–d) HR-TEM images of the Pd1Sn3–TaN/C-m-alkaline catalyst (the enlarged image of the red rectangular frame). (e–m) The EDX elemental mapping images. | ||
In order to further investigate the surface/interface structures of the PdxSny–TaN/C catalysts, XPS characterization is performed to reveal the valence state information of the surface. As a reference, all the spectra are calibrated based on 284.8 eV of C 1s. The high-resolution Pd 3d spectrum in Fig. 3(a) can be deconvoluted into Pdδ+ (∼337.0 eV for Pd 3d5/2 and ∼342.0 eV for Pd 3d3/2) and element Pd (∼335.0 eV for Pd 3d5/2 and ∼340.0 eV for Pd 3d3/2). On the one hand, the synthesis parameters and/or compositions of the three catalysts (including the Pd1Sn3–TaN/C-m-alkaline, Pd–TaN/C-m-alkaline and Pd1Sn3/C-m-alkaline catalysts) are similar except for whether Sn and TaN are added or not. As shown in Fig. 3(a), it is found that the binding energy of Pd 3d for Pd1Sn3–TaN/C-m-alkaline (335.19 eV) is slightly lower than that of Pd–TaN/C-m-alkaline (335.98 eV) and Pd1Sn3/C-m-alkaline (335.29 eV), suggesting that both the addition of TaN and Sn will transfer electrons to Pd. Specifically, the negative shift brought about by Sn is larger than that of TaN, implying that the donation of Sn added is larger than that of TaN. On the other hand, the electronic structure information of Pd 3d is different from that of the corresponding catalysts with changed synthesis parameters and compositions, as shown in Table S2† and Fig. 3(a). The binding energies of PdxSny–TaN/C catalysts decrease with the increase of Sn contents (from 335.36 eV to 335.19 eV). With the increase of the pH value under the synthesis conditions, the binding energies of Pd 3d for the PdxSny–TaN/C catalysts decrease firstly and then enhance with the increase of KOH contents during the synthesis process. Briefly, the Pd1Sn3–TaN/C-m-alkaline catalyst (335.19 eV) shows the lowest binding energy of Pd 3d among the catalysts, indicating that it has the most electron-rich Pd and a strong interaction with TaN and Sn. Moreover, to some extent, the integrated areas can be used to compare the concentration of different metallic valence states. As shown in Table S2,† it is found that the Pd atoms mainly exist in the element state in the PdxSny–TaN/C and Pd1Sn3/C-m-alkaline catalysts. Compared to the Pd–TaN/C-m-alkaline catalyst, all the PdxSny–TaN/C catalysts display an increased percentage of Pdδ+, which may result from the high oxophilicity of the second metal Sn.50 The percentages of Pdδ+ in PdxSny–TaN/C catalysts follow the order of Pd1Sn1–TaN/C-m-alkaline (16.01%) < Pd1Sn3–TaN/C-w-alkaline (21.49%) < Pd1Sn3–TaN/C-m-alkaline (25.19%) < Pd1Sn3–TaN/C-s-alkaline (44.87%). The results indicate that the increase of Sn contents in the catalysts is beneficial for the formation of Pdδ+. As the synthesis conditions become more and more alkaline, the concentration of Pdδ+ in the Pd1Sn3–TaN/C catalyst also significantly increases, which is responsible for the altered active sites to activate methanol or formic acid molecules in alkaline media. Thus, it can be proposed that introducing a rational component and regulating microenvironment of synthesis for catalysts can change the corresponding electronic structure.
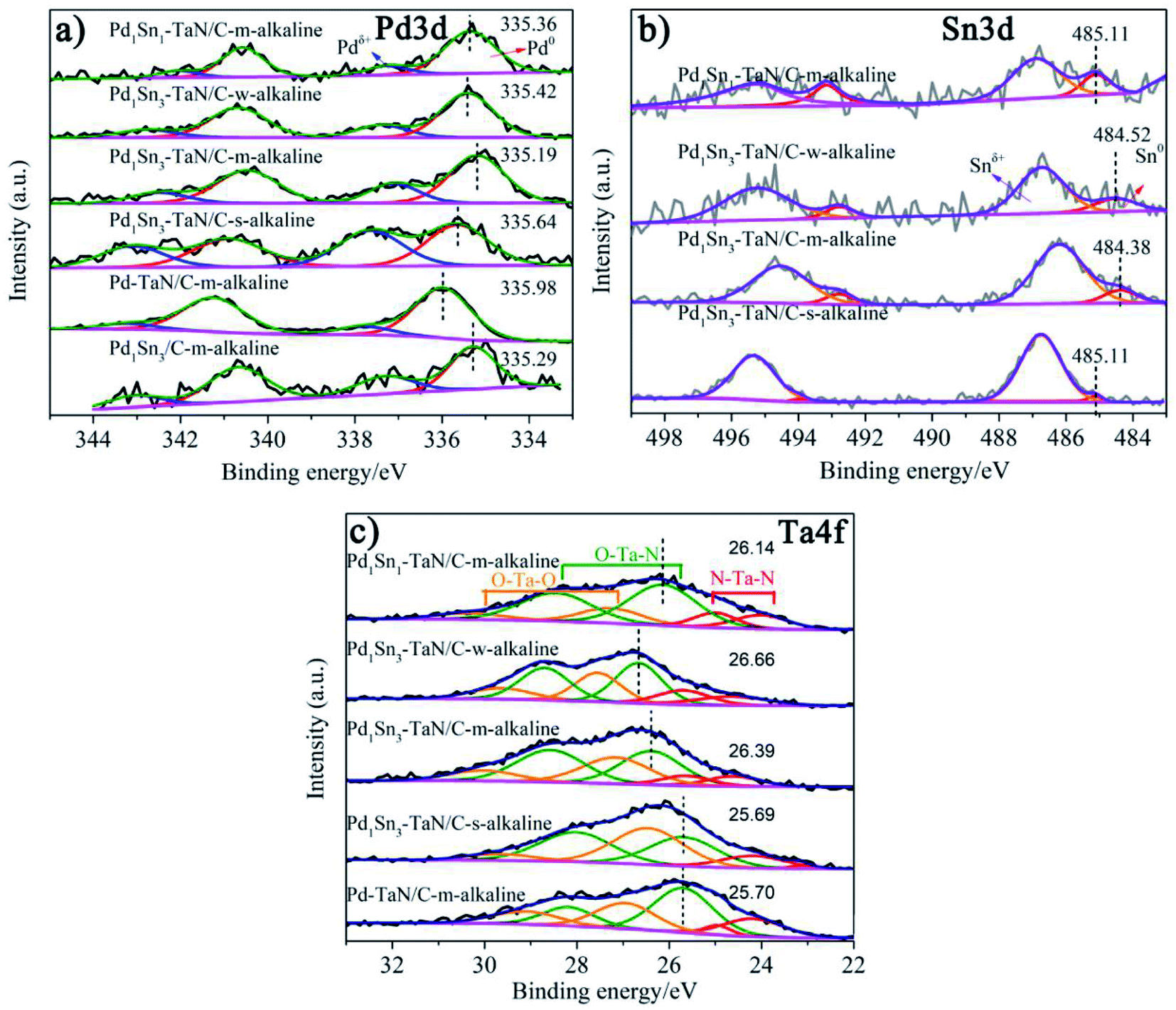 | ||
| Fig. 3 High-resolution XPS spectra of (a) Pd 3d, (b) Sn 3d and (c) Ta 4f core levels of the PdxSny–TaN/C and Pd1Sn3/C-m-alkaline catalysts (the black dashed lines represent the corresponding specific peak positions). | ||
The high-resolution Sn 3d spectrum in Fig. 3(b) can be deconvoluted into Snδ+ (∼486.7 eV) and element Sn (∼484.9 eV). Similar to Pd, the binding energies of Sn 3d for the PdxSny–TaN/C catalysts decrease with the increase of Sn contents (from 485.11 eV to 484.38 eV), indicating that their electronic structures are changed, as shown in Table S3.† Also, similar to the trend of the percentages of Pdδ+ for PdxSny–TaN/C catalysts, the percentages of Snδ+ for PdxSny–TaN/C catalysts also follow the order of Pd1Sn1–TaN/C-m-alkaline (72.67%) < Pd1Sn3–TaN/C-w-alkaline (83.25%) < Pd1Sn3–TaN/C-m-alkaline (87.63%) < Pd1Sn3–TaN/C-s-alkaline (93.13%). Especially for the Pd1Sn3–TaN/C-s-alkaline catalyst, almost no element Sn is observed, indicating that Sn mainly exists in the oxidized state on the surface. The results indicate that the alkaline synthesis conditions are beneficial to the production of Pdδ+ as well as Snδ+.
The core level Ta 4f spectrum in Fig. 3(c) can be deconvoluted into N–Ta–N (∼23.5/25.1 eV), N–Ta–O (∼26.3/28.7 eV) and O–Ta–O (∼27.7/29.9 eV).23,51–53 On the one hand, as shown in Table S4,† compared with the Pd–TaN/C-m-alkaline catalyst (25.70 eV), the Pd1Sn3–TaN/C-m-alkaline catalyst (26.39 eV) shows a significantly positive shift of 0.6 eV, indicating that TaN will transfer electrons to Sn. Combined with the shift of Pd 3d in Fig. 3(a) in the Pd1Sn3–TaN/C-m-alkaline catalyst after adding Sn, it is found that TaN transfers electrons to Sn, and Sn transfers electrons to Pd. Meanwhile, combined with the shift of Pd 3d in Fig. 3(a) in the Pd1Sn3–TaN/C-m-alkaline catalyst after adding TaN, it is found that TaN also transfers electrons to Pd. Therefore, it is concluded that TaN transfers electrons to Sn and Pd by dual channels, and at the same time, Sn also transfers electrons to Pd, confirming a strong interaction among Pd, Sn and TaN. On the other hand, by comparing Pd–TaN/C-m-alkaline (25.70 eV, 49.07%), Pd1Sn1–TaN/C-m-alkaline (26.14 eV, 63.58%) and Pd1Sn3–TaN/C-m-alkaline (26.39 eV, 54.61%) catalysts in Fig. 3(c) and Table S4,† it is found that the doped Sn brings about not only an increase of the percentage of N–Ta–O, but also a positive shift of N–Ta–O binding energy. This may result from the fact that more Pd takes part in the bonding of N–Ta–O to form the Pd–TaON interface with the added Sn, as observed by HRTEM, because Pd has a larger electronegativity than Ta.54 A possible explanation for the above results is that the interface may be formed via the Pd–TaON bond between Pd and TaN.
3.2. MOR performances of the PdxSny–TaN/C catalysts
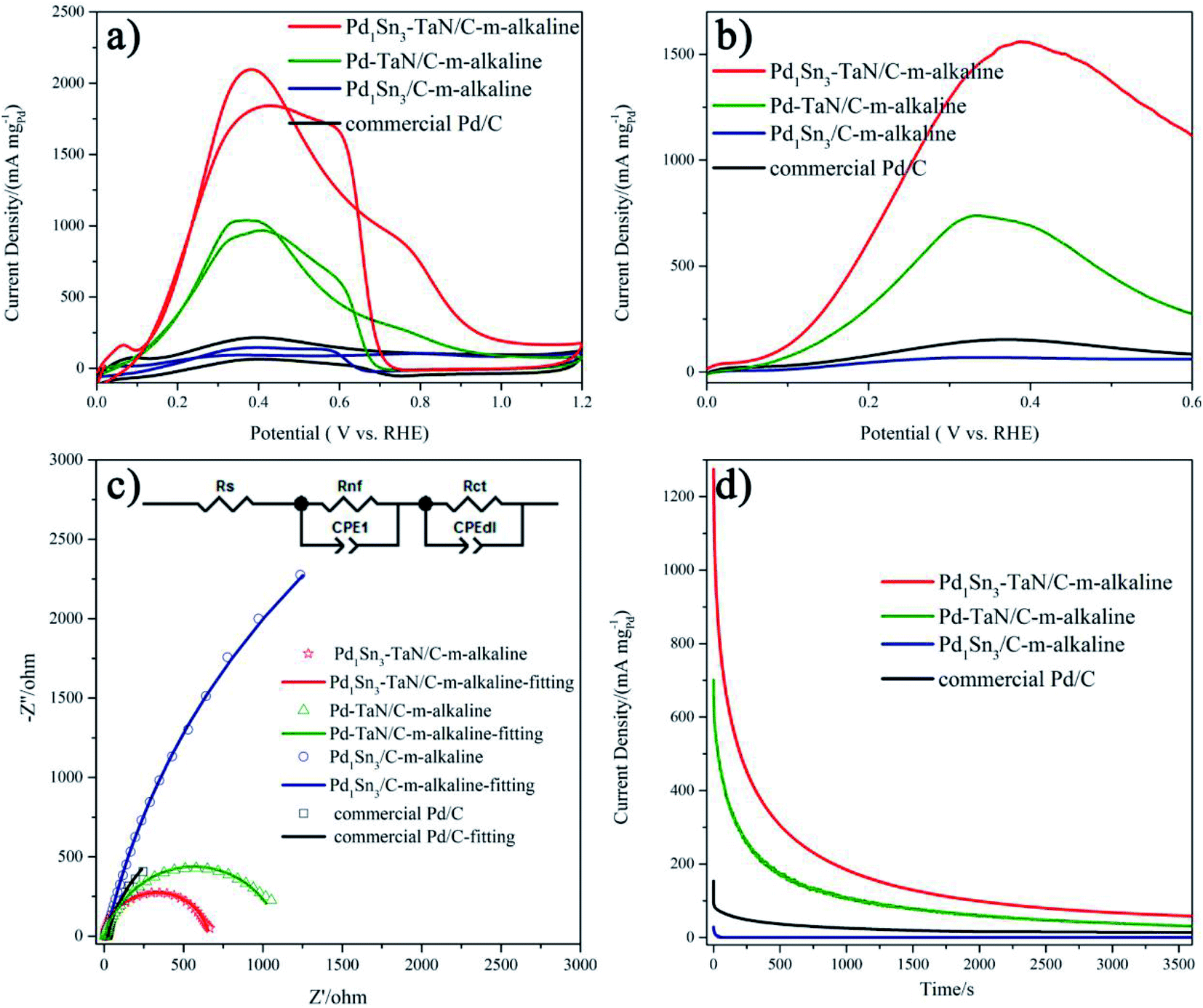
Inspired by the unique structural properties of the PdxSny–TaN/C catalysts, we further study their electrochemical performances, as shown in Fig. 4, in which the mass activity is based on the mass of Pd and the specific activity is based on the ECSAs calculated by the CO stripping technique in the work. The obtained ECSAs for the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts are listed in Table 1. The order of the ECSAs is commercial Pd/C (42.23) < Pd1Sn3/C-m-alkaline (70.18) < Pd–TaN/C-m-alkaline (119.45) < Pd1Sn1–TaN/C-m-alkaline (134.78) < Pd1Sn3–TaN/C-s-alkaline (139.32) < Pd1Sn3–TaN/C-w-alkaline (152.23) < Pd1Sn3–TaN/C-m-alkaline (203.99). Relative to the Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts, the PdxSny–TaN/C catalysts show larger ECSAs, indicating that the presence of TaN is beneficial to expose more Pd active sites, which may be due to the unique core–shell like structures of PdxSny–TaN/C that provide more interfacial interconnection, revealed by HRTEM and XPS results. Pd1Sn3–TaN/C-m-alkaline shows the smallest particle size, in agreement with the larger ECSA.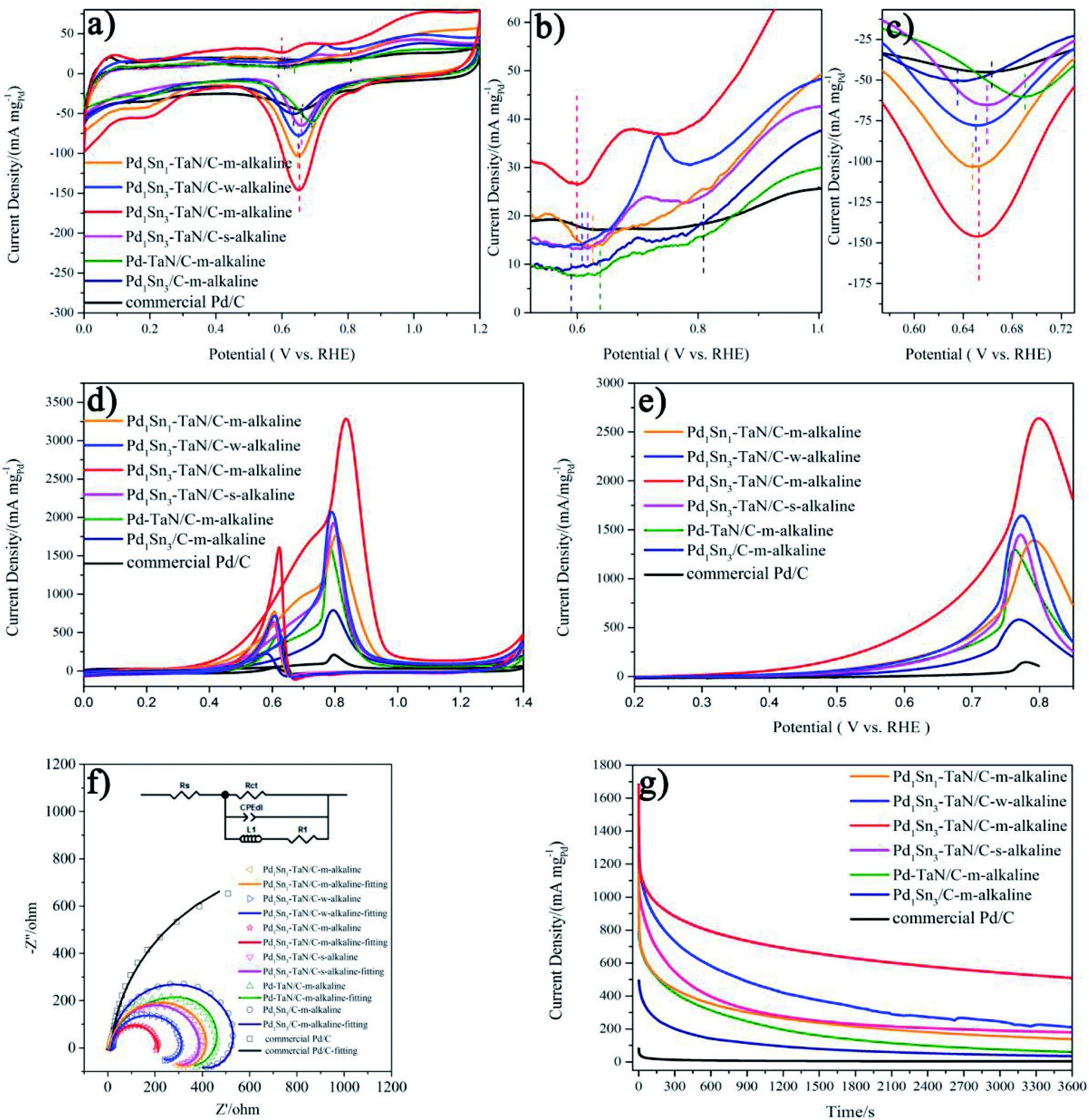 | ||
| Fig. 4 (a) CV curves and (b and c) the enlarged CV curves of the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts in Ar-saturated 1 M KOH at 20 mV s−1 (the dashed lines in (a) and (b) represent the onset potential of Pd–OHad, and the dashed lines in (c) represent the peak potential of the PdO reduction peak). (d) CV curves at 50 mV s−1, (e) LSV curves at 10 mV s−1, (f) Nyquist plots with simplified Randles equivalent circuits at 0.7 V and (g) CA curves at 0.74 V for the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts in an Ar-saturated 1 M KOH/1 M CH3OH solution. | ||
| Materials | ECSA (m2 g−1) | MOR | |
|---|---|---|---|
| I p(m) (mA mgPd−1) | I p(sa) (mA cmPd−2) | ||
| Pd1Sn1–TaN/C-m-alkaline | 134.78 | 1770.33 | 1.31 |
| Pd1Sn3–TaN/C-w-alkaline | 152.23 | 2081.34 | 1.37 |
| Pd1Sn3–TaN/C-m-alkaline | 203.99 | 3293.46 | 1.62 |
| Pd1Sn3–TaN/C-s-alkaline | 139.32 | 1929.82 | 1.35 |
| Pd–TaN/C-m-alkaline | 119.45 | 1674.64 | 1.39 |
| Pd1Sn3/C-m-alkaline | 70.18 | 797.45 | 1.14 |
| Commercial Pd/C | 42.23 | 210.53 | 0.49 |
At first, Fig. 4(a)–(c) present the CV curves of the PdxSny–TaN/C catalysts in an Ar-saturated 1 M KOH solution. It consists of three regions: the hydrogen adsorption/desorption peak (0–0.35 V), the double layer charge (0.35–0.55 V) and the formation/reduction of Pd oxide (Pd–OHad and Pd–O, 0.55–1.4 V).55–61 As we all know, the definite mechanisms of the formation/reduction of Pd oxide are still under discussion, while the possible mechanism is generally recognized as follows.55,60–62
| Pd + OH− → Pd–OHads + e− | (3) |
| Pd–OHads + OH− → Pd–O + H2O + e− | (4) |
| Pd–OHads + Pd–OHads → Pd–O + H2O | (5) |
| Pd–O + H2O + 2e− → Pd + OH− | (6) |
For one thing, as shown in Fig. 4(b), the onset potentials of Pd–OHad adsorption follow the order of Pd1Sn3/C-m-alkaline (0.590 V) < Pd1Sn3–TaN/C-m-alkaline (0.599 V) < Pd1Sn3–TaN/C-w-alkaline (0.608 V) < Pd1Sn3–TaN/C-s-alkaline (0.618 V) < Pd1Sn1–TaN/C-m-alkaline (0.626 V) < Pd–TaN/C-m-alkaline (0.638 V) < commercial Pd/C (0.809 V). Relative to Pd–TaN/C-m-alkaline, as shown in Fig. 4(b), the onset potentials of Pd–OHad adsorption of the PdxSny–TaN/C catalysts are negatively shifted, suggesting facile Pd–OHad adsorption occurs on the PdxSny–TaN/C catalyst surfaces. This may be due to the fact that Sn exhibits more oxophilic nature than Pd.63 For another, as mentioned in previous reports, the reduction potential of Pd oxide is a qualitative indicator of oxophilicity.64–66 Among these catalysts, as shown in Fig. 4(c), the PdO reduction peak for Pd1Sn3/C-m-alkaline shows up at the most negative potential, while that of Pd–TaN/C-m-alkaline shows up at the most positive potential. The PdO reduction peaks of the PdxSny–TaN/C catalysts fall in the range of those of Pd1Sn3/C-m-alkaline and Pd–TaN/C-m-alkaline. As shown in Fig. 4(c), the PdO reduction peaks follow the order of Pd1Sn3/C-m-alkaline (0.635 V) < Pd1Sn1–TaN/C-m-alkaline (0.648 V) < Pd1Sn3–TaN/C-w-alkaline (0.650 V) < Pd1Sn3–TaN/C-m-alkaline (0.653 V) < Pd1Sn3–TaN/C-s-alkaline (0.659 V) < Pd–TaN/C-m-alkaline (0.690 V). Interestingly, it is also found that the order of the PdO reduction peak of the PdxSny–TaN/C catalysts agrees well with the trend of the increasing concentration of Pdδ+ as well as Snδ+. Therefore, the oxophilic nature of the PdxSny–TaN/C catalysts is dependent on the synthesis parameters and Sn contents to some extent.
Then, the electrocatalytic performances of the PdxSny–TaN/C catalysts for the MOR in alkaline medium are evaluated. Fig. 4(d) presents the CV curves of the PdxSny–TaN/C catalysts in a 1 M KOH/1 M CH3OH solution saturated with Ar. The Pd1Sn3–TaN/C-m-alkaline catalyst exhibits a mass activity of 3293.46 A gPd−1, higher than those of the Pd1Sn3/C-m-alkaline (797.45 A gPd−1), Pd1Sn3–TaN/C-w-alkaline (2081.34 A gPd−1), Pd1Sn3–TaN/C-s-alkaline (1929.82 A gPd−1), Pd1Sn1–TaN/C-m-alkaline (1770.33 A gPd−1), Pd–TaN/C-m-alkaline (1674.64 A gPd−1) and commercial Pd/C (210.53 A gPd−1) catalysts, suggesting a remarkable improvement of the mass activity for the MOR with the introduction of TaN and Sn. Also, the Pd1Sn3–TaN/C-m-alkaline catalyst also presents the most outstanding specific activity among these catalysts, as listed in Table 1. The catalytic activity of Pd1Sn3–TaN/C-m-alkaline towards the MOR in alkaline medium is also much higher than those of other Pd-based catalysts previously reported, as shown in Table S5,† indicating that the unique structure and interface in Pd1Sn3–TaN/C-m-alkaline bring about an evident enhancement of catalytic activity for the MOR. In addition, as shown in linear scanning voltammetry (LSV) curves in Fig. 4(e), Pd1Sn3–TaN/C-m-alkaline presents the lowest MOR onset potential in alkaline medium among these catalysts, indicating that the reaction kinetics of methanol molecules and the intermediates at the interface are dramatically improved. To further study the charge transfer kinetics at the interface, EIS is conducted. The corresponding Nyquist plots with simplified Randles equivalent circuits and Bode plots are shown in Fig. 4(f) and S3.†Rs indicates the solution resistance. Rct and CPEdl indicate the charge transfer resistance at the interface near the electrode surface and constant-phase element that corresponds to the double layer capacitance and pseudocapacitance, respectively.67 As shown in Table S6,† the results indicate that the Rct follows the order of Pd1Sn3–TaN/C-m-alkaline (200.2 Ω) < Pd1Sn3–TaN/C-w-alkaline (302.2 Ω) < Pd1Sn3–TaN/C-s-alkaline (334.9 Ω) < Pd1Sn1–TaN/C-m-alkaline (427.6 Ω) < Pd–TaN/C-m-alkaline (477.2 Ω) < Pd1Sn3/C-m-alkaline (645.2 Ω) < commercial Pd/C (1690.0 Ω). Pd1Sn3–TaN/C-m-alkaline displays the smallest Rct, indicating the lowest charge transfer resistance.
Subsequently, to investigate the stability of different catalysts for the MOR in alkaline medium, chronoamperometry (CA) measurement is performed for 3600 s at 0.74 V in an Ar-saturated 1 M KOH/1 M CH3OH solution (Fig. 4(g)). For all the catalysts, in the beginning, the mass activities decrease rapidly and then decrease slowly, which may mainly result from the accumulation of oxygen-containing substances.68 Throughout the entire testing period, these PdxSny–TaN/C catalysts still show higher mass activity than other catalysts in the work. After 3600 s, it is demonstrated that Pd1Sn3–TaN/C-m-alkaline with unique electronic structures has the best catalytic stability and anti-poisoning ability. This may due to the facilely formed Pd–OHad, which improves the anti-poisoning ability, as revealed by CV results. In short, these results confirm that Pd1Sn3–TaN/C-m-alkaline acts as an outstanding catalyst for the MOR in alkaline medium.
3.3. In situ observation of intermediates of the MOR in alkaline media
In order to identify the intermediates of the MOR in alkaline medium at the molecular level and clarify the reaction mechanism, in the voltammetry measurement process, in situ ATR-SEIRAS studies are performed for the Pd1Sn3–TaN/C-m-alkaline (abbreviated as PdSn–TaN/C later in the section of in situ ATR-SEIRAS), Pd–TaN/C, Pd1Sn3/C-m-alkaline (abbreviated as PdSn/C later in the section of in situ ATR-SEIRAS) and commercial Pd/C catalysts in a 1 M KOH/1 M CH3OH solution, which are recorded from 0 to 1.4 V and shown in Fig. 5. The band assignments in in situ ATR-SEIRAS spectra are summarized in Table 2. Fig. 5(a), (c), (e) and (g) display the bands at wave numbers between 3000 and 4000 cm−1. The band at around 3000 cm−1 can be assigned to the C–H stretching vibration of adsorbed CH3OH and/or CH3O.69,70 The band at around 3600 cm−1 can be assigned to the OH stretching vibration, as reported previously.71 A detailed and quantitative fitting analysis of the band at around 3600 cm−1 can be seen later. Moreover, from a qualitative point of view, as the potential increases, it is evident that the ν(O–H) bands of PdSn–TaN/C and Pd–TaN/C shift to positive positions, while almost no shift is observed for commercial Pd/C. According to the study by Chen et al., the potential dependence of ν(O–H) band positions indicates that the OH species are adsorbed on the catalyst surface with co-adsorbed COad.72 Therefore, both the presence and shifts of ν(O–H) bands are considered to be beneficial for the oxidation and removal of COad. | ||
| Fig. 5 In situ ATR-SEIRAS spectra of PdSn–TaN/C (a and b), Pd–TaN/C (c and d), PdSn/C (e and f) and commercial Pd/C (g and h) in a 1 M KOH/1 M CH3OH solution (the reference spectra of (a), (c), (e) and (g) are at 1.4 V; the reference spectra of (b), (d), (f) and (h) are at 0 V; the dashed lines in (a), (c), (e) and (g) represent the deconvoluted bands at around 3600, 3450 and 3270 cm−1). | ||
| Wavenumbers/cm−1 | Assignments |
|---|---|
| a ν, stretching vibration; δ, bending vibration. | |
| ∼1050 | ν(C–O) of surface CH3OH and/or CH3O |
| ∼1440 | δ(C–H) of surface CH3OH and/or CH3O |
| ∼1580 | ν(OCO) of asymmetric HCOOad |
| ∼1650 | δ(H–O–H) of interfacial H2O |
| 1680–1860 | ν(C–O) of COad (multi-bonded CO and bridge-bonded CO band) |
| 2030–2090 | ν(C–O) of COad (linear-bonded CO band) |
| 2500–3000 | ν(C–H) stretching of adsorbed CH3OH and/or CH3O |
| 3000–3700 | ν(O–H) of H2O and interfacial water |
To clearly study the intermediates generated from the MOR in alkaline medium, the reference spectra of Fig. 5(b), (d), (f) and (h) are recorded at 0 V, where the MOR is suppressed.73,74 The band at around 1650 cm−1 in Fig. 5(b), (d), (f) and (h) can be regarded as the bending vibration of interfacially absorbed H2O.75 The altered intensity of the H2O band with the variation of the applied potential may be due to the bending deformations of the interfacial H2O.75–77 However, the strong signal from interfacial H2O makes it hard to distinguish the band near the H2O band, i.e. stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at near 1630 cm−1 (ref. 75 and 78) and the HCO3− band at around 1673 cm−1.76,79 The band at around 1580 cm−1 in Fig. 5(b), (d), (f) and (h) can be assigned to the asymmetric stretching vibration of the adsorbed HCOOad, consistent with the band observed in reported studies.69,71,75,79–83 As shown in Fig. 5(b), (d), (f) and (h), for PdSn–TaN/C and Pd–TaN/C, the intensity of the HCOOad band grows with the increase of the potential, while the position of the band hardly changes with the altered potential, similar to previous reports.74 Specifically, it is also clearly observed that PdSn–TaN/C exhibits a higher intensity of the HCOOad band than Pd–TaN/C, indicating that a larger amount of HCOOad as the intermediate is generated on PdSn–TaN/C. However, for commercial Pd/C and PdSn/C, no obvious HCOOad band is observed, implying that HCOOad is not the intermediate. In Fig. 5(b), (d), (f) and (h), two small bands at around 2030–2090 cm−1 correspond to the linear-bonded CO band, which may be caused by the CO species adsorbed on the different active sites, such as terraces or step sites.3,72,79,84,85 The bands at 1680–1860 cm−1 in Fig. 5(b), (d), (f) and (h) are assigned to the multi-bonded CO and bridge-bonded CO band.38,71,74,79,84,86,87 These bands can also be displayed in Fig. S4(a), (c), (e) and (g).† The reference spectrum is at 1.4 V, where COad is completely oxidized and no adsorbed CO is observed on the surface.72,88 This makes it easy to analyze the varied trend of COad and calculate the integrated absorbance of COad for the subsequent quantitative analysis in the study, consistent with the rules in previous reports.71,72,74 In Fig. S4(a), (c), (e) and (g),† the adsorbed COad (i.e. COB plus COL) species initially appear at low potentials, in line with reported results.71,74,79 The COad species on the Pd catalysts at lower potentials are produced by the dissociative adsorption of methanol.3,71,74,79,89 Yang et al.71 also confirmed that the stepwise dehydrogenation of methanol was easy to occur on the Pd surface in alkaline media, as shown in the following eqn (7)–(11), whereas it hardly occurred in acidic media.
O at near 1630 cm−1 (ref. 75 and 78) and the HCO3− band at around 1673 cm−1.76,79 The band at around 1580 cm−1 in Fig. 5(b), (d), (f) and (h) can be assigned to the asymmetric stretching vibration of the adsorbed HCOOad, consistent with the band observed in reported studies.69,71,75,79–83 As shown in Fig. 5(b), (d), (f) and (h), for PdSn–TaN/C and Pd–TaN/C, the intensity of the HCOOad band grows with the increase of the potential, while the position of the band hardly changes with the altered potential, similar to previous reports.74 Specifically, it is also clearly observed that PdSn–TaN/C exhibits a higher intensity of the HCOOad band than Pd–TaN/C, indicating that a larger amount of HCOOad as the intermediate is generated on PdSn–TaN/C. However, for commercial Pd/C and PdSn/C, no obvious HCOOad band is observed, implying that HCOOad is not the intermediate. In Fig. 5(b), (d), (f) and (h), two small bands at around 2030–2090 cm−1 correspond to the linear-bonded CO band, which may be caused by the CO species adsorbed on the different active sites, such as terraces or step sites.3,72,79,84,85 The bands at 1680–1860 cm−1 in Fig. 5(b), (d), (f) and (h) are assigned to the multi-bonded CO and bridge-bonded CO band.38,71,74,79,84,86,87 These bands can also be displayed in Fig. S4(a), (c), (e) and (g).† The reference spectrum is at 1.4 V, where COad is completely oxidized and no adsorbed CO is observed on the surface.72,88 This makes it easy to analyze the varied trend of COad and calculate the integrated absorbance of COad for the subsequent quantitative analysis in the study, consistent with the rules in previous reports.71,72,74 In Fig. S4(a), (c), (e) and (g),† the adsorbed COad (i.e. COB plus COL) species initially appear at low potentials, in line with reported results.71,74,79 The COad species on the Pd catalysts at lower potentials are produced by the dissociative adsorption of methanol.3,71,74,79,89 Yang et al.71 also confirmed that the stepwise dehydrogenation of methanol was easy to occur on the Pd surface in alkaline media, as shown in the following eqn (7)–(11), whereas it hardly occurred in acidic media.
| Pd + CH3OH ⇔ Pd–CH3OHad | (7) |
| Pd–CH3OHad → Pd–CH2OHad + H+ + e− | (8) |
| Pd–CH2OHad + Pd → Pd2–CHOHad + H+ + e− | (9) |
| Pd2–CHOHad + Pd → Pd3–COHad + H+ + e− | (10) |
| Pd3–COHad → 2Pd + Pd–COad + H+ + e− | (11) |
At high potentials, for one thing, it is observed that the intensities of the COad bands for all the catalysts decrease as a function of increasing potential. This indicates that the oxidative removal of COad exceeds the formation process of COad arising from both the dissociative adsorption of methanol and the subsequent methanol oxidation.72,79 For another, it is also found that as the potential increases firstly, the positions of the COad bands shift to higher wave numbers relative to that at lower potentials, which can be explained by the general Stark effect.71,73,74,79 However, the opposite result occurs at higher potentials, which may result from the decrease of the COad lateral interaction.74 Different from PdSn–TaN/C and Pd–TaN/C, for commercial Pd/C and PdSn/C, there are no obvious HCOOad bands or other intermediates except for CO bands, indicating that methanol oxidation follows the indirect CO pathway on commercial Pd/C and PdSn/C. Furthermore, the band at around 1440 cm−1 in Fig. 5(b), (d), (f) and (h) represents the bending vibration band of C–H scissoring of CH3OH and/or CH3O.71,80 In Fig. S4(b), (d), (f) and (h),† the band at around 1050 cm−1 corresponds to the C–O stretching vibration of surface CH3OH and/or CH3O.78 These bands are typical CH3OH molecules, consistent with previous reports.80
Specifically, to shed more light on the possible mechanism, from the perspective of quantitative evaluation, the potential-dependence of the integrated absorbance of COad, HCOOad and OH bands of PdSn–TaN/C, Pd–TaN/C, PdSn/C and commercial Pd/C catalysts acquired from the in situ ATR-SEIRAS results (Fig. 5 and S4†) are studied and shown in Fig. 6. As shown in Fig. 6(a), (b) and S5,† in the entire potential range, commercial Pd/C shows the largest integrated COad band intensity and no integrated HCOOad band intensity. PdSn/C shows the smallest integrated COad band intensity and no integrated HCOOad band intensity. PdSn–TaN/C shows the largest integrated HCOOad band intensity and a relatively small integrated COad band intensity. Pd–TaN/C displays moderate integrated HCOOad band and COad band intensities. These results in Fig. 6(a) and (b) indicate that both PdSn–TaN/C and Pd–TaN/C follow the dual-channel pathways including a large amount of HCOOad and a small amount of COad towards the MOR in alkaline medium, while commercial Pd/C and PdSn/C only follow the COad pathway. Therefore, it may be proposed that, compared with commercial Pd/C and PdSn/C, the presence of TaN in PdSn–TaN/C and Pd–TaN/C alters the reaction pathway by opening a HCOOad pathway based on the initial CO pathway. The effect of Sn further enhances the adsorbed HCOOad intensity. Besides, for PdSn–TaN/C in Fig. 6(a), it is also found that the integrated absorbance of the HCOOad band versus the potential is in accord with the activity sequence of the electrocatalytic behaviour in Fig. 4, indicating that HCOOad may be an important intermediate of the MOR in alkaline medium.38,90 Based on previous studies, it has been found that the adsorbed formate can be identified as an intermediate, which is beneficial for the subsequent oxidation due to the fact that its C–H bond is easier to cleave and produce the final CO2 products, consistent with reported HPLC and NMR results.38,71,81,90,91 Moreover, it is generally recognized that COad is a poisoning intermediate because it is hard to oxidize on the Pd surface to relieve the active Pd sites for subsequent oxidation.3,73,79
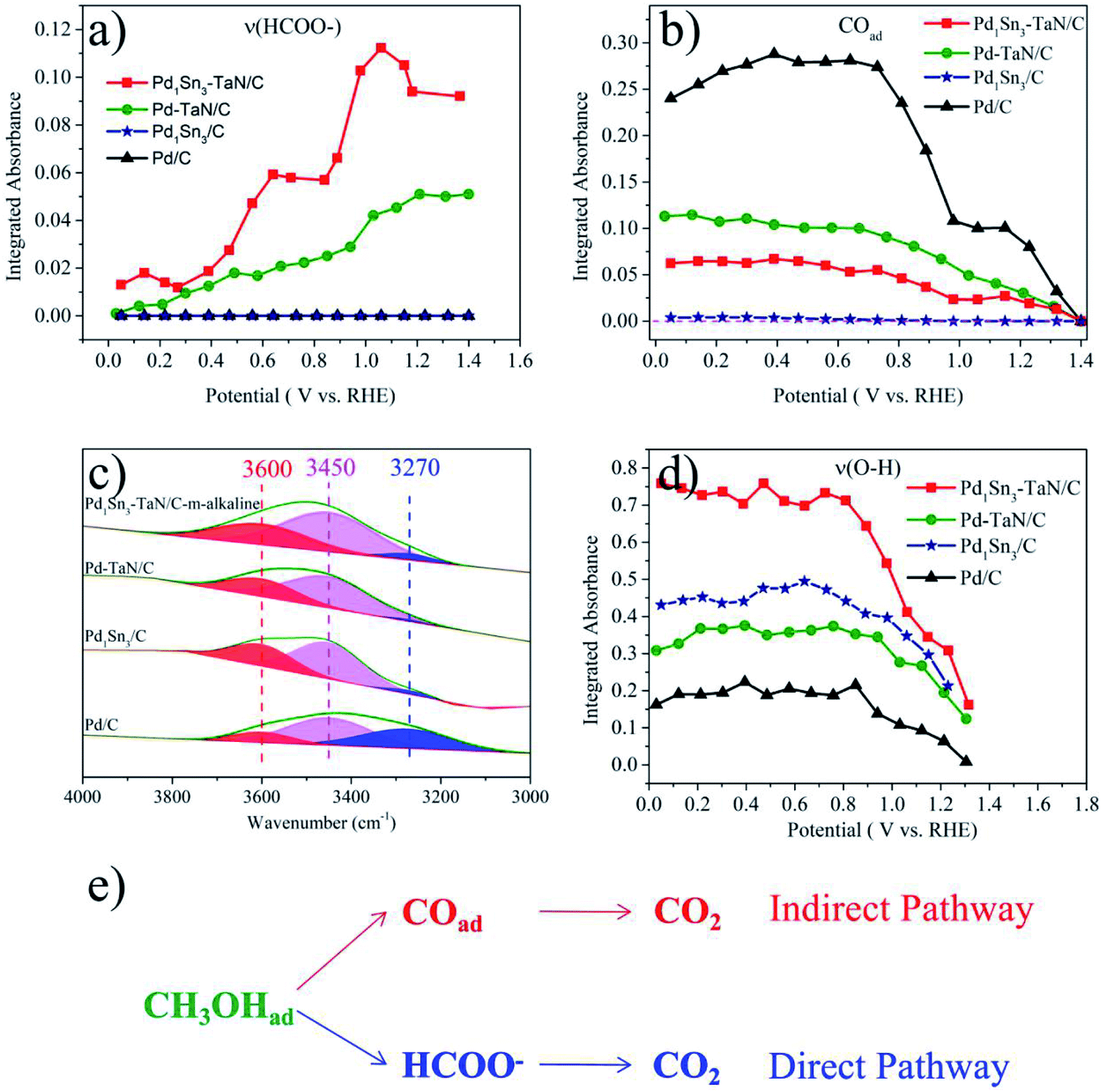 | ||
| Fig. 6 The potential dependence of the intensities of the CO band (a) and formate (b) in in situ ATR-SEIRAS spectra for PdSn–TaN/C, Pd–TaN/C, PdSn/C and commercial Pd/C. The fitting results of the H2O feature band in the range of 3000 to 3800 cm−1 at 0.4 V (c) and the potential dependence of the intensities of the OH band (d) for PdSn–TaN/C, Pd–TaN/C, PdSn/C and commercial Pd/C. The proposed mechanism of the MOR on PdSn–TaN/C in alkaline medium (e). | ||
In addition to the above possible intermediates, the variation of the interfacial adsorbed H2O feature band in the wavenumber range of 3000 to 3700 cm−1 is also investigated. The detailed fitting analysis and potential dependence at 3600 cm−1 can be seen in Fig. 6(c), (d), S6–S8 and Tables S7–S10.† According to previous literature, the deconvoluted bands at around 3600, 3450 and 3270 cm−1 are associated with the dangling O–H bond, trihedrally coordinated water and tetrahedrally coordinated water, respectively, where the first high frequency band is attributed to the stretching vibration of non-hydrogen-bonded adsorbed water molecules (or called isolated interfacial water molecules) adsorbed together with CO and two low frequency bands are attributed to hydrogen-bonded water molecules adsorbed together with CO.70–72,85,87,88,92–95 More specifically, the tetrahedrally coordinated water and trihedrally coordinated water correspond to the strongly hydrogen-bonded ice-like water molecules and disordered weakly hydrogen-bonded water molecules.89 As proposed by Hanawa and coworkers,92 the production of adsorbed OH species from the dissociation of non-hydrogen-bonded adsorbed water molecules is easier than that from hydrogen-bonded adsorbed water molecules, thereby resulting in more accessible non-hydrogen-bonded adsorbed water molecules to provide the adsorbed OH species.
According to bi-functional theory, the OH group helps to remove the adsorbed COad by facilitating the subsequent oxidation of COad on the basis of the Langmuir–Hinshelwood (L–H) mechanism.32,38,72,74
| H2O → OHad + H+ + e− | (12) |
| Pd–COad + OHad → Pd + CO2 + H+ + e− | (13) |
On the one hand, it can be seen from Fig. 6(d) that PdSn–TaN/C shows a rather stronger OH band than Pd–TaN/C, PdSn/C and commercial Pd/C. Besides, as shown in Fig. S7 and Tables S7–S10,† compared with commercial Pd/C, it can be found that the presence of TaN and Sn decreases the proportion of the strongly hydrogen-bonded ice-like water molecules of PdSn/C, PdSn–TaN/C and Pd–TaN/C, indicating that TaN and Sn weaken the hydrogen network interaction of the interfacial water. It also means that the OH species are easily generated from the dissociation of water on the surfaces of PdSn/C, PdSn–TaN/C and Pd–TaN/C. This may be due to the fact that TaN favors the exposure of interfacial active sites by the special core–shell like structure. Meanwhile, the OH band intensity of PdSn–TaN/C and PdSn/C in Fig. 6(d) is stronger than that of Pd–TaN/C, which may be ascribed to the highly oxophilic nature of Sn. Thus, it can be confirmed that the additions of TaN and Sn can be responsible for the variation in the interfacial water structure by modulating the adsorption behaviors, that is, the interaction with the interfacial water via the hydrogen bond. In short, the presence of TaN and Sn can increase the adsorption of the dangling OH species free from hydrogen bonds. Furthermore, noticeably, the combination of Fig. 4(d) and 6(d) indicates that PdSn/C shows a higher OH band intensity than Pd–TaN/C; however PdSn/C shows a lower catalytic activity than Pd–TaN/C, which can be explained by the smaller ECSA of PdSn/C compared with Pd–TaN/C. This also implies the effectiveness of the interface interaction provided by TaN.
Furthermore, as shown in Fig. 6(d), S8 and Tables S7–S10,† it can be found that the integrated OH band intensities initially remain almost unchanged followed by a rapid decrease with the increase of the potential. Interestingly, the trend of the integrated COad band intensities is approximately consistent with that of the OH band as a function of the potential, which implies that OH takes part in the oxidative removal of COad.72,92 As proposed by Xu et al.,88 a similar varied trend of the OH band at 3600 cm−1 and the COad band suggested that the OH stretching of the non-hydrogen-bonded adsorbed water molecules (or called isolated interfacial water molecules) was promoted by CO-adsorption, in which the OH species were embedded in the CO adsorption layer. Therefore, it can be concluded that the OH band associated with non-hydrogen-bonded adsorbed water molecules (at 3600 cm−1) will react with the adsorbed CO and the anti-CO poisoning ability will be enhanced on PdSn–TaN/C. The non-hydrogen-bonded adsorbed water molecules are considered to be the main sources of the adsorbed OH species, consistent with previous results.89,92
On the other hand, for PdSn–TaN/C, Pd–TaN/C and PdSn/C, the adsorbed H2O feature band positions shift toward the positive direction with the increase of the potential, whereas almost no shifts of the H2O bands are observed for commercial Pd/C. The positive shifts of the H2O feature band positions can be attributed to the formation of more non-hydrogen-bonded adsorbed water molecules co-adsorbed with COad on the surface. This further confirms the enhanced anti-CO poisoning ability of PdSn–TaN/C and Pd–TaN/C.
In a word, compared with Pd–TaN/C, PdSn/C and commercial Pd/C, for PdSn–TaN/C, TaN helps the MOR to proceed along the HCOOad route and CO route, and the Sn further enhances the intensity of HCOOad in the dual-channel pathways and promotes the adsorption of OH species. Based on the above analysis, a possible mechanism of the MOR in alkaline media on PdSn–TaN/C is proposed and displayed in Fig. 6(e), where the formate is the active intermediate.
3.4. DFT calculations
DFT calculations are used to confirm in situ ATR-SEIRAS and electrochemical results and study the effect of OH on the catalytic performance for the MOR. The binding energies of CO and OH are calculated on the Pd(111), Pd4/TaN(001) and Pd4–SnO2/TaN(001) surfaces. The adsorption configurations are shown in Fig. 7(a). It can be indicated that the CO binding energy is only −0.85 eV on the Pd4–SnO2/TaN(001) surface, lower than −2.18 and −1.36 eV on Pd(111) and Pd4/TaN(001). The result suggests that the existence of TaN and Sn is beneficial for the desorption of COad on the catalyst surface, which is in agreement with the trend of COad band intensity (Fig. 6(b)). For the OH group, the binding energy is up to −4.42 eV, higher than −2.26 and −3.86 eV on Pd(111) and Pd4/TaN(001), implying that the interaction between OH and the catalyst surface is enhanced evidently, confirmed by the results of integrated OH band intensities in in situ ATR-SEIRAS (Fig. 6(d)). Thus, doping TaN and Sn into Pd-based catalysts can bring about a weakened CO binding energy and enhanced OH binding energy, suggesting that the anti-CO poisoning ability of Pd4–SnO2/TaN(001) is enhanced for the MOR, which is consistent with the electrochemical and in situ ATR-SEIRAS measurements. | ||
| Fig. 7 (a) The optimized adsorption configurations of CO and OH on the Pd(111), Pd4/TaN(001) and Pd4–SnO2/TaN(001) surfaces; (b) the potential energy profiles of COad + OHad to COOHad on the Pd(111), Pd4/TaN(001) and Pd4–SnO2/TaN(001) surfaces. | ||
According to the in situ ATR-SEIRAS results (Fig. 6), it can be found that the OH group helps to remove the adsorbed COad by facilitating the subsequent oxidation of COad on the catalyst surface. So, we further calculate the energy barriers of the reaction CO + OH → COOH on the Pd(111), Pd4/TaN(001) and Pd4–SnO2/TaN(001) surfaces. The configurations of the initial states, transition states and final states are shown in Fig. S9–S11.† The potential energy profiles of the reaction are shown in Fig. 7(b). Compared with Pd(111), it can be found that the energy barriers of the reaction CO + OH → COOH are reduced evidently on the Pd4/TaN(001) and Pd4–SnO2/TaN(001) surfaces. The activation of CO + OH to COOH only require 0.58 eV on Pd4–SnO2/TaN(001). Compared with the desorption energy of CO, it can be found that the existence of OH species can promote the CO oxidation to CO2 on the three surfaces. Thus, the OH concentration dispersed on the catalyst surface is important for CO conversion. Thus, both the DFT calculations and the in situ ATR-SEIRAS results confirm that the high concentration of OH should bring about CO removal on the PdSn/TaN catalysts. It can be indicated that the energy barrier of COOH formation is 0.54 eV on the Pd4–SnO2/TaN(001) surface, lower than the values of 0.90 and 1.13 eV on Pd/TaN(001) and Pd(111). In other words, the coadsorption intensity and amount of OH provide a chance for CO conversion into CO2 on Pd4–SnO2/TaN(001) and then bring about the enhancement of anti-CO poisoning ability.
3.5. Brief discussion
Considering that pure TaN or Sn is not active for the MOR (Fig. S12†), it is confirmed that the interconnection among Pd, Sn and TaN is an important part of the improvement of catalytic performance. For one thing, relative to the Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts, the Pd–TaN/C catalysts show larger ECSAs, indicating that the presence of TaN is beneficial to expose more Pd active sites, which may be due to the fact that the unique core–shell like structures provide more interfacial interconnection, revealed by HRTEM and XPS results. For another, on the basis of the Pd–TaN catalysts, the synthesis parameters and Sn contents are further elaborately controlled to adjust the oxophilicity properties of PdxSny–TaN/C. The results show that the optimized Pd1Sn3–TaN/C-m-alkaline shows the highest mass activity of 3293.46 A gPd−1 towards the MOR in alkaline media, which is 15.64 times that of the commercial Pd/C catalyst (210.53 A gPd−1) and also superior to that of other catalysts. Here, to gain insight into the mechanism for the enhanced catalytic performance of PdxSny–TaN/C from the inherent electronic structure, the valence state spectra of XPS characterization are recorded and shown in Fig. 8(a), consistent with our previous work.36,96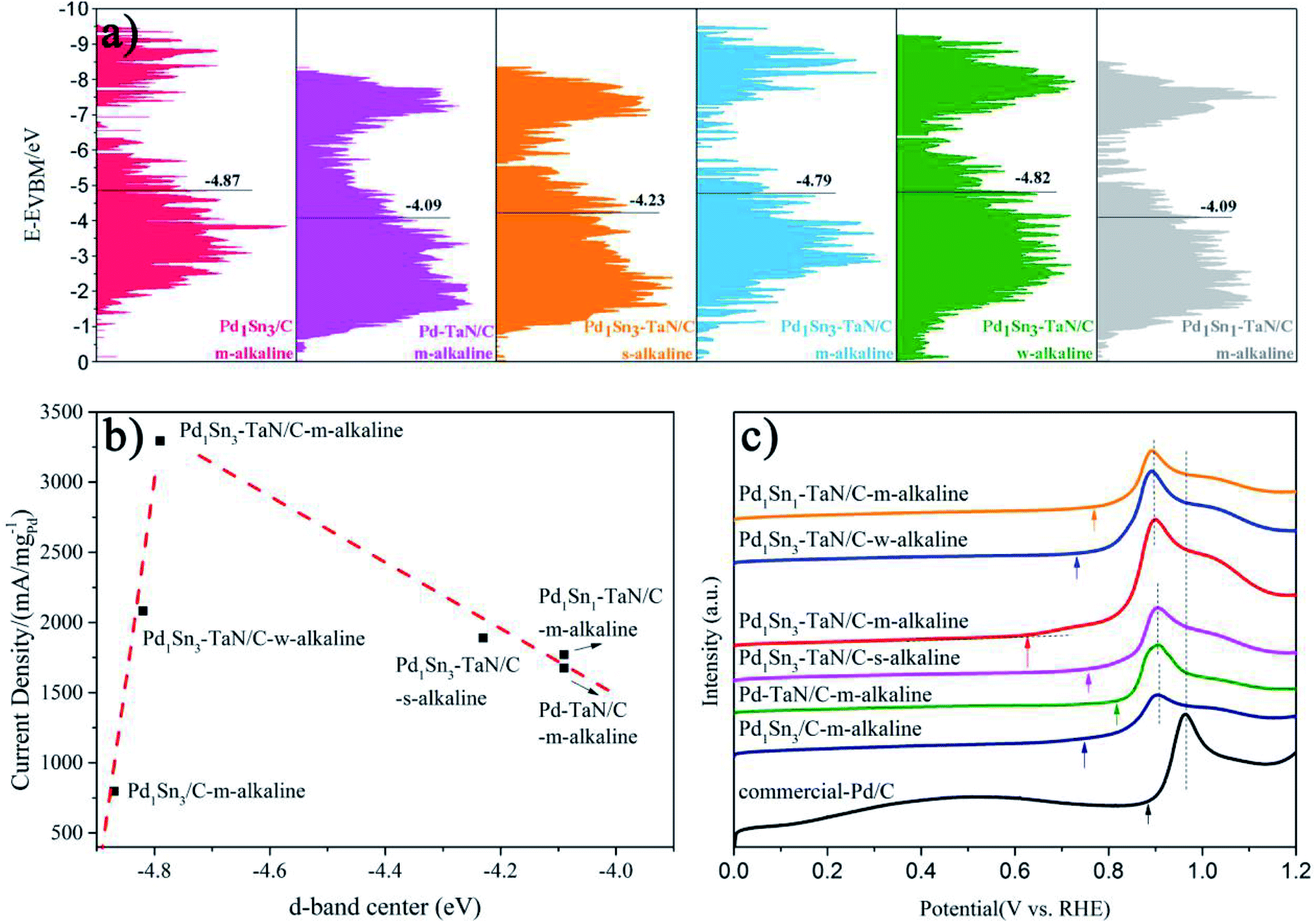 | ||
| Fig. 8 (a) Valence band spectra relative to the valence band maximum (after the subtraction of the Shirley background), (b) d-band center vs. catalytic performance for the MOR in alkaline media and (c) the CO stripping voltammograms of the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts (the arrows in (c) represent the onset potential of CO oxidation, and the dashed lines in (c) represent the peak potential of CO oxidation). | ||
Based on d-band center theory, the position of the d-band center is responsible for the binding strength between intermediates and catalysts.97,98 Specifically, when adsorbates adsorb on the metal surface, hybridization between the adsorbates and catalysts will occur. Fully filled bonding orbits as well as the partially filled anti-bonding orbits* are formed, where the high-energy anti-bonding orbits* are responsible for the locations of the d-band center and the binding strength.97–102 It is observed that the d-band center of Pd1Sn3/C-m-alkaline is far away from the Fermi level, whereas that of Pd–TaN/C-m-alkaline is located at the nearest position. The d-band centers of PdxSny–TaN/C fall in between them. The d-band centers shift up towards the Fermi level in the following order of Pd1Sn3/C-m-alkaline (−4.87) < Pd1Sn3–TaN/C-w-alkaline (−4.82) < Pd1Sn3–TaN/C-m-alkaline (−4.79) < Pd1Sn3–TaN/C-s-alkaline (−4.23) < Pd1Sn1–TaN/C-m-alkaline (−4.09) ≈ Pd–TaN/C-m-alkaline (−4.09). With the increase of alkaline contents, the d-band centers with respect to the Fermi level increase significantly by comparing the three Pd1Sn3–TaN/C catalysts. This can be attributed to the increasing number of the high-valence Sn and Pd states, which provides fewer electrons to fill the anti-bonding orbitals.97–99 Interestingly, a volcano-like correlation between the d-band center and the catalytic performances of the MOR in alkaline medium is observed as shown in Fig. 8(b), in which Pd1Sn3–TaN/C-m-alkaline is located at the summit of the volcano. The optimized d-band electronic structure of Pd1Sn3–TaN/C-m-alkaline reveals that the introduction of Sn can further tailor the interfacial electronic structures. The downshifted d-band center is vital to weaken the binding strength of CO, thereby enhancing the CO tolerance.101 This can be further demonstrated by the above in situ ATR-SEIRAS and subsequent CO oxidation results.
To evaluate the tolerance to carbonaceous (i.e. CO-like) intermediates for the MOR, CO stripping tests are carried out. As shown in Fig. 8(c), compared with Pd/C, the onset potentials of CO oxidation for the PdxSny–TaN/C catalysts are obviously shifted negatively, revealing that the introduction of Sn or TaN is effective to remove the CO species on the catalyst surfaces. The order of the onset potentials of CO oxidation for PdxSny–TaN/C is Pd1Sn3–TaN/C-m-alkaline (0.626) < Pd1Sn3–TaN/C-w-alkaline (0.732) < Pd1Sn3–TaN/C-s-alkaline (0.757) < Pd1Sn1–TaN/C-m-alkaline (0.769) < Pd–TaN/C-m-alkaline (0.817) < commercial Pd/C (0.884). The results indicate that Pd1Sn3–TaN/C-m-alkaline has the most negative onset potential of CO oxidation among these catalysts, indicating that the CO oxidation easily occurs on Pd1Sn3–TaN/C-m-alkaline at low potential. There are two reasons for the enhancement of the anti-CO poisoning ability. For one thing, as depicted in the d-band center results, the decreased d-band center of Pd1Sn3–TaN/C-m-alkaline results in the weakened adsorption of CO species, confirmed by DFT calculations. For another, Sn has more oxophilicity than Pd, which is beneficial for the formation of OHad that can react with COad, as confirmed by CV (Fig. 4), in situ ATR-SEIRAS (Fig. 5) and DFT results. It accelerates the removal of CO-like poisoning intermediates as a result of the dual-functional effect on the basis of the Langmuir–Hinshelwood mechanism.68,74,103 Besides, the enhanced anti-CO poisoning abilities of PdxSny–TaN/C also contribute to the release of the active sites, thereby improving their catalytic stabilities, in agreement with the CA results.
What's more, to further reveal the intrinsic mechanism of the tunable catalytic performance after the addition of Sn to Pd–TaN/C, the mass activities of the PdxSny–TaN/C catalysts are studied as a function of the valence states of Pd and Sn. A volcano-shaped correlation is observed between the mass activities and the ratios of the valence states for Pd and Sn, shown in Fig. 9(a) and (b), indicating that the appropriate ratios of oxidation states of Pd and Sn contribute to the enhanced catalytic activity of the MOR in alkaline medium. Specifically, the increase of KOH and Sn contents in the synthesis process can bring about the improvement of Pdδ+ and Snδ+ in the PdxSny–TaN/C catalysts, which can be quantitatively evaluated by the potential of PdO in CV tests in KOH solution. It is observed that Pdδ+ and Snδ+ in PdxSny–TaN/C are highly consistent with the potential of PdO for the PdxSny–TaN/C catalysts, as shown in Fig. 9(c), indicating that the oxophilicity can be precisely controlled. Furthermore, it is found that the onset potential of CO stripping and Pd–OHad for the PdxSny–TaN/C catalysts display similar volcanic trends, as shown in Fig. 9(d) and (e). Notably, there exists a nearly linear correlation between the onset potential of CO stripping and Pd–OHad adsorption, implying that the presence of Sn in the PdxSny–TaN/C catalysts promotes the tolerance to CO-like poisoning species by a dual-functional mechanism, in accord with the above in situ ATR-SEIRAS characterization results. The results also indicate that the catalytic performance of PdxSny–TaN/C catalysts is modulated by the oxophilicity of Sn, in which OHad determines the anti-CO poisoning ability.
 | ||
| Fig. 9 (a) The MOR performance in alkaline media, (b) the valence states of Pd and Sn, (c) the peak potential of PdO, (d) the onset potential of CO stripping and (e) the onset potential of Pd–OHad for the PdxSny–TaN/C catalysts and Pd–TaN/C catalysts. | ||
As a result, the finely tailored Pd1Sn3–TaN/C-m-alkaline catalyst exhibits good catalytic performances for the MOR, which can be ascribed to the interfacial structures and bi-functional effects. Briefly, the core–shell like structure can provide an effective Pd–TaN/C interface, and the introduction of Sn can synergistically modulate the interfacial electronic structures, thereby significantly promoting the MOR performances. Besides, in view of the reaction pathway, compared with commercial Pd/C, PdSn–TaN/C goes through the dual-channel pathways including a large amount of HCOOad and a small amount of COad towards MOR in alkaline medium. With the assistance of Sn, the process is accompanied by an enhanced adsorption of the dangling OH species associated with the non-hydrogen-bonded adsorbed interfacial H2O.
Interestingly, the PdxSny–TaN/C catalysts also show remarkable FAOR catalytic performance in acidic media. As depicted in Fig. 10(a) and Table S11,† the Pd1Sn3–TaN/C-m-alkaline catalyst shows the highest mass activity of 2097.29 A gPd−1, which is 9.8 times that of the commercial Pd/C catalyst (215.79 A gPd−1) and is also superior to that of other catalysts (Table S12†). Also, Pd1Sn3–TaN/C-m-alkaline displays the lowest onset potential and the smallest Rct for the FAOR in acidic media among these catalysts in LSV and Nyquist plots (Fig. 10(b), (c), S13 and Table S13†), indicating that the formic acid molecule is easily oxidized on the Pd1Sn3–TaN/C-m-alkaline catalyst. As shown in Fig. 10(d), the catalytic durability is also investigated at 0.25 V in an Ar-saturated 0.5 M HCOOH/0.5 M H2SO4 solution. It is found that Pd1Sn3–TaN/C-m-alkaline shows a higher current density than other catalysts from the beginning to the end, indicating that its catalytic stability for the FAOR is also enhanced to some extent. This may be due to the facile Pd–OHad adsorption, which is beneficial for the removal of oxygen-containing species. Overall, the above results confirm that the Pd1Sn3–TaN/C-m-alkaline catalyst may be a promising bi-functional electrocatalyst in appropriate solutions.
 | ||
| Fig. 10 (a) CV curves at 50 mV s−1, (b) LSV curves at 10 mV s−1, (c) Nyquist plots with simplified Randles equivalent circuits at 0.1 V and (d) CA curves at 0.25 V of the PdxSny–TaN/C, Pd1Sn3/C-m-alkaline and commercial Pd/C catalysts in an Ar-saturated 0.5 M HCOOH/0.5 M H2SO4 solution. | ||
4. Conclusion
In this work, a series of PdxSny–TaN/C with varied oxophilicity have been successfully prepared via a facile solvothermal method, in which the synthesis parameters and Sn contents are elaborately regulated. As demonstrated by HRTEM and XPS, the optimized Pd1Sn3–TaN/C-m-alkaline catalyst shows a special core–shell like interfacial structure accompanied by precisely controlled oxophilicity. Based on electrochemical measurements, Pd1Sn3–TaN/C-m-alkaline with a unique structure shows the highest mass activity of 3293.46 A gPd−1 towards the MOR in alkaline media, which is 15.64 times that of the commercial Pd/C catalyst (210.53 A gPd−1) and also superior to that of other catalysts, suggesting a remarkable improvement of the activity for the MOR with the introduction of TaN and Sn. In the meantime, the catalytic durability and anti-CO poisoning abilities towards the MOR in alkaline medium of Pd1Sn3–TaN/C-m-alkaline are also superior to those of other catalysts, which can be due to the negative shift of the d-band center by weakening the adsorption of CO. Specifically, the core–shell like structure can provide an effective Pd–TaN/C interface related to the large ECSA, and the introduction of Sn can further synergistically modulate the interfacial electronic structures, thereby significantly promoting the MOR performances, as revealed by HRTEM, XPS and electrochemical results. In other words, TaN is responsible for providing effective active sites for the MOR in alkaline medium. And after the subsequent introduction of Sn, the catalytic performances of PdxSny–TaN/C catalysts can be further controlled by the varied valence states related to the oxophilic nature. In situ ATR-SEIRAS measurements show that the difference of the catalytic performance is attributed to the change of the reaction pathway on PdSn–TaN/C, Pd–TaN/C and Pd/C. For PdSn–TaN/C and Pd–TaN/C, they proceed via dual pathways with a large amount of formate as the important intermediate. In other words, except the CO pathway, methanol oxidation on PdSn–TaN/C and Pd–TaN/C can also occur via the HCOOad pathway on PdSn–TaN/C followed by the facile cleavage of the C–H bond to produce CO2 subsequently. However, only the COad pathway occurs on the commercial Pd/C and PdSn/C catalysts. At the same time, the CO adsorptions of PdSn–TaN/C, Pd–TaN/C and PdSn/C are lower than that of commercial Pd/C. The reason can be ascribed to the fact that the promoted adsorption intensity of OH from the introduction of TaN and Sn helps to remove COad, confirmed by the enhanced adsorption of the dangling OH species related to non-hydrogen-bonded adsorbed interfacial H2O in in situ ATR-SEIRAS, electrochemical and DFT results. Meanwhile, the optimized Pd1Sn3–TaN/C-m-alkaline also shows the highest mass activity of 2097.29 A gPd−1 towards the FAOR in acidic media, 9.8 times that of the commercial Pd/C catalyst (215.79 A gPd−1), which further confirms that Pd1Sn3–TaN/C-m-alkaline may be a promising bi-functional electrocatalyst in appropriate media. This work lays the foundation and provides fundamental guidance for the design and preparation of efficient and inexpensive TaN-modified Pd-based catalysts for direct liquid fuel cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075225, 21706203 and 22038011), Special Guidance Funds for the Construction of World-Class Universities (Disciplines) and Characteristic Development in Central Universities (PY3A076), China Postdoctoral Science Foundation (2020T130508, 2019M660258 and 2019M663731), Natural Science Basic Research Plan in Shaanxi Province of China (2017JQ2030 and 2020JQ023), Fundamental Research Funds for the Central Universities (cxtd2017004, xjj2018035 in Xi'an Jiaotong University), Shaanxi Creative Talents Promotion Plan-Technological Innovation Team (2019TD-039), research funding from the Joint Laboratory of Xi'an Jiaotong Univ. and Shaanxi Coal Chemical Industry Technology Research Institute Co. Ltd. The authors would like to thank Dr Jiao Li, Engineer Jiamei Liu and Engineer Yu Wang at the Instrument Analysis Center of Xi'an Jiaotong University, who provided guidance of characterization.References
- J. Fan, H. Du and Y. Zhao, et al., Recent Progress on Rational Design of Bimetallic Pd Based Catalysts and Their Advanced Catalysis, ACS Catal., 2020, 10, 13560–13583 CrossRef CAS.
- V. L. Marinho, E. Antolini and M. J. Giz, et al., Ethylene glycol oxidation on carbon supported binary PtM (M = Rh, Pd an Ni) electrocatalysts in alkaline media, J. Electroanal. Chem., 2021, 880, 114859 CrossRef CAS.
- J. Zhang, X. Qu and Y. Han, et al., Engineering PtRu bimetallic nanoparticles with adjustable alloying degree for methanol electrooxidation: Enhanced catalytic performance, Appl. Catal., B, 2020, 263, 118345 CrossRef CAS.
- F. Gao, Y. Zhang and F. Ren, et al., Universal Surfactant-Free Strategy for Self-Standing 3D Tremella-Like Pd–M (M = Ag, Pb, and Au) Nanosheets for Superior Alcohols Electrocatalysis, Adv. Funct. Mater., 2020, 30, 2000255 CrossRef CAS.
- C. Su, W. Wang and Z. Shao, Cation-Deficient Perovskites for Clean Energy Conversion, Acc. Mater. Res., 2021, 2, 477–488 CrossRef CAS.
- H. C. Lau, S. Ramakrishna and K. Zhang, et al., The Role of Carbon Capture and Storage in the Energy Transition, Energy Fuels, 2021, 35, 7364–7386 CrossRef CAS.
- P. Gabrielli, M. Gazzani and M. Mazzotti, The Role of Carbon Capture and Utilization, Carbon Capture and Storage, and Biomass to Enable a Net-Zero-CO2 Emissions Chemical Industry, Ind. Eng. Chem. Res., 2020, 15, 7033–7045 CrossRef.
- T. Shen, S. Chen and R. Zeng, et al., Tailoring the Antipoisoning Performance of Pd for Formic Acid Electrooxidation via an Ordered PdBi Intermetallic, ACS Catal., 2020, 10, 9977–9985 CrossRef CAS.
- G. Liu, J. Shi and F. Zhang, et al., A Tantalum Nitride Photoanode Modified with a Hole-Storage Layer for Highly Stable Solar Water Splitting, Angew. Chem., Int. Ed., 2014, 53, 7295–7299 CrossRef CAS PubMed.
- M. Yang, A. R. Van Wassen and R. Guarecuco, et al., Nano-structured ternary niobium titanium nitrides as durable non-carbon supports for oxygen reduction reaction, Chem. Commun., 2013, 49, 10853 RSC.
- M. Yang, A. R. Van Wassen and R. Guarecuco, et al., Nano-structured ternary niobium titanium nitrides as durable non-carbon supports for oxygen reduction reaction, Chem. Commun., 2013, 49, 10853 RSC.
- F. Yu, Y. Xie and H. Tang, et al., Platinum decorated hierarchical porous structures composed of ultrathin titanium nitride nanoflakes for efficient methanol oxidation reaction, Electrochim. Acta, 2018, 264, 216–224 CrossRef CAS.
- J. Zhang, L. Ma and M. Gan, et al., TiN@nitrogen-doped carbon supported Pt nanoparticles as high-performance anode catalyst for methanol electrooxidation, J. Power Sources, 2016, 324, 199–207 CrossRef CAS.
- Z. Cui, M. Yang and F. J. Disalvo, MO2N/C hybrid material as a promising support for the electro-oxidation of methanol and formic acid, Electrochem. Commun., 2013, 33, 63–67 CrossRef CAS.
- T. D. C. Nguyen, T. P. L. C. Nguyen and H. T. T. Mai, et al., Novel photocatalytic conversion of CO2 by vanadium-doped tantalum nitride for valuable solar fuel production, J. Catal., 2017, 352, 67–74 CrossRef CAS.
- T. Pham, B. Lee and V. N. Nguyen, et al., RETRACTED: Novel photocatalytic activity of vanadium-doped tantalum nitride sensitized/protected by polyaniline for efficient visible light water splitting, J. Catal., 2017, 352, 13–21 CrossRef CAS.
- M. Yang, Z. Cui and F. J. Disalvo, Mesoporous vanadium nitride as a high performance catalyst support for formic acid electrooxidation, Chem. Commun., 2012, 48, 10502–10504 RSC.
- S. Jing, L. Luo and S. Yin, et al., Tungsten nitride decorated carbon nanotubes hybrid as efficient catalyst supports for oxygen reduction reaction, Appl. Catal., B, 2014, 147, 897–903 CrossRef CAS.
- S. Yin, P. Wang and J. Lu, et al., Tungsten nitride decorated CNTs as efficient hybrid supports for PtRh alloys in electrocatalytic ethanol oxidation, Int. J. Hydrogen Energy, 2017, 42, 22805–22813 CrossRef CAS.
- J. Shi, Z. Pu and Q. Liu, et al., Tungsten nitride nanorods array grown on carbon cloth as an efficient hydrogen evolution cathode at all pH values, Electrochim. Acta, 2015, 154, 345–351 CrossRef CAS.
- Y. Xiao, Z. Fu and G. Zhan, et al., Increasing Pt methanol oxidation reaction activity and durability with a titanium molybdenum nitride catalyst support, J. Power Sources, 2015, 273, 33–40 CrossRef CAS.
- X. Pan, X. Song and S. Lin, et al., A facile route to graphite-tungsten nitride and graphite-molybdenum nitride nanocomposites and their ORR performances, Ceram. Int., 2016, 42, 16017–16022 CrossRef CAS.
- J. Masud, M. T. Alam and Z. Awaludin, et al., Electrocatalytic oxidation of methanol at tantalum oxide-modified Pt electrodes, J. Power Sources, 2012, 220, 399–404 CrossRef CAS.
- S. Jing, L. Luo and S. Yin, et al., Tungsten nitride decorated carbon nanotubes hybrid as efficient catalyst supports for oxygen reduction reaction, Appl. Catal., B, 2014, 147, 897–903 CrossRef CAS.
- X. Tian, J. Luo and H. Nan, et al., Transition Metal Nitride Coated with Atomic Layers of Pt as a Low-Cost, Highly Stable Electrocatalyst for the Oxygen Reduction Reaction, J. Am. Chem. Soc., 2016, 138, 1575–1583 CrossRef CAS PubMed.
- M. Roca-Ayats, G. García and J. L. Galante, et al., TiC, TiCN, and TiN Supported Pt Electrocatalysts for CO and Methanol Oxidation in Acidic and Alkaline Media, J. Phys. Chem. C, 2013, 117, 20769–20777 CrossRef CAS.
- E. L. Clark, C. Hahn and T. F. Jaramillo, et al., Electrochemical CO2 Reduction over Compressively Strained CuAg Surface Alloys with Enhanced Multi-Carbon Oxygenate Selectivity, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- S. Yan, Y. Huang and C. Yang, et al., Enhanced activity of ethanol oxidation reaction on PtM (M = Au, Ag and Sn): The importance of oxophilicity and surface oxygen containing species, Electrochim. Acta, 2018, 259, 733–741 CrossRef CAS.
- R. Subbaraman, D. Tripkovic and K. Chang, et al., Trends in activity for the water electrolyser reactions on 3d M (Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- W. Du, G. Yang and E. Wong, et al., Platinum-Tin Oxide Core-Shell Catalysts for Efficient Electro-Oxidation of Ethanol, J. Am. Chem. Soc., 2014, 136, 10862–10865 CrossRef CAS PubMed.
- S. M. Alia, B. S. Pivovar and Y. Yan, Platinum-Coated Copper Nanowires with High Activity for Hydrogen Oxidation Reaction in Base, J. Am. Chem. Soc., 2013, 135, 13473–13478 CrossRef CAS PubMed.
- M. Arenz, K. J. J. Mayrhofer and V. Stamenkovic, et al., The Effect of the Particle Size on the Kinetics of CO Electrooxidation on High Surface Area Pt Catalysts, J. Am. Chem. Soc., 2005, 127, 6819–6829 CrossRef CAS PubMed.
- F. Wu, L. Zhang and J. Lai, et al., Modulating the oxophilic properties of inorganic nanomaterials for electrocatalysis of small carbonaceous molecules, Nano Today, 2019, 29, 100802 CrossRef CAS.
- Z. Liang, L. Song and S. Deng, et al., Direct 12-Electron Oxidation of Ethanol on a Ternary Au(core)-PtIr(Shell) Electrocatalyst, J. Am. Chem. Soc., 2019, 141, 9629–9636 CrossRef CAS PubMed.
- Z. Qi, C. Xiao and C. Liu, et al., Sub-4 nm PtZn Intermetallic Nanoparticles for Enhanced Mass and Specific Activities in Catalytic Electrooxidation Reaction, J. Am. Chem. Soc., 2017, 139, 4762–4768 CrossRef CAS PubMed.
- N. Ye, Y. Bai and Z. Jiang, et al., Design the PdCu/TaN C electrocatalyst with core-shell structure having high efficiency for methanol and formic acid oxidation reactions, Electrochim. Acta, 2021, 383, 138365 CrossRef CAS.
- K. P. Kepp, A Quantitative Scale of Oxophilicity and Thiophilicity, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- Y. Zhou, Y. Chen and K. Jiang, et al., Probing the enhanced methanol electrooxidation mechanism on platinum-metal oxide catalyst, Appl. Catal., B, 2021, 280, 119393 CrossRef CAS.
- Y. Katayama, T. Okanishi and H. Muroyama, et al., Enhanced Supply of Hydroxyl Species in CeO2-Modified Platinum Catalyst Studied by in Situ ATR-FTIR Spectroscopy, ACS Catal., 2016, 6, 2026–2034 CrossRef CAS.
- S. Zhu, X. Qin and Y. Yao, et al., pH-Dependent Hydrogen and Water Binding Energies on Platinum Surfaces as Directly Probed through Surface-Enhanced Infrared Absorption Spectroscopy, J. Am. Chem. Soc., 2020, 142, 8748–8754 CrossRef PubMed.
- J. Heyes, M. Dunwell and B. Xu, CO2 Reduction on Cu at Low Overpotentials with Surface-Enhanced in Situ Spectroscopy, J. Phys. Chem. C, 2016, 120, 17334–17341 CrossRef CAS.
- H. Miyake, S. Ye and M. Osawa, Electroless deposition of gold thin films on silicon for surface-enhanced infrared spectroelectrochemistry, Electrochem. Commun., 2002, 4, 973–977 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- H. Wang, Z. Liu and S. Ji, et al., Ethanol oxidation activity and structure of carbon-supported Pt-modified PdSn-SnO2 influenced by different stabilizers, Electrochim. Acta, 2013, 108, 833–840 CrossRef CAS.
- X. Ji, X. Song and J. Li, et al., Size Control of Gold Nanocrystals in Citrate Reduction: The Third Role of Citrate, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS PubMed.
- Q. Wang, C. Xu and W. Liu, et al., Coordination engineering of iridium nanocluster bifunctional electrocatalyst for highly efficient and pH-universal overall water splitting, Nat. Commun., 2020, 11, 4246 CrossRef CAS PubMed.
- A. Henglein and M. Giersig, Reduction of Pt(II) by H2: Effects of Citrate and NaOH and Reaction Mechanism, J. Phys. Chem. B, 2000, 104, 6767–6772 CrossRef CAS.
- Y. Zhu, L. Bu and Q. Shao, et al., Structurally Ordered Pt3Sn Nanofibers with Highlighted Antipoisoning Property as Efficient Ethanol Oxidation Electrocatalysts, ACS Catal., 2020, 10, 3455–3461 CrossRef CAS.
- W. Du, G. Yang and E. Wong, et al., Platinum-Tin Oxide Core-Shell Catalysts for Efficient Electro-Oxidation of Ethanol, J. Am. Chem. Soc., 2014, 136, 10862–10865 CrossRef CAS PubMed.
- Y. He, P. Ma and S. Zhu, et al., Photo-Induced Performance Enhancement of Tantalum Nitride for Solar Water Oxidation, Joule, 2017, 1, 831–842 CrossRef CAS.
- M. Wassner, M. Eckardt and C. Gebauer, et al., Synthesis and electrocatalytic performance of spherical core-shell tantalum (oxy)nitride@nitrided carbon composites in the oxygen reduction reaction, Electrochim. Acta, 2017, 227, 367–381 CrossRef CAS.
- L. Wang, B. Zhang and Q. Rui, Plasma-Induced Vacancy Defects in Oxygen Evolution Cocatalysts on Ta3N5 Photoanodes Promoting Solar Water Splitting, ACS Catal., 2018, 8, 10564–10572 CrossRef CAS.
- M. Xiao, Z. Wang and B. Luo, et al., Enhancing photocatalytic activity of tantalum nitride by rational suppression of bulk, interface and surface charge recombination, Appl. Catal., B, 2019, 246, 195–201 CrossRef CAS.
- H. Lin, M. Muzzio and K. Wei, et al., PdAu Alloy Nanoparticles for Ethanol Oxidation in Alkaline Conditions: Enhanced Activity and C1 Pathway Selectivity, ACS Appl. Energy Mater., 2019, 2, 8701–8706 CrossRef CAS.
- S. Guo, S. Dong and E. Wang, Pt/Pd bimetallic nanotubes with petal-like surfaces for enhanced catalytic activity and stability towards ethanol electrooxidation, Energy Environ. Sci., 2010, 3, 1307 RSC.
- Y. Katayama, T. Okanishi and H. Muroyama, et al., Enhanced Supply of Hydroxyl Species in CeO2-Modified Platinum Catalyst Studied by in Situ ATR-FTIR Spectroscopy, ACS Catal., 2016, 6, 2026–2034 CrossRef CAS.
- Y. Yang, M. Tian and Q. Li, et al., Ethanol Electrooxidation Catalyzed by Tungsten Core@Palladium Shell Nanoparticles, ACS Appl. Mater. Interfaces, 2019, 11, 30968–30976 CrossRef CAS PubMed.
- T. Hu, Y. Wang and H. Xiao, et al., Shape-control of super-branched Pd-Cu alloys with enhanced electrocatalytic performance for ethylene glycol oxidation, Chem. Commun., 2018, 54, 13363–13366 RSC.
- Z. Zhang, Y. Gong and D. Wu, et al., Facile fabrication of stable PdCu clusters uniformly decorated on graphene as an efficient electrocatalyst for formic acid oxidation, Int. J. Hydrogen Energy, 2019, 44, 2731–2740 CrossRef CAS.
- R. Jayarajan, R. Kumar and J. Gupta, et al., Fabrication of an amyloid fibril-palladium nanocomposite: a sustainable catalyst for C-H activation and the electrooxidation of ethanol, J. Mater. Chem. A, 2019, 7, 4486–4493 RSC.
- I. Feliciano-Ramos, E. O. Ortiz-Quiles and L. Cunci, et al., Unsupported palladium nanoparticles for ethanol cyclic voltammetric sensing in alkaline media, J. Solid State Electrochem., 2016, 20, 1011–1017 CrossRef CAS.
- S. Lu and Z. Zhuang, Investigating the Influences of the Adsorbed Species on Catalytic Activity for Hydrogen Oxidation Reaction in Alkaline Electrolyte, J. Am. Chem. Soc., 2017, 139, 5156–5163 CrossRef CAS PubMed.
- M. Arenz, K. J. J. Mayrhofer and V. Stamenkovic, et al., The Effect of the Particle Size on the Kinetics of CO Electrooxidation on High Surface Area Pt Catalysts, J. Am. Chem. Soc., 2005, 127, 6819–6829 CrossRef CAS PubMed.
- H. A. Gasteiger, S. S. Kocha and B. Sompalli, et al., Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Mukerjee and J. Mcbreen, Effect of particle size on the electrocatalysis by carbon-supported Pt electrocatalysts: an in situ XAS investigation, J. Electroanal. Chem., 1998, 448, 163–171 CrossRef CAS.
- V. Nguyen, Q. C. Tran and N. D. Quang, et al., N-doped C dot/PtPd nanonetwork hybrid materials as highly efficient electrocatalysts for methanol oxidation and formic acid oxidation reactions[J], J. Alloys Compd., 2018, 766, 979–986 CrossRef CAS.
- Z. Chen, Y. Liu and C. Liu, et al., Engineering the Metal/Oxide Interface of Pd Nanowire@CuOx Electrocatalysts for Efficient Alcohol Oxidation Reaction, Small, 2020, 16, 1904964 CrossRef CAS PubMed.
- M. C. Figueiredo, R. M. Arán-Ais and J. M. Feliu, et al., Pt catalysts modified with Bi: Enhancement of the catalytic activity for alcohol oxidation in alkaline media, J. Catal., 2014, 312, 78–86 CrossRef CAS.
- Q. Nian, J. Wang and S. Liu, et al., Aqueous Batteries Operated at −50 °C, Angew. Chem., Int. Ed., 2019, 58, 16994–16999 CrossRef CAS PubMed.
- Y. Yang, J. Ren and H. Zhang, et al., Infrared Spectroelectrochemical Study of Dissociation and Oxidation of Methanol at a Palladium Electrode in Alkaline Solution, Langmuir, 2013, 29, 1709–1716 CrossRef CAS PubMed.
- D. Chen and Y. J. Tong, An in situ electrochemical IR investigation of solution CO electro-oxidation on a polycrystalline Au surface in an alkaline electrolyte: Identification of active reaction intermediates, J. Electroanal. Chem., 2017, 800, 39–45 CrossRef CAS.
- F. Zhu, K. Tu and L. Huang, et al., High selectivity PtRh/RGO catalysts for ethanol electro-oxidation at low potentials: Enhancing the efficiency of CO2 from alcoholic groups, Electrochim. Acta, 2018, 292, 208–216 CrossRef CAS.
- Q. S. Chen, S. G. Sun and Z. Y. Zhou, et al., CoPt nanoparticles and their catalytic properties in electrooxidation of CO and CH3OH studied by in situ FTIRS, Phys. Chem. Chem. Phys., 2008, 10, 3645–3654 RSC.
- J. L. Bott-Neto, T. S. Martins and S. A. S. Machado, et al., Electrocatalytic Oxidation of Methanol, Ethanol, and Glycerol on Ni(OH)2 Nanoparticles Encapsulated with Poly[Ni(salen)] Film, ACS Appl. Mater. Interfaces, 2019, 11, 30810–30818 CrossRef CAS PubMed.
- P. A. Christensen, A. Hamnett and D. Linares-Moya, The electro-oxidation of formate ions at a polycrystalline Pt electrode in alkaline solution: an in situ FTIR study, Phys. Chem. Chem. Phys., 2011, 13, 11739–11747 RSC.
- K. Ataka, T. Yotsuyanagi and M. Osawa, Potential-Dependent Reorientation of Water Molecules at an Electrode/Electrolyte Interface Studied by Surface-Enhanced Infrared Absorption Spectroscopy, J. Phys. Chem., 1996, 100, 10664–10672 CrossRef CAS.
- M. Li, D. A. Cullen and K. Sasaki, et al., Ternary Electrocatalysts for Oxidizing Ethanol to Carbon Dioxide: Making Ir Capable of Splitting C–C Bond, J. Am. Chem. Soc., 2012, 135, 132–141 CrossRef PubMed.
- Z. Zhou, N. Tian and Y. Chen, et al., In situ rapid-scan time-resolved microscope FTIR spectroelectrochemistry: study of the dynamic processes of methanol oxidation on a nanostructured Pt electrode, J. Electroanal. Chem., 2004, 573, 111–119 CAS.
- G. Orozco, M. C. Pérez and A. Rincón, et al., Electrooxidation of methanol on silver in alkaline medium, J. Electroanal. Chem., 2000, 495, 71–78 CrossRef CAS.
- G. García, C. Stoffelsma and P. Rodriguez, et al., Influence of beryllium cations on the electrochemical oxidation of methanol on stepped platinum surfaces in alkaline solution, Surf. Sci., 2015, 631, 267–271 CrossRef.
- T. Haisch, F. Kubannek and S. Baranton, et al., The influence of adsorbed substances on alkaline methanol electro-oxidation, Electrochim. Acta, 2019, 295, 278–285 CrossRef CAS.
- P. A. Christensen and D. Linares-Moya, The Role of Adsorbed Formate and Oxygen in the Oxidation of Methanol at a Polycrystalline Pt Electrode in 0.1 M KOH: An In Situ Fourier Transform Infrared Study, J. Phys. Chem. C, 2010, 114, 1094–1101 CrossRef CAS.
- W. Wang, X. Chen and X. Zhang, et al., Quatermetallic Pt-based ultrathin nanowires intensified by Rh enable highly active and robust electrocatalysts for methanol oxidation, Nano Energy, 2020, 71, 104623 CrossRef CAS.
- M. Watanabe, T. Sato and K. Kunimatsu, et al., Temperature dependence of co-adsorption of carbon monoxide and water on highly dispersed Pt/C and PtRu/C electrodes studied by in situ ATR-FTIRAS, Electrochim. Acta, 2008, 53, 6928–6937 CrossRef CAS.
- Y. Yan, Y. Yang and B. Peng, et al., Study of CO Oxidation on Polycrystalline Pt Electrodes in Acidic Solution by ATR-SEIRAS, J. Phys. Chem. C, 2011, 115, 16378–16388 CrossRef CAS.
- Y. G. Yan, Q. X. Li and S. J. Huo, et al., Ubiquitous strategy for probing ATR surface-enhanced infrared absorption at platinum group metal-electrolyte interfaces, J. Phys. Chem. B, 2005, 109, 7900–7906 CrossRef CAS PubMed.
- Q. Xu, A. Berná and I. V. Pobelov, et al., ATR-SEIRAS study of CO adsorption and oxidation on Rh modified Au(111-25 nm) film electrodes in 0.1 M H2SO4, Electrochim. Acta, 2015, 176, 1202–1213 CrossRef CAS.
- S. X. Liu, L. W. Liao and Q. Tao, et al., The kinetics of CO pathway in methanol oxidation at Pt electrodes, a quantitative study by ATR-FTIR spectroscopy, Phys. Chem. Chem. Phys., 2011, 13, 9725–9735 RSC.
- Y. X. Chen, A. Miki and S. Ye, et al., Formate, an Active Intermediate for Direct Oxidation of Methanol on Pt Electrode, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed.
- X. Qin, H. Li and S. Xie, et al., Mechanistic Analysis-Guided Pd-Based Catalysts for Efficient Hydrogen Production from Formic Acid Dehydrogenation, ACS Catal., 2020, 10, 3921–3932 CrossRef CAS.
- H. Hanawa, K. Kunimatsu and H. Uchida, et al., In situ ATR-FTIR study of bulk CO oxidation on a polycrystalline Pt electrode, Electrochim. Acta, 2009, 54, 6276–6285 CrossRef CAS.
- Q. Zhang, Y. Ma and Y. Lu, et al., Modulating electrolyte structure for ultralow temperature aqueous zinc batteries, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- L. F. Shen, B. A. Lu and Y. Y. Li, et al., Interfacial Structure of Water as a New Descriptor of the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2020, 59, 22397–22402 CrossRef CAS PubMed.
- C. Li, J. Le and Y. Wang, et al., In situ probing electrified interfacial water structures at atomically flat surfaces, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- N. Ye, Z. Jiang and T. Fang, Assembling the PdCu/rGO catalysts for methanol oxidation reaction in alkaline media by tuning the electronic structure, Electrochim. Acta, 2020, 352, 136473 CrossRef CAS.
- C. Wei, Y. Sun and G. G. Scherer, et al., Surface Composition Dependent Ligand Effect in Tuning the Activity of Nickel-Copper Bimetallic Electrocatalysts toward Hydrogen Evolution in Alkaline, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS PubMed.
- L. Bai, X. Wang and Q. Chen, et al., Explaining the Size Dependence in Platinum-Nanoparticle-Catalyzed Hydrogenation Reactions, Angew. Chem., 2016, 128, 15885–15890 CrossRef.
- Y. Zheng, Y. Jiao and M. Jaroniec, et al., Advancing the Electrochemistry of the Hydrogen-Evolution Reaction through Combining Experiment and Theory, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- X. Du, J. Huang and J. Zhang, et al., Modulating Electronic Structures of Inorganic Nanomaterials for Efficient Electrocatalytic Water Splitting, Angew. Chem., 2019, 131, 4532–4551 CrossRef.
- Z. Yang, S. Pedireddy and H. K. Lee, et al., Manipulating the d-Band Electronic Structure of Platinum-Functionalized Nanoporous Gold Bowls: Synergistic Intermetallic Interactions Enhance Catalysis, Chem. Mater., 2016, 28, 5080–5086 CrossRef CAS.
- P. D. C. King, T. D. Veal and A. Schleife, et al., Valence-band electronic structure of CdO, ZnO, and MgO from x-ray photoemission spectroscopy and quasi-particle-corrected density-functional theory calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 205205 CrossRef.
- S. Kwon, D. J. Ham and T. Kim, et al., Active Methanol Oxidation Reaction by Enhanced CO Tolerance on Bimetallic Pt/Ir Electrocatalysts Using Electronic and Bifunctional Effects, ACS Appl. Mater. Interfaces, 2018, 10, 39581–39589 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07382f |
| ‡ These authors contributed equally to this work and should be regarded as co-first authors. |
| This journal is © The Royal Society of Chemistry 2022 |