
Electronic engineering of amorphous Fe–Co–S sites in hetero-nanoframes for oxygen evolution and flexible Al–air batteries†
Min
Lu
a,
Li
An
a,
Jie
Yin
a,
Jing
Jin
a,
Rui
Yang
a,
Bolong
Huang
 *b,
Yang
Hu
a,
Yong-Qing
Zhao
a and
Pinxian
Xi
*a
*b,
Yang
Hu
a,
Yong-Qing
Zhao
a and
Pinxian
Xi
*a
aState Key Laboratory of Applied Organic Chemistry, Frontiers Science Center for Rare Isotopes, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, China. E-mail: xipx@lzu.edu.cn
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hum, Kowloon, Hong Kong SAR, China. E-mail: bhuang@polyu.edu.hk
First published on 23rd February 2022
Abstract
The electrochemical oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) are key electrochemical processes in metal–air batteries and water splitting devices. Aluminium–air batteries, as an important type of metal–air battery, have been considered to be promising power candidates for flexible electronics. Here, we describe electronically engineered amorphous Fe–Co–S sites embedded in Prussian blue analogue (FeCoSx-PBA) hetero-nanoframes. The experimental results and DFT calculations reveal the critical role of the introduced FeCoSx layer to PBA, which enhances the electron transfer and alleviates the overbinding effect of OH* during the OER. The FeCoSx-PBA hybrid system supplies an optimized electronic structure for the alkaline OER, which is also confirmed by the much-lowered overpotential (266 mV at 10 mA cm−2) for the alkaline OER. Furthermore, a flexible Al–air battery based on an FeCoSx-PBA cathode catalyst exhibits a high peak power density (58.3 mW cm−2) and energy density (1483 W h kgAl−1), and outstanding stability for more than 50 h of operation under bending or stretching conditions, demonstrating its potential in the practical application of flexible electronic devices. Our results may provide a new strategy of modulating the electronic structure of air electrode catalysts to efficiently promote the reactivity of alkaline OER and Al–air battery processes.
 Pinxian Xi | Pinxian Xi received his PhD degree in 2010 from Lanzhou University. From 2009 to 2010 he worked as a visiting scholar at Brown University. In 2018, he was appointed professor by the College of Chemistry and Chemical Engineering of Lanzhou University. His research focuses on the controlled synthesis of functional materials (including transition metal sulfide, rare earth functional materials) and developing their applications in the electrocatalytic and energy fields. |
Introduction
The rapid development of flexible and wearable electronic equipment is promoting the exploitation of highly efficient energy storage and conversion devices such as metal–air batteries. It has remarkable advantages of high theoretical energy density, excellent safety, and low cost compared to traditional batteries (such as lithium-ion batteries). The reaction involving oxygen is the most crucial process for metal–air batteries in portable devices and electric vehicles.1–3 However, the related electro-process involving oxygen is usually restricted by the intrinsically sluggish kinetics for multiple-step proton-coupled electron transfer steps.4,5 Therefore, developing highly efficient catalysts with excellent activity and predominant durability is critical for practical applications. Although noble metal and metal oxides (Pt, Ir, and IrO2) exhibit state-of-the-art catalytic activities for the process involving oxygen, their multiple disadvantages, including high cost, scarcity, and poor durability, limit their application on a commercial scale.6 For this purpose, the development of low-cost and highly active transition metal-based catalysts is essential.7–9Hetero-nanoframes consisting of hollow frame structures have been considered an essential morphology in nanocatalysts due to their abundant exposed active sites, enhanced mass/charge transfer velocity, reduced possibility of aggregation, and favorable interface effects.10–12 However, the poor intrinsic catalytic performance is still a challenge for their application in electrocatalysis. Cation and anion exchange is an efficient approach to modify the inherent electronic structure and create new active sites through the introduction of hetero-atoms, thus improving the intrinsic catalytic activity. After the introduction of heteroatoms, the surface electron density of active sites can be changed due to the variational coordination environment among neighboring atoms accompanied by the mismatch between electronegativity and ionic radius.13 Accordingly, a tailored electronic configuration with an optimized band structure and abundant defects can be adjusted into catalysts through a precise ion-exchange strategy, which is favorable for promoting catalytic activity.14–16
In our work, an amorphous FeCoSx embedded in Prussian blue analogue hetero-nanoframes (FeCoSx-PBA) was fabricated through an ultrasound-assisted ion-exchange method. By engineering the electronic structure through hetero-ion introduction (Fe2+ and S2−) and amorphization, the FeCoSx-PBA catalyst has enhanced electron transfer capability and optimized binding strength with intermediates. Besides, its unique hetero-nanoframe morphology also provides more exposed active sites. The as-obtained material exhibits an excellent oxygen evolution performance with a low overpotential of 266 mV at 10 mA cm−2 and superior durability. A dynamic OER study unravels the important role of S atoms in the OER with the FeCoSx-PBA catalyst. Through DFT calculations, the obvious change in the electronic structures was unraveled, which indicates the boosted electronic conductivity to promote electron/charge transfer during the OER. Meanwhile, the surface-unsaturated FeCoSx layer demonstrates the optimized binding strength of OH, leading to the alleviated energy barrier of the rate-determining step for the OER. In addition, when serving as an air cathode catalyst in Al–air batteries, FeCoSx-PBA shows enhanced battery performance. A remarkably high discharge power density of 58.3 mW cm−2, a large energy density of 1483 W h kg−1 and excellent long-term stability for more than 50 h of operation after mechanical recharge were achieved. More importantly, all-solid-state Al–air batteries with excellent bendability and stretchability were assembled and applied to charge a smartphone successfully.
Experimental section
Materials and methods
Structural characterizations
X-ray diffraction (XRD) experiments were conducted with an X'Pert Pro X-ray diffractometer with Cu Kα radiation (λ = 0.1542 nm) from 10° to 90° under a constant voltage of 40 kV. Field-emission scanning electron microscopy (FESEM) was used to investigate the morphologies of the samples at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) were performed under an acceleration voltage of 200 kV with a JEOL JEM 2100 TEM. X-ray photoelectron spectroscopy (XPS) analyses were undertaken with a VG ESCALAB 220I-XL device and corrected with the C 1s line at 284.8 eV. The Mössbauer spectrum was recorded in transmission mode with a57Co source in a rhodium matrix. The Mössbauer spectrometer was of the electromechanical type with a fixed absorber and operating source in constant acceleration mode, which was calibrated using an α-Fe foil. The Mössbauer spectrum was least-squares fitted, providing the values of the hyperfine field (Hhf), isomer shift (δiso), electric quadrupole splitting (ΔEQ), and relative area of Fe ions. The Raman spectra were recorded on a HORIBA LabRAM HR Evolution with laser wavelength 532 nm. The EELS spectra were recorded on a probe aberration-corrected STEM (Cubed Titan G2 60-300, FEI, USA).Electrochemical measurements
All the electrochemical measurements were carried out using a CHI 760 E Electrochemical Workstation (CHI Instruments, Shanghai Chenhua Instrument Corp., China) using a glass carbon electrode (geometric area = 0.071 cm2) as the working electrode, and Pt and Hg/HgO (1 M KOH) as auxiliary and reference electrodes in 1.0 M KOH solution. The potentials were referenced to a reversible hydrogen electrode (RHE) through the equation (E(RHE) = E(Hg/HgO) + 0.0951pH + 0.098). All electrochemical experiments were conducted at 20 ± 0.2 °C. The Ir/C (20%) catalysts were prepared according to a reported method.25,31 The loading amount of the catalysts was 0.2 mg cm−2. Electrochemical impedance spectroscopy (EIS) measurements were performed by applying an AC voltage with 5 mV amplitude in a frequency range from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 0.01 Hz and recorded at 1.58 V vs. RHE. The electrical double layer capacitance (Cdl) and surface roughness factor (Rf) of the as-synthesized materials were measured from double-layer charging curves using cyclic voltammograms (CVs) in a small potential range of 1.17–1.27 V.
000 to 0.01 Hz and recorded at 1.58 V vs. RHE. The electrical double layer capacitance (Cdl) and surface roughness factor (Rf) of the as-synthesized materials were measured from double-layer charging curves using cyclic voltammograms (CVs) in a small potential range of 1.17–1.27 V.
To investigate the reaction mechanism for the OER, rotating ring-disk electrode (RRDE) voltammograms were conducted on an RRDE configuration (RRDE-3A, Japan) consisting of a glassy carbon disk electrode and a Pt ring electrode. The loading amount was 0.2 mg cm−2. A scan rate of 2 mV s−1 and a rotation rate of 1600 rpm were applied for RRDE tests. Specifically, to ensure that the oxidation current originates from oxygen evolution rather than other side reactions, and to calculate the faradaic efficiency of the system, the ring potential was held constant at 0.40 V versus RHE to reduce the O2 formed from the catalyst on the disk electrode in N2-saturated 1 M KOH solution. The faradaic efficiency (ε) was calculated as follows:
| e = Ir/(IdN) |
The electron transfer number (n) during the ORR was calculated from the Koutecky–Levich (K–L) equations:20
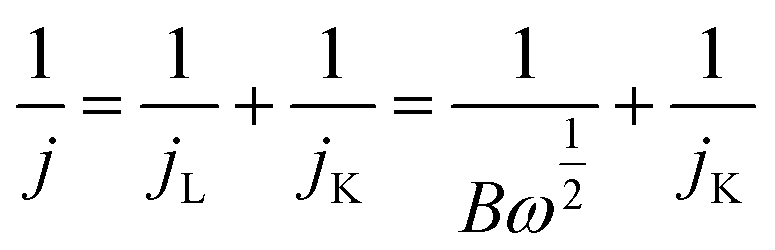
where j, jL, and jK correspond to the measured, diffusion-limiting, and kinetic current densities, respectively; ω is the rotation rate (rpm), F is the Faraday constant (96
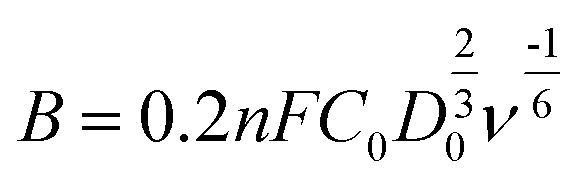 485C mol−1), C0 is the bulk concentration of oxygen (1.26 × 10−6 mol cm−3), D0 is the diffusion coefficient of oxygen (1.9 × 10−5 cm2 s−1), and n is the kinetic viscosity (0.01 cm2 s−1).
485C mol−1), C0 is the bulk concentration of oxygen (1.26 × 10−6 mol cm−3), D0 is the diffusion coefficient of oxygen (1.9 × 10−5 cm2 s−1), and n is the kinetic viscosity (0.01 cm2 s−1).
Al–air battery assembly
An aluminium sheet and FeCoSx-PBA/carbon black loaded on carbon cloth (2 mg cm−2) served as the anode and air cathode, respectively. All as-prepared electrodes were directly used as the air cathodes and aluminium plates were polished to be used as the anodes. The liquid Al–air battery was assembled in 2 M KOH with ZnO and Na2SnO3 as an additive was used as the electrolyte to slow down the corrosion of aluminium.21 The alkaline gel electrolyte was prepared by dissolving KOH, PVA, ZnO and Na2SnO3 in deionized water at 95 °C under vigorous stirring until the solution became clear. Flexible aluminium–air batteries were assembled by separating the air cathode and Al anode with the alkaline gel polymer. Galvanostatic discharge and charge measurements were carried out with the LAND CT2001A multichannel battery testing system.Computational method
In this work, simplified rotationally invariant DFT + U calculations22 within the CASTEP code23 were applied for all the calculations. The GGA and PBE exchange-correlation functionals were selected and the plane-wave cutoff energy was set to 750 eV.24,25 Meanwhile, for all the geometric optimizations, the algorithm of Broyden–Fletcher–Goldfarb–Shanno (BFGS) was selected.26 In addition, the ensemble DFT (EDFT) by Marzari et al.27 was applied during the electronic-minimization process, which aims to improve the convergence quality of the transition metal in the FeCoSx-PBA composite system.By utilizing the OPIUM code in the Kleinman–Bylander projector form,28 the norm-conserving pseudopotentials of Fe, Co, C, N, O, and H were generated with the incorporation of non-linear partial core correction29 and a scalar relativistic averaging scheme.30 The valence states of Fe, Co, C, N, O, and H were chosen as (3d, 4s, 4p), (3d, 4s, 4p), (2s, 2p), (2s, 2p), (2s, 2p) and (1s), respectively. The RRKJ method was applied for the optimization of the pseudopotentials.31
Two different models of FeCoSx-PBA were constructed based on two different bonding situations at the interface: S bonding dominant and Fe–S bonding dominant. The vacuum space of FeCoSx-PBA was set to 15 Å, which guarantees sufficient space for all the geometric optimizations. Considering the DFT computational cost, Monkhorst–Pack reciprocal space integration was performed using Gamma-center-off special k-points with a mesh of 2 × 2 × 2,32 which was guided by the initial convergence test. The overall total convergence settings were set so that the total energy for each step was less than 5.0 × 10−7 eV per atom while the Hellmann–Feynman forces on the atom should not exceed 0.001 eV Å−1.
Results and discussion
FeCoSx-PBA hetero-nanoframes were synthesized by a sequential ultrasound-assisted ion-exchange method. The initial PBA nanocubes were ultrasonicated in dilute HCl solution containing Fe2+, leading to the partial replacement of Co2+ with Fe2+ in the PBA surface and the formation of Fe/Co mixed Prussian blue analogue (MPBA) nanocubes, which provided a basis for the further synthesis of an FeCoSx structure. Further solvothermal treatment was performed to achieve S2− ion exchange with Fe(CN)63−.The crystal structures of PBA, MPBA, and FeCoSx-PBA (Fig. 1a) were studied by X-ray diffraction (XRD). The results of all three samples match well with the standard diffraction pattern representing a Prussian blue structure. Furthermore, after the introduction of S into MPBA, FeCoSx-PBA also shows the same XRD pattern as MPBA, demonstrating that the major component of FeCoSx-PBA is still MPBA. During the solvothermal reaction process, a low concentration of S2− was released from thioacetamide (TAA). Then S2− inactivated by ethanol favors the incomplete decomposition of MPBA, resulting in the partial retention of the Prussian blue structure.17–19 It is worth noting that the (111) diffraction peaks are 17.80°, 17.57° and 17.43° in PBA, MPBA, and FeCoSx-PBA, respectively (Fig. 1a). The continuous negative shift of the XRD diffraction peak reveals the expansion in lattice distance caused by the successful introduction of hetero cation (Fe2+) and anion (S2−).
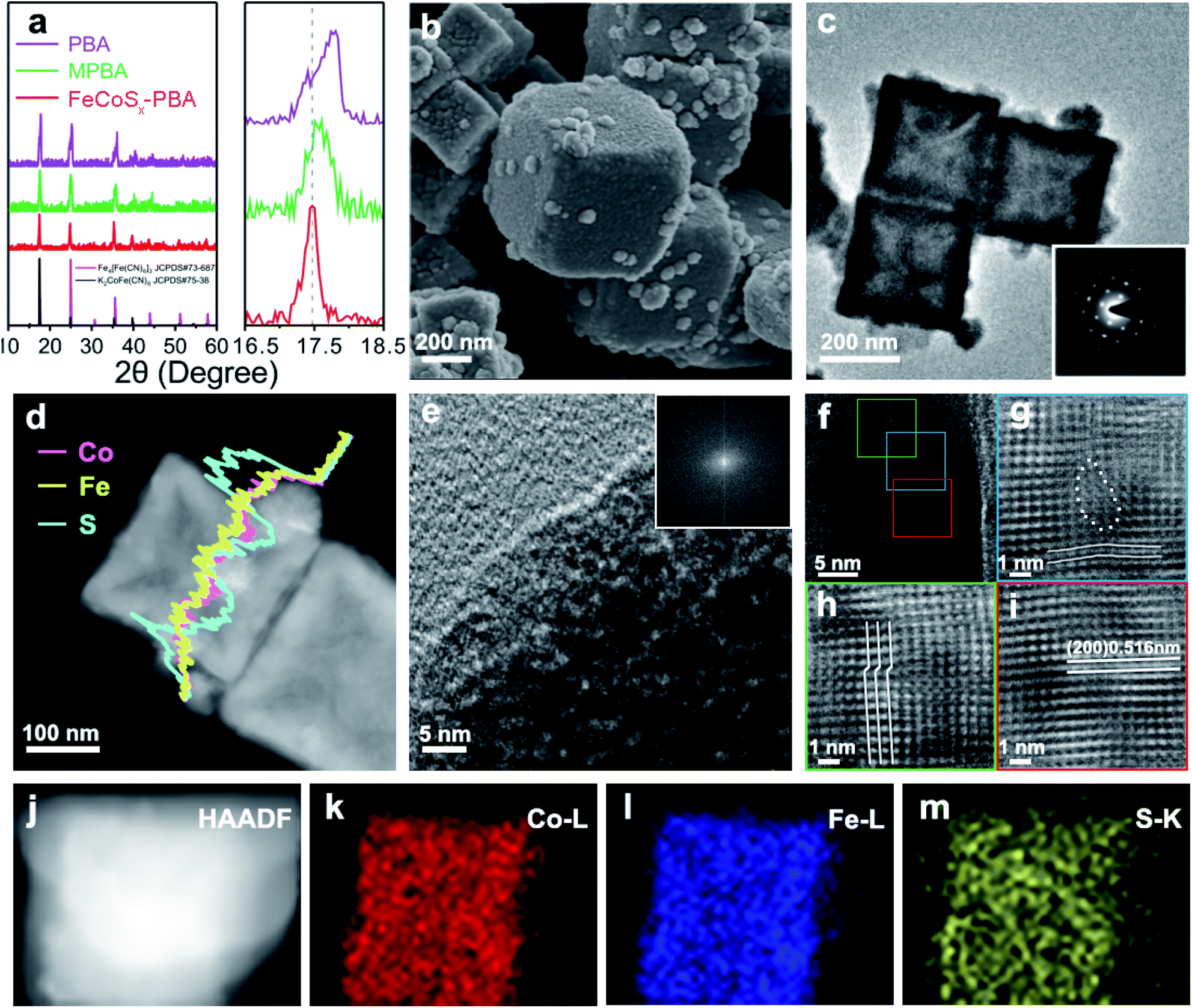 | ||
| Fig. 1 (a) Powder XRD patterns of PBA, MPBA, and FeCoSx-PBA (left). The corresponding XRD patterns range from 16° to 19° (right). (b) SEM image of FeCoSx-PBA. (c) TEM image of FeCoSx-PBA, and the corresponding SAED image (inset). (d) EDX line scan of a single FeCoSx-PBA hetero-nanoframe. (e) HRTEM image of a nanoparticle in the surface of FeCoSx-PBA, and the corresponding FFT image (inset). (f) HRTEM image of the skeleton part in FeCoSx-PBA. (g–i) Magnified HRTEM images extracted by inverse Fourier filtering of blue, red and green boxes in (f). EDX mapping of FeCoSx-PBA in (j) HAADF-image, (k) Co L edge, (i) Fe L edge, (m) S K edge. | ||
The morphology was study by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The results show PBA exhibits a nanocube morphology with a diameter of about 250 nm (Fig. S1†). After Fe2+ ion exchange, the MPBA nanocubes had a nearly unchanged diameter and morphology (Fig. S2†). The Fe2+ ion exchange process was also studied by the same ultrasonic experiment without the addition of Fe2+. As can be seen, the PBA nanocubes were corroded and broken into porous nanocubes (Fig. S3†). The above results indicate that Fe2+ has stronger bonding than Co2+ with [Fe(CN)6]3− and can resist corrosion by HCl, which drives the Fe2+ ion-exchange process. After ion exchange with S2−, the SEM image of FeCoSx-PBA in Fig. 1b clearly shows a typical cubic shape with numerous small nanoparticles attached on the surface. The TEM image further confirms the hollow cube-like structure of FeCoSx-PBA with an outer diameter of about 250 nm and a skeleton thickness of 20–40 nm. The attached surface nanoparticles ranged from 30 nm to 50 nm. The Brunauer–Emmett–Teller (BET) results (Fig. S4 and Table S1†) also support the hollow structure of FeCoSx-PBA, which has a significantly increased BET surface area from ∼8.9 m2 g−1 to ∼28.9 m2 g−1.
To determine the structural changes caused by the introduction of S, TEM was further performed on FeCoSx-PBA. The remarkable single crystal structure with a cubic system was also confirmed by the selected area electron diffraction (SAED) images in the inset of Fig. 1c. Combined with the XRD results, this indicated that the hollow cube is mainly an MPBA structure. The energy-dispersive X-ray spectroscopy (EDX) element line scan profile demonstrates the uniform distribution of Fe and Co of FeCoSx-PBA (Fig. 1d). The S content is significantly higher in the surface nanoparticles than in the skeleton part. This suggests that the surface nanoparticles may be a FeCoSx sulfide. High-resolution transmission electron microscopy (HRTEM) was used to study the lattice structure of FeCoSx-PBA further. As shown in Fig. 1e, no lattice fringe can be observed for the surface particle. Its corresponding fast Fourier transform (FFT) image (insert of Fig. 1e) also shows no diffraction spots, revealing the amorphous feature of FeCoSx. The amorphous structure was also evidenced by a contrast experiment, where MPBA reacted with Na2S instead of TAA under the same experimental conditions to fully convert Prussian blue to sulfide. The as-obtained materials show no XRD peak (Fig. S5†), revealing the amorphous structure.
The skeleton part of FeCoSx-PBA displayed legible lattice fringes, which can be ascribed to the (200) planes of K2CoFe(CN)6 and Fe4[Fe(CN)6]3 in Fig. 1f and S6.† Three parts marked in green, blue, and red frames were chosen to remove background noise by the inverse Fourier filtering method and are shown in Fig. 1g–i. Fig. 1g shows significant lattice distortions marked by lines and a large area of vacancy defects marked by dots. Fig. 1h displays characteristic lattice dislocations of ∼0.23 nm, in which the periodic arrangement of atoms mismatches and slides. Fig. 1i illustrates a representative HRTEM image, in which the marked lattice spacing value is 0.516 nm that is assigned to the (200) plane of the Prussian blue structure. Compared to PBA and MPBA with lattice distances of 0.504 nm and 0.506 nm, respectively, FeCoSx-PBA shows a small lattice expansion of 0.01 nm, which may be attributed to the flexible and framework-like lattice structure of ferricyanide. This S2− doping and the resulting abundant lattice imperfections were proved to exist in FeCoSx-PBA and could be expected to promote the electrocatalytic performance.33–35 The element mappings in Fig. 1j–m also demonstrate the distributions of Fe, Co, and S in FeCoSx-PBA.
The above results indicated that the S atom has two primary forms in FeCoSx-PBA. One is S2− in FeCoSx, and the other is doped S in Prussian blue (Fig. 2a). X-ray photoelectron spectroscopy (XPS) was performed to provide in-depth evidence and trace the differences in electronic structure among PBA, MPBA, and FeCoSx-PBA (Fig. 2b–d, S7 and S8). Along with the introduction of S, the Co 2p spectrum of FeCoSx-PBA in Fig. 2b was deconvoluted into two pairs of peaks at binding energies of 778.3 eV, 781.1 eV, 793.3 eV, and 797.3 eV, which were attributed to Co–S and Co–N species, respectively.36,37 The Fe 2p spectrum of FeCoSx-PBA (Fig. 2c) shows a similar structure to Co. The peaks at 708.1 eV and 720.7 eV belong to Fe–C species, while the peaks at 708.5 eV and 721.8 eV belong to Fe–S species.38,39 Moreover, the S 2p spectrum of FeCoSx-PBA (Fig. 2d) can be identified as two pairs of peaks with binding energies of 161.8 eV, 163.0 eV, 163.9 eV, and 165.0 eV. The former two peaks can be assigned to Fe/Co–S groups, and the latter two belong to C–S groups.33–35 From these results, it can be found that doped S atom in FeCoSx-PBA bonds with the C atom of the cyanide ligands to form a C–S structure. This suggestion was further confirmed by C 1s XPS spectra, where a new peak centered at 285.6 eV is observed for FeCoSx-PBA and can be assigned to the C–S structure (Fig. S8†).40 Other peaks at 284.6 eV, 286.7 eV, and 288.7 eV are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C, C–O/C–N, and CO, respectively.41,42 In addition, N coordination was verified by the N 1s spectra. Three types of nitrogen peaks of FeCoSx-PBA were fitted at 397.1 eV, 398.3 eV, and 401.8 eV, which are attributed to Fe/Co–N, C–N, and N–H species.43,44 Furthermore, a significant peak shift was observed after the introduction of S. The original Co–N and Fe–C peaks appear to have negative shifts of 0.8 eV and 0.5 eV, indicating the increased electron density of the Co and Fe atoms. The electrical effect of the S atoms was further spread to the adjacent N and C atoms. The negative shift and positive peak shift of N and C, respectively, revealed that the electron density in the C and N atoms decreased and increased separately. Besides, a significantly enhanced N–H peak at 401.8 eV observed in FeCoSx-PBA also demonstrates that N atoms become more electronegative and more accessible for combining with hydrogen molecules in the atmosphere to form N–H-like species.43,44 The above results further support the two primary forms of S in FeCoSx-PBA.
C–C, C–O/C–N, and CO, respectively.41,42 In addition, N coordination was verified by the N 1s spectra. Three types of nitrogen peaks of FeCoSx-PBA were fitted at 397.1 eV, 398.3 eV, and 401.8 eV, which are attributed to Fe/Co–N, C–N, and N–H species.43,44 Furthermore, a significant peak shift was observed after the introduction of S. The original Co–N and Fe–C peaks appear to have negative shifts of 0.8 eV and 0.5 eV, indicating the increased electron density of the Co and Fe atoms. The electrical effect of the S atoms was further spread to the adjacent N and C atoms. The negative shift and positive peak shift of N and C, respectively, revealed that the electron density in the C and N atoms decreased and increased separately. Besides, a significantly enhanced N–H peak at 401.8 eV observed in FeCoSx-PBA also demonstrates that N atoms become more electronegative and more accessible for combining with hydrogen molecules in the atmosphere to form N–H-like species.43,44 The above results further support the two primary forms of S in FeCoSx-PBA.
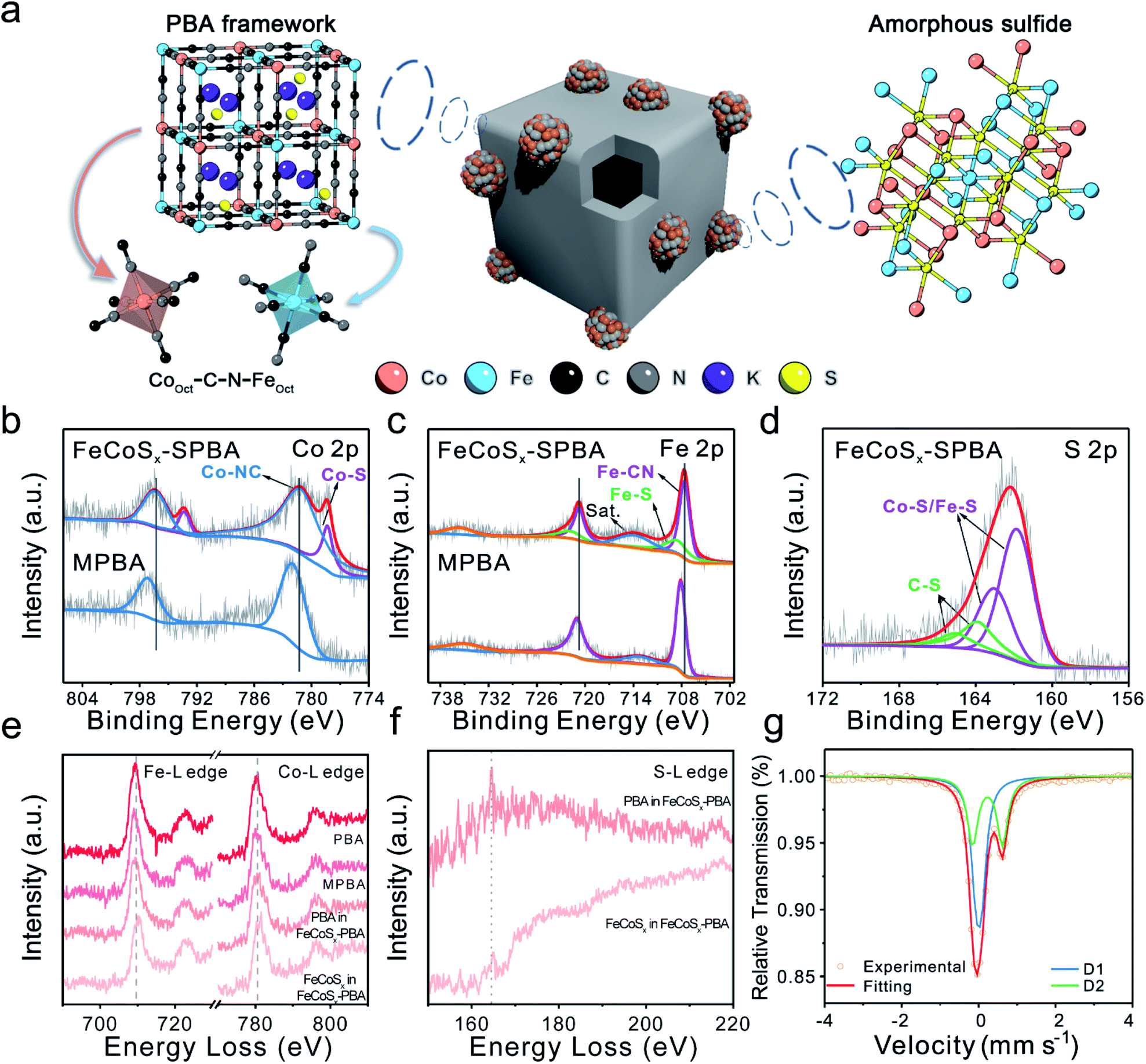 | ||
| Fig. 2 (a) Schematic diagrams of FeCoSx-PBA. (b) Co 2p and (c) Fe 2p XPS spectra of FeCoSx-PBA and MPBA. (d) S 2p XPS spectra of FeCoSx-PBA. (e) Co L2,3 and Fe L2,3 edge EELS spectra of PBA, MPBA, and FeCoSx-PBA. (f) S L2,3 edge EELS spectra of FeCoSx-PBA. (g) Mössbauer spectrum pattern of FeCoSx-PBA. | ||
To provide further detail about the local electronic structure, electron energy loss spectroscopy (EELS) was performed. Especially for FeCoSx-PBA, the PBA frame region and amorphous FeCoSx particles region were studied separately (Fig. S9†). First, the Fe L2,3 white lines (Fig. 2e) show a 0.5 eV negative shift from PBA to MPBA, which indicates a slight decrease in the valence state of Fe. This is consistent with the introduction of Fe2+ into MPBA. Thus, the Co and Fe L2,3 spectra of the PBA frame region in FeCoSx-PBA indicate a small change compared with MPBA. This shows that the doped S influences the valence state of Co and Fe in the PBA framework. But the Co and Fe L2,3 spectra of the amorphous FeCoSx region in FeCoSx-PBA exhibit a 1 eV positive shift compared with the PBA frame region. This represents a significant structural difference between the PBA structure and the sulfide structure.45 The intensity of the S L2,3 white lines (Fig. 2f) in the PBA frame region is lower than in the amorphous FeCoSx region and has a 0.5 eV difference. This also shows the different forms of S in FeCoSx-PBA.46,47 Mössbauer spectroscopy was further employed to analyze different Fe species. The Mössbauer spectrum of FeCoSx-PBA (Fig. 2g) was fitted with a single and a doublet assigned to Fe(II)–C/N and Fe(II)–S species, respectively. The Mössbauer spectra of PBA and MPBA (Fig. S10 and S11†) were fitted with a single and a doublet but assigned to low-spin Fe(II) and high-spin Fe(III) species, respectively.48,49 After introducing Fe2+, the region ratio of single:doublet in MPBA increased, showing an increased Fe2+/Fe3+ ratio from 1.46 to 2.91. The corresponding fitting parameters and the relative peak area for the catalysts are summarized in Table S2.†
Then we introduced DFT calculations to unravel the significant improvements in OER performance in the FeCoSx-PBA hybrid systems. Although the cage structure of PBA endows them with a stable structure with a high surface area, the low electronic conductivity still leads to limited application in the electrocatalyst. With the introduction of FeCoSx on the surface with different bonding situations, the electroactivity of PBA has been activated on the surface-defective FeCoSx layer with large exposure of active sites. The bonding and anti-bonding orbitals near the Fermi level (EF) are mostly dominated by the surface FeCoSx layers. Meanwhile, the remarkable stability of the PBA structural frame determines the long-term durability of the composite electrocatalyst (Fig. 3a). Thus, the projected partial density of states (PDOS) of S, Fe, Co, and –CN functional groups in FeCoSx-PBA are illustrated. Except for the surface, S sites demonstrate broad S-3p orbitals, covering from EV −8.0 eV to EV +3.5 eV (EV = 0 eV). However, the surface-unsaturated S shows a strong delocalization feature with the dominant peak at EV −4.7 eV, which effectively suppresses the overbinding effect of –OH in the alkaline environment (Fig. 3b). From the bulk to the surface, the eg of Fe-3d orbitals significantly shifts from EV +4.5 eV to EV +0.6 eV. The downshifting of the d-band center of surface Fe is also noted, which boosts both electron transfer and binding with the OH group. Meanwhile, the surface Fe indicates the evident crossing of EF, supporting the reduction of valence state with improved electronic conductivity. This significantly compensates for the poor electron transfer capability in pristine PBA (Fig. 3c). For Co-3d orbitals, we notice the evident splitting of t2g and eg in PBA-Co and the interfacial Co. Only Co sites in the surface FeCoSx layer demonstrate the increased electron density of 3d orbitals near EF to further enhance the electron transfer (Fig. 3d), which is consistent with the XPS results. In addition, the CN groups in PBA were also investigated regarding their electronic structures. From the bulk PBA to the interfacial region binding with FeCoSx, the s,p orbitals of –CN groups demonstrate a gradual downshift away from the EF, becoming more electronegative, which confirms the experimental characterizations. This also means that the introduction of the surface FeCoSx layers not only leads to modified –CN functional groups with improved stability but also the overbinding poisoning of OH groups during the OER (Fig. 3e).
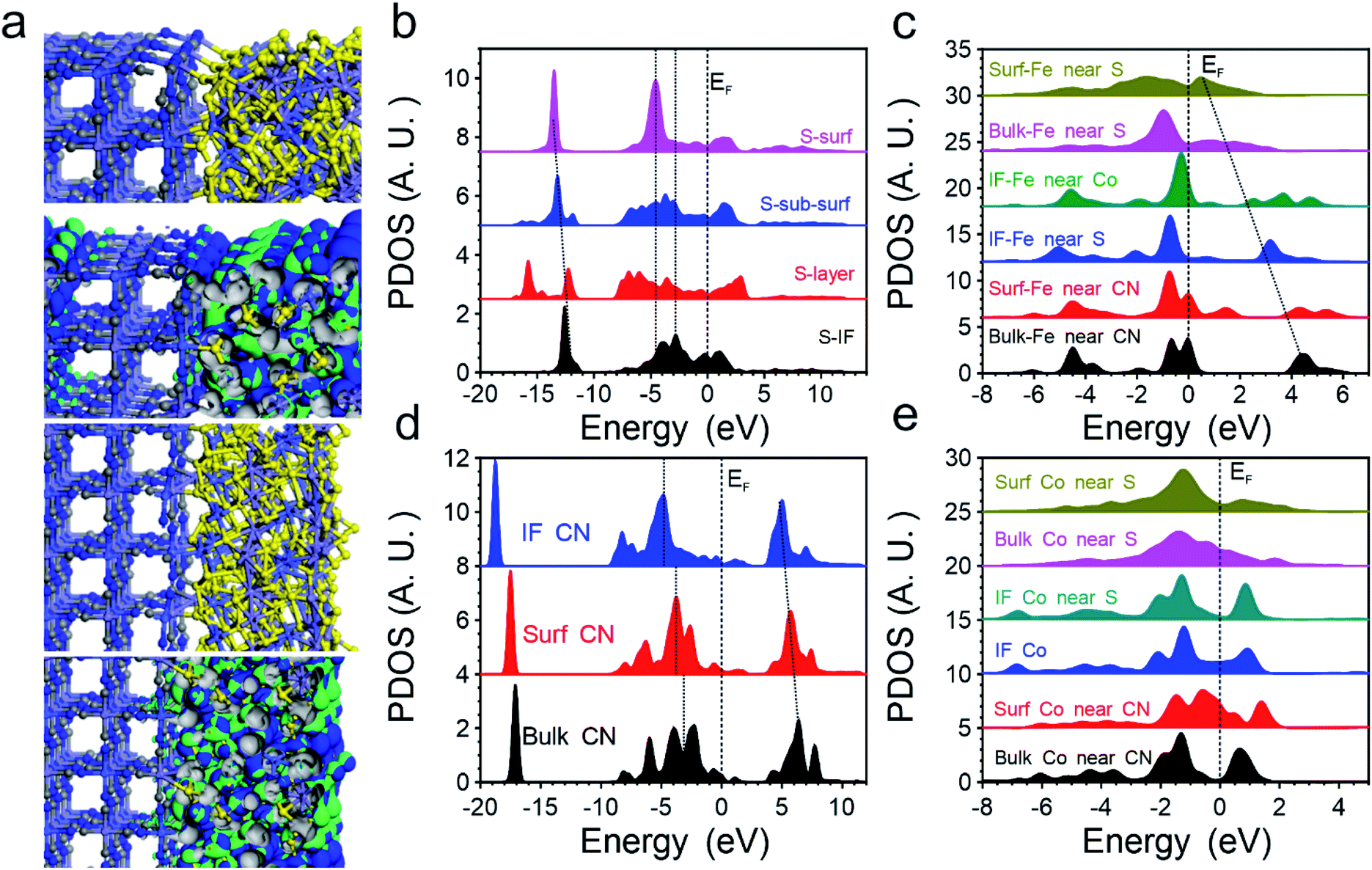 | ||
| Fig. 3 The electronic structures of FeCoSx-PBA. (a) Structural configurations and real spatial 3D orbital contour plots of two different FeCoSx-PBA systems. (b) Site-dependent PDOSs of S in FeCoSx-PBA. (c) Site-dependent PDOSs of Fe in FeCoSx-PBA. (d) Site-dependent PDOSs of Co in FeCoSx-PBA. (e) Site-dependent PDOSs of –CN in FeCoSx-PBA. | ||
The OER activity was studied in an alkaline medium (1.0 M KOH) through linear sweep voltammetry (LSV) (Fig. 4a) for the as-prepared three catalysts and benchmark Ir/C (20%) which were prepared on glassy carbon electrodes. After modifying the outermost surface of PBA by Fe2+, the as-obtained MPBA has an obviously enhanced catalytic performance with 75 mV increased overpotential at 10 mA cm−2 compared with PBA. Furthermore, FeCoSx-PBA with further optimization of the electronic structure shows the best OER activity among all these samples, including commercial Ir/C. Its overpotential at a current density of 10 mA cm−2 is approximately 266 mV, much lower than those of Ir/C (320 mV), PBA (409 mV), or MPBA (334 mV). The Tafel plot is an inherent parameter for evaluating OER catalytic kinetics. The lowest Tafel slope of 33 mV dec−1 (Fig. 4b) for FeCoSx-PBA suggested the fastest OER dynamics. Long-term stability is another crucial criterion in evaluating the catalytic property. Hence, the chronoamperometric response was carried out at a constant current density of ∼8 mA cm−2. After 15 hours of testing, FeCoSx-PBA still retains 91.4% current, indicating its superior durability (Fig. S12†). To eliminate the influence of corrosion current, a faradaic efficiency study was performed on a rotating ring-disk electrode (RRDE) in N2-saturated 1 M KOH (Fig. S13†). With the disk current held constant at 200 μA, a ring current of ∼38.3 μA was detected, which proved that the observed OER current catalyzed by FeCoSx-PBA could be mainly attributed to the OER with a high faradaic efficiency of 95.7%.
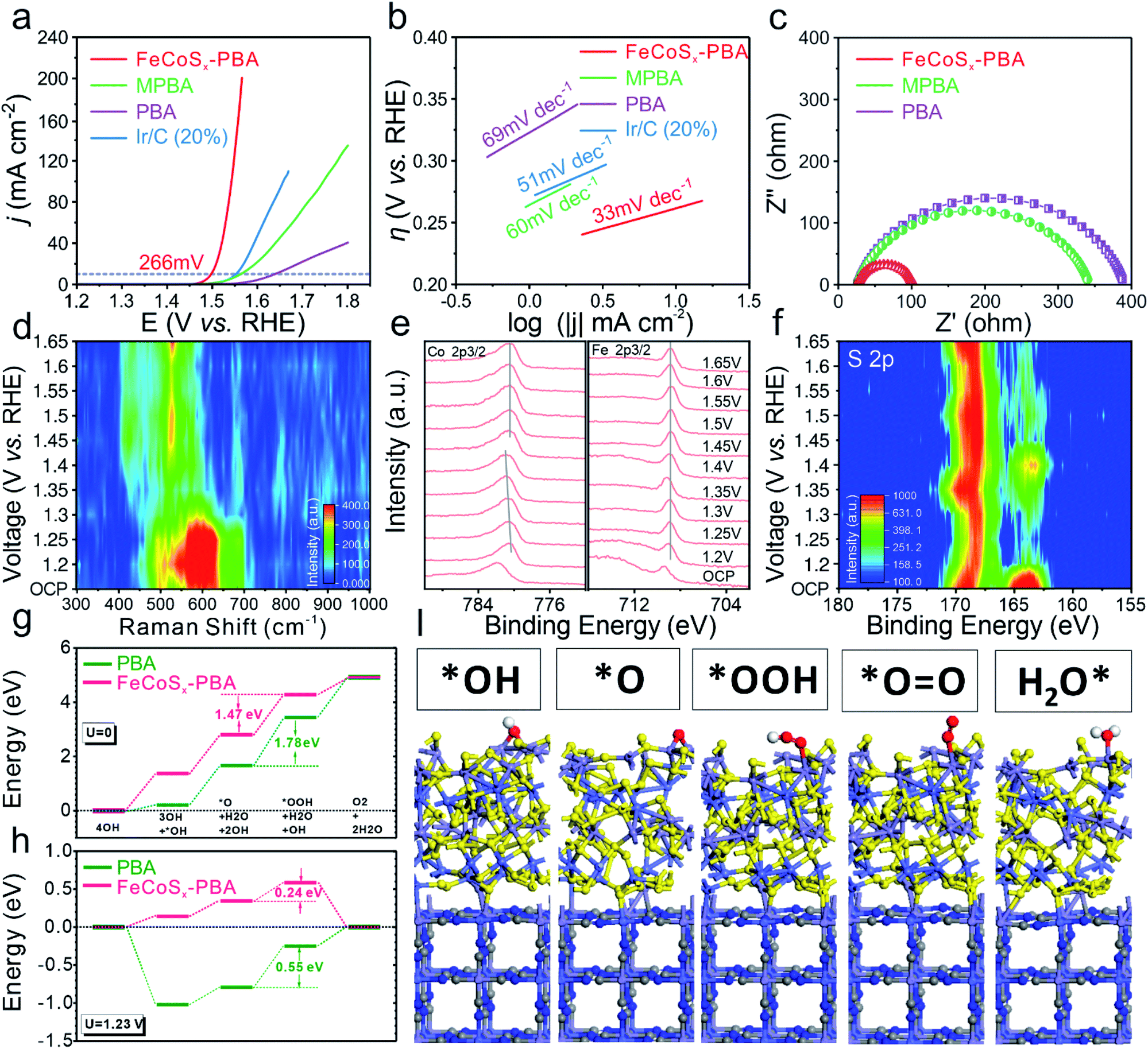 | ||
| Fig. 4 (a) Polarization curves and (b) Tafel plots of PBA, MPBA, FeCoSx-PBA and commercial Ir/C (20%) in an O2-saturated 1 M KOH solution (scan rate 2 mV s−1). (c) EIS spectra of PBA, MPBA and FeCoSx-PBA. (d) In situ Raman spectra of FeCoSx-PBA during the OER. (e) Quasi-in situ Co 2p3/2 and Fe 2p3/2 XPS spectra of FeCoSx-PBA during the OER. (f) Quasi-in situ S 2p XPS spectra of FeCoSx-PBA during the OER. (g) The OER reaction pathways of FeCoSx-PBA and PBA in the alkaline environment at the equilibrium state (U = 0 V). (h) The OER reaction pathways of FeCoSx-PBA and PBA in the alkaline environment with an applied potential of 1.23 V. (i) The structural configurations of adsorption of key intermediates on FeCoSx-PBA. | ||
To gain deeper insight into the OER activity of FeCoSx-PBA, the electrochemical impedance spectra (EIS) were taken and an electrochemical double-layer capacitance (Cdl) test was performed. The Cdl of FeCoSx-PBA, MPBA, and PBA (Fig. S14†) were confirmed to be 24.3 mF cm−2, 11.8 mF cm−2, and 8.2 mF cm−2, respectively. Combined with BET results (Fig. S4†), FeCoSx-PBA performed with better exposure and enhanced electroactive sites. In order to clarify the intrinsic activity differences of the above materials, the catalytic current was normalized to electrode geometric area, BET surface area, and ECSA, respectively. As shown in Fig. S15,† after eliminating the effect of surface area, FeCoSx-PBA still performs with the best OER activity. This demonstrates that the superior OER activity of FeCoSx-PBA is due to the co-optimization of electronic structure and morphology. In the Nyquist plots (Fig. 4c), PBA displays the largest charge transfer resistance (Rct), and MPBA shows a 12% reduced Rct value. Undoubtedly, FeCoSx-PBA has the lowest Rct of all three catalysts. It is only approximately a quarter of PBA, which suggests the faster electron transfer ability during the OER in FeCoSx-PBA. This result further proved that the electron structure was modified from PBA to FeCoSx-PBA along with the decreasing Rct. The important role of PBA frameworks in the OER was also unraveled by a contrast experiment. As shown in Fig. S16,† amorphous FeCoSx shows poor OER activities compared to FeCoSx-PBA. In addition, the stability test result of amorphous FeCoSx without a PBA framework is shown in Fig. S16.† This clearly indicates poor stability. The above results proved that an FeCoSx layer combined with a PBA structural frame determines the long-term durability of the composite electrocatalyst.
OER dynamic study is another vital tool for understanding the OER process and mechanism. In situ Raman and quasi-in situ XPS spectra were performed on FeCoSx-PBA during the OER catalysis process. The in situ Raman spectrum (Fig. 4d and S17†) shows the reconstruction of a catalyst at 1.35 V (vs. RHE) and the formation of CoFe oxy-hydroxides. The low reconstruction voltage is favorable for catalyzing the OER and could be caused by the large amount of defects in the amorphous structure. Further quasi-in situ XPS results unravel the electronic structure evolution of different elements at different applied OER voltages. As shown in Fig. 4e, with increased voltage, the valence state of Co also grows and becomes stable at a high value. But Fe offers a very steady valence state during the OER process. These results show that the Co sites facilitate site-to-site electron transfer and stabilize the Fe sites. Moreover, the S 2p quasi-in situ XPS result (Fig. 4f and S18†) shows the existence of residual sulfur in the catalysts during the OER. The increased adsorbed SO42− peak also indicated that lattice S in FeCoSx-PBA is partially oxidized to SO42− during the OER. The SO42− would be adsorbed on the catalyst surface and promote the OER process.50–53
Then we further unravel the improved electroactivity of the FeCoSx-PBA hybrid system based on the energetic reaction pathway. In the equilibrium state, the OER reaction pathway shows a continuous trend for both PBA and FeCoSx-PBA composite systems. The reaction [*O + H2O + 2OH] → [*OOH + H2O + OH] is the rate-determining step for the OER due to its having the largest energy barrier. For pure PBA, the energy barrier is 1.78 eV, which is much larger than the 1.47 eV of the FeCoSx-PBA composite system, confirming the enhanced electroactivity for the OER (Fig. 4g). With an applied standard potential of 1.23 V, the evident reaction trend is displayed. For pristine PBA, the initial OH adsorption of the alkaline OER is very strong, which leads to a nearly 1 eV energy drop. However, such a strong overbinding effect of OH also hinders the subsequent reactions, resulting in a 0.55 eV overpotential for the OER. In comparison, although the slightly weaker adsorption of OH on the FeCoSx-PBA composite shows the initial uphill trend for the OER, the overall overpotential has been greatly lowered to 0.24 eV, which is highly consistent with the electrochemical performances (Fig. 4h). The stable adsorption of key intermediates shows that the O-related species prefer the Fe sites on the surface with mild adsorption strength to maintain efficient intermediate transformation. Meanwhile, the Co sites facilitate the site-to-site electron transfer from the surface, and S sites balance the optimal binding strength with OH groups within the alkaline environment. The stable structure of PBA after adsorption supports the long-term durability performances in the experiments (Fig. 4i). The above results are also consistent with the quasi-in situ XPS results (Fig. 4e, f and S18†).
To evaluate the Al–air battery performance of FeCoSx-PBA, the oxygen reduction reaction (ORR) performance was evaluated first in Fig. S19.† FeCoSx-PBA indicates an optimized half-wave potential and diffusion-limited current density. The electron transfer number of FeCoSx-PBA is 3.3. Then a liquid Al–air battery was assembled with 2 M KOH. Polarization curves (Fig. 5b) indicated that the maximum power density of FeCoSx-PBA is 58.3 mW cm−2. The capacity property was studied by galvanostatic discharge curves. As shown in Fig. 5c, FeCoSx-PBA generates a large specific capacity of 1259 mA h g−1 at a current density 1 mA cm−2, and the energy density was calculated as 1483 W h kg−1. When the current density increased to 5 mA cm−2, the specific capacity and energy density also decreased to 1091 mA h g−1 and 1199 W h kg−1, respectively. Furthermore, at a large discharge current density of 10 mA cm−2, this Al–air battery still retains 1002 mA h g−1 capacity and 970 W h kg−1 energy density. These battery parameters of FeCoSx-PBA materials are better than those of other materials in the literature to the best of our knowledge and indicate their great potential to serve as efficient air cathodes for clean energy storage and conversion applications. Due to the extremely high reduction potential of Al in aqueous solution, Al cannot be electrodeposited in aqueous electrolytes. Thus, the Al–air battery was not rechargeable. Hence, to assess the long-term stability of the FeCoSx-PBA cathode, a mechanical recharge through replacing the Al sheet in the battery was performed. 10 hours was chosen as the period, and in all 5 periods, the Al–air battery retains a nearly unchanged discharge voltage, as shown in Fig. 5d. This result demonstrated that the FeCoSx-PBA cathode exhibits superior long-term stability. Further, a blue-light-emitting diode was successfully lit by two series-connected Al–air batteries (Fig. S20†). Also, the Ragone diagram plotting the energy and power densities of the fabricated device compared to those reported Al–air batteries are depicted in Fig. 5e and Table S4.†
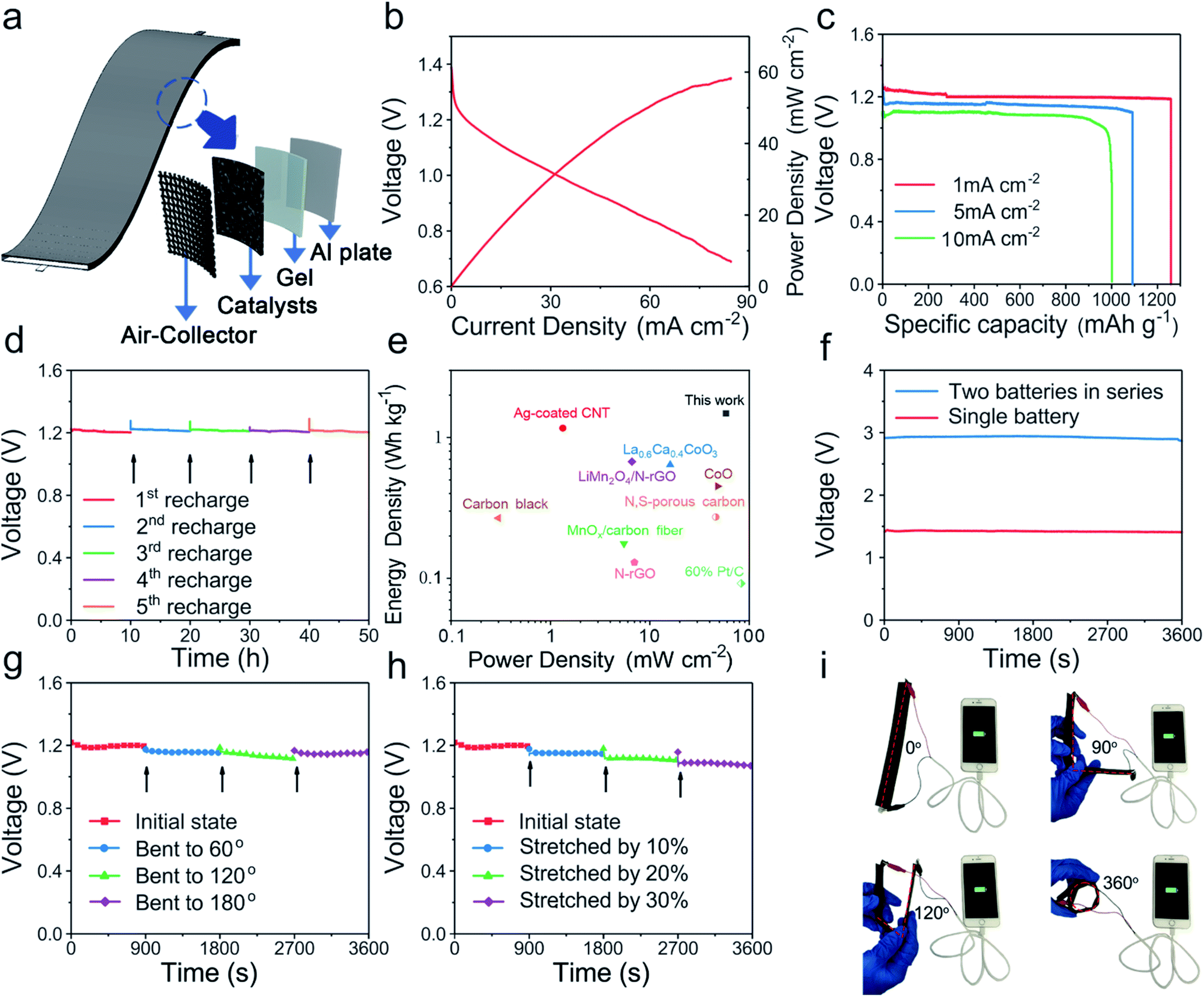 | ||
| Fig. 5 (a) Schematic diagram of flexible Al–air batteries. (b) Polarization and power density curves of the Al–air batteries. (c) Galvanostatic discharge curves at 1 mA cm−2, 5 mA cm−2, 10 mA cm−2 of Al–air batteries. (d) Discharge curves at 1 mA cm−2 and recharged by replacing Al foil. (e) Ragone plots of the Al–air batteries. The values reported for other energy storage devices are added for comparison. (f) Open circuit voltage–time curves of one and two solid Al–air batteries. (g–h) Discharge curves of wearable Al–air batteries at different bending angles or stretching ratios at a discharge current of 1 mA. (i) Photographs of four batteries in series charging a smartphone with bending angles 0°, 90°, 120°, and 360°. | ||
Driven by the increasing demand for flexible and wearable electronic devices, solid Al–air batteries were further fabricated with FeCoSx-PBA/carbon black material supported on carbon cloth as the air cathode, aluminium sheet as the metal anode, and alkaline polyvinyl alcohol/polyethylene glycol as the gel electrolyte (Fig. 5a). The fabricated battery exhibited outstanding bendability and stretchability. Fig. 5f shows a single battery displaying an open circuit voltage of 1.44 V, and two batteries in series exhibiting an open-circuit voltage of 2.94 V. The hydrogel used in the battery endows it with excellent flexibility (Fig. S21†). At a discharge current of 1 mA, this solid Al-battery was bent to 60, 120, and 180° and its discharge performance was almost constant because its discharge voltage only slightly decreased (Fig. 5g). When the battery was stretched by 10%, 20%, and 30%, it did not break upon elongation. After stretching 10% to 30%, the battery had a gradually decreasing discharge voltage at 1 mA cm−2, but the amplitude of the voltage decrease is small. Even up to 30%, the battery output voltage was also maintained above 1 V during stretching (Fig. 5h). It is easy to understand that stretched batteries will increase charge and mass transfer distance and hinder battery performance. From the above results, this solid flexible Al–air battery displays remarkable bendability and stretchability. The actual application shows that a device with four batteries connected in series can charge a smartphone from 20% electricity to full charge (Fig. 5i) under various bending conditions from 0° to 360°.
Conclusions
In summary, we have successfully developed a unique hollow FeCoSx-PBA hetero-nanoframe catalyst via an ultrasound-assisted ion-exchange procedure. The introduction of hetero Fe and S atoms modifies the intrinsic electronic structure of the Co site. DFT calculations further confirm that the surface FeCoSx layer significantly enhances the electron transfer capability of PBA to achieve an efficient OER process. The suppression of OH overbinding also guarantees the efficient transformation of the intermediate. Meanwhile, the stable structure of PBA protected by the surface FeCoSx layer is the critical factor for the long-term durability of the electrocatalyst. The above morphological and structural advantages provide more exposed active sites and an optimized intrinsic electronic structure, which dramatically enhances the catalytic activity. Moreover, the FeCoSx-PBA cathode can drive a solid Al–air battery with remarkable bendability, stretchability, and a high specific capacity of 1259 mA h g−1 at 1 mA cm−2. This work may expand the toolbox of strategies to design and synthesize high-performance energy-related catalysts in terms of morphology and structure through electronic engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the National Natural Science Foundation of China (No. 21931001 and 21922105), the Special Fund Project of Guiding Scientific and Technological Innovation Development of Gansu Province (2019ZX-04) and the 111 Project (B20027). We also acknowledge support by the Fundamental Research Funds for the Central Universities (lzujbky-2021-pd04, lzujbky-2021-it12 and lzujbky-2021-37). B. H. acknowledges the support of the Natural Science Foundation of China (NSFC) (No. 21771156) and the Early Career Scheme (ECS) fund (Grant PolyU 253026/16P) from the Research Grant Council (RGC) in Hong Kong. J. Y. acknowledges the support of the China Postdoctoral Science Foundation (2021M691375) and the China National Postdoctoral Program for Innovative Talents (BX20200157).References
- C. Steven and A. Majumdar, Nature, 2012, 488, 294 CrossRef PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and S. H. Yang, Nat. Chem., 2011, 3, 546 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072 CrossRef CAS PubMed.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624 CrossRef CAS PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and S. H. Yang, J. Phys. Chem., 2012, 3, 399 CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and S. H. Yang, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC.
- H. Zhang, J. Wang, Q. Cheng, P. Saha and H. Jiang, Green Energy Environ., 2020, 5, 492 CrossRef.
- X. Y. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. D. Lou and U. Paik, Adv. Mater., 2016, 28, 9006 CrossRef CAS PubMed.
- J. Nai, Y. Lu, L. Yu, X. Wang and X. W. D. Lou, Adv. Mater., 2017, 29, 1703870 CrossRef PubMed.
- R. Wang, Y. Wei, L. An, R. Yang, L. Guo, Z. Weng, P. Da, W. Chen, J. Jin, J. Li and P. Xi, Chin. J. Chem., 2020, 38, 772 CrossRef CAS.
- S. Liu, M. Wang, X. Sun, N. Xu, J. Liu, Y. Wang, T. Qian and C. Yan, Adv. Mater., 2018, 30, 12876 Search PubMed.
- V. R. Stamenkovic, B. Fowler, B. Simon Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211 CrossRef CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- L. Han, X. Y. Yu and X. W. Lou, Adv. Mater., 2016, 28, 4601 CrossRef CAS PubMed.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Adv. Mater., 2015, 127, 5421 Search PubMed.
- G. Yilmaz, K. M. Yam, C. Zhang, H. J. Fan and G. W. Ho, Adv. Mater., 2017, 29, 1606814 CrossRef PubMed.
- X. Zhong, M. Oubla, X. Wang, Y. Huang, H. Zeng, S. Wang, K. Liu, J. Zhou, L. He, H. Zhong, N. Alonso-Vante, C.-W. Wang, W.-B. Wu, H.-J. Lin, C.-T. Chen, Z. Hu, Y. Huang and J. Ma, Nat. Commun., 2021, 12, 3136 CrossRef CAS PubMed.
- Y. Xu, Y. Zhao, J. Ren, Y. Zhang and H. Peng, Angew. Chem., Int. Ed., 2016, 128, 8111 CrossRef.
- I. A. Vladimir, F. Aryasetiawan and A. I. Lichtenstein, J. Phys.: Condens. Matter, 1997, 9, 767 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982 CrossRef CAS.
- J. D. Head and M. C. Zerner, Chem. Phys. Lett., 1985, 122, 264 CrossRef CAS.
- N. Marzari, D. Vanderbilt and M. C. Payne, Phys. Rev. Lett., 1997, 79, 1337 CrossRef CAS.
- L. Kleinman and D. M. Bylander, Phys. Rev. Lett., 1982, 48, 1425 CrossRef CAS.
- S. G. Louie, S. Froyen and M. L. Cohen, Phys. Rev. B, 1982, 26, 1738 CrossRef CAS.
- I. Grinberg, N. J. Ramer and A. M. Rappe, Phys. Rev. B, 2000, 62, 2311 CrossRef CAS.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B, 1990, 41, 1227 CrossRef PubMed.
- M. I. J. Probert and M. C. Payne, Phys. Rev. B, 2003, 67, 075204 CrossRef.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131 CrossRef CAS PubMed.
- Y. Dou, T. Liao, Z. Ma, D. Tian, Q. Liu, F. Xiao, Z. Sun, J. Ho Kim and S. Xue Dou, Nano Energy, 2016, 30, 267 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xiao, S. Wei, B. Ye and Y. Xie, J. Am. Chem. Soc., 2014, 136, 15670 CrossRef CAS PubMed.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076 CrossRef CAS PubMed.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. Zhang, J. Am. Chem. Soc., 2016, 138, 6924 CrossRef CAS PubMed.
- Y. Chen, R. Gokhale, A. Serov, K. Artyushkova and P. Atanassov, Nano Energy, 2017, 38, 201 CrossRef CAS.
- Y. Li, Y. Wang, B. Pattengale, J. Yin, L. An, F. Cheng, Y. Li, J. Huang and P. Xi, Nanoscale, 2017, 9, 9230 RSC.
- D. R. Mullins and P. F. Lyman, J. Phys. Chem., 1993, 97, 9226 CrossRef CAS.
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, J. Am. Chem. Soc., 2016, 138, 14546 CrossRef CAS PubMed.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143 CrossRef CAS PubMed.
- J. Yin, Y. Li, F. Lv, Q. Fan, Y. Q. Zhao, Q. Zhang, W. Wang, F. Cheng, P. Xi and S. Guo, ACS Nano, 2017, 11, 2275 CrossRef CAS PubMed.
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 2, 869 CrossRef CAS.
- T.-H. Shen, L. Spillane, J. Vavra, T. H. M. Pham, J. Peng, Y. Shao-Horn and V. Tileli, J. Am. Chem. Soc., 2020, 142, 15876 CrossRef CAS PubMed.
- W. Sohn, K. C. Kwon, J. M. Suh, T. H. Lee, K. C. Roh and H. W. Jang, Nano Convergence, 2021, 8, 11 CrossRef CAS PubMed.
- K. Urita, T. Fujimori, H. Notohara and I. Moriguchi, ACS Appl. Energy Mater., 2018, 1, 807 CrossRef CAS.
- W. Wang, J. Jiang, T. Ding, C. Wang, J. Zuo and Q. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 2235 CrossRef CAS PubMed.
- U. I. Kramm, M. Lefevre, N. Larouche, D. Schmeisser and J. P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329 CrossRef CAS PubMed.
- B. S. Yeo and A. T. Bell, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901 CrossRef CAS PubMed.
- N. Kornienko, J. Resasco, N. Becknell, C.-M. Jiang, Y.-S. Liu, K. Nie, X. Sun, J. Guo, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2015, 137, 7448 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00191h |
| This journal is © The Royal Society of Chemistry 2022 |