
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transfer mediated triplet excited state formation in donor–acceptor–donor BODIPY: Application for recording of holographic structures in photopolymerizable glass†
Tatsiana
Mikulchyk
 a,
Safakath
Karuthedath
b,
Catherine S. P.
De Castro
b,
Andrey A.
Buglak
cd,
Aimee
Sheehan
e,
Aaron
Wieder
e,
Frédéric
Laquai
b,
Izabela
Naydenova
a and
Mikhail A.
Filatov
*e
a,
Safakath
Karuthedath
b,
Catherine S. P.
De Castro
b,
Andrey A.
Buglak
cd,
Aimee
Sheehan
e,
Aaron
Wieder
e,
Frédéric
Laquai
b,
Izabela
Naydenova
a and
Mikhail A.
Filatov
*e
aCentre for Industrial and Engineering Optics, School of Physics and Clinical and Optometric Sciences, College of Science and Health, Technological University Dublin, City Campus, Grangegorman, Dublin 7, Ireland
bKing Abdullah University of Science and Technology (KAUST), KAUST Solar Center (KSC), Physical Sciences and Engineering Division (PSE), Material Science and Engineering Program (MSE), Thuwal, 23955-6900, Kingdom of Saudi Arabia
cFaculty of Physics, Saint-Petersburg State University, University Emb. 7-9, 199034, St. Petersburg, Russia
dInstitute of Physics, Kazan Federal University, Kremlevskaya 18, Kazan, 420008, Russia
eSchool of Chemical and Pharmaceutical Sciences, Technological University Dublin, City Campus, Grangegorman, Dublin 7, Ireland. E-mail: mikhail.filatov@tudublin.ie
First published on 18th July 2022
Abstract
Donor–acceptor–donor BODIPY triads bearing anthracene or pyrene as electron donating subunits were prepared through a stepwise synthesis. Photoinduced electron transfer and formation of long-lived triplet excited states via spin–orbit charge transfer intersystem crossing (SOCT-ISC) was studied by steady-state and ultrafast pump-probe spectroscopy and further supported by DFT computations. New BODIPYs were found to form triplet states and sensitize singlet oxygen in both polar and non-polar solvents which is unusual for photosensitizers operating via SOCT-ISC. BODIPY-anthracene triad (ABA) was used as a photosensitizer component in a photopolymerizable glass that was prepared by a four-step sol–gel process. ABA in combination with N-phenylglycin (NPG) showed the ability to initiate a free-radical polymerization of methacrylate monomers under 532 nm irradiation thus allowing for holographic recording of diffractive structures. High diffraction efficiency (up to 87%) obtained for ABA-NPG containing glass as compared to a reference diiodo-BODIPY (I2BDP) demonstrates for the first time that heavy-atom-free SOCT-ISC photosensitizers can efficiently operate in the solid state.
Introduction
The development of photonic technologies for health, energy and communication creates a need for novel functional dyes with optimized excited state properties to meet new and demanding criteria. Triplet photosensitizers (PSs) are organic dyes showing rapid intersystem crossing (ISC) and forming long-lived excited states.1 Such dyes have found multiple uses in photocatalysis,2 photodynamic therapy (PDT),3 photovoltaics4 and triplet–triplet annihilation up-conversion (TTA-UC),5 where long-lived triplet excited states are required for efficient harvesting of light energy. A common approach for the enhancement of ISC in organic chromophores relies on the introduction of heavy atoms into the structure, such as halogens or transition metals, which promote ISC via spin–orbital interactions.6 However, the incorporation of heavy atoms results in high cost of synthesis, increased toxicity and shortening of the triplet state lifetimes.7 Therefore, replacing of costly transition metal complexes with heavy-atom-free organic PSs has drawn considerable attention and alternative approaches for promoting ISC in organic chromophores have been actively studied.8The formation of triplet excited states in electron donor–acceptor molecules via the process of spin–orbit charge transfer intersystem crossing (SOCT-ISC) has attracted particular attention. In these systems, photoinduced electron transfer between the donor and acceptor subunits leads to formation of a charge-transfer state (1CT), which further undergoes charge recombination into the lowest triplet excited state (T1). SOCT-ISC has been observed in donor–acceptor dyads based on BODIPYs9 and other difluoroboron complexes,10 metal dipyrrins,11 biphenyls,12 naphthalimides,13 perylenimides,14 Nile Red derivatives15 and spiro rhodamines.16 Many reported molecular systems undergoing SOCT-ISC exhibit triplet quantum yields (ΦT) that are comparable or even higher than those of transition metal complexes and halogenated dyes. Other important advantages include synthetic accessibility, low toxicity, high singlet oxygen quantum yields (ΦΔ), long triplet excited state lifetimes and intense visible light absorption. Due to these advantages, heavy-atom-free donor–acceptor PSs gained increasing attention in applications where contamination with transition metals may not be suitable, e.g., biomedical and electronic applications. In particular, such dyes have been proven to be successful as catalysts for photocontrolled polymerization through an oxidative quenching mechanism.17
Volume holographic optical elements are critically important for information storage,18,19 augmented reality displays,20,21 solar energy harvesting,22,23 and LED light management technologies.24 Photopolymerization is one of the most convenient processes for creating holographic microstructures in the volume of photosensitive materials as it can be spatially and temporally controlled.25–27
Microstructures created using this method represent holographic optical elements with high efficiency (up to 100%) and tunable selectivity (angular bandwidth varying in the range from a fraction of a degree to tens of degrees and spectral bandwidth varying from a few to hundreds of nanometres).28,29 A widely used type of photoinitiating system for holographic recording relies on the combination of a photosensitizer and a co-initiator component, which forms free radicals under light irradiation. Photosensitizers, such as methylene blue30 and xanthene dyes,31 are regularly employed in photoinitiating systems. However, their performance is not always satisfactory in terms of diffraction efficiency of the resulting holographic structures, presumably due to insufficient photostability and short excited state lifetimes.32 Taking into account the SOCT-ISC photosensitizers’ excellent optical characteristics and tunable properties, their use for holographic recording could allow to enhance photosensitivity of the photopolymerizable material and improve the resulting diffraction efficiency. To the best of our knowledge, such dyes have not yet been tested in this process.
Among potential limitations for using SOCT-ISC dyes in photopolymerization is the inherent dependence of triplet state formation via this mechanism on parameters of the environment, in particular its polarity33 and viscosity.34 Although, solvent-independent PSs operating via SOCT-ISC are highly desirable, so far, only a few examples of such systems have been reported. Zhao and co-workers described carbazole–anthracene–BODIPY triads in which the carbazole unit was used as the secondary electron donor besides the anthryl moiety.35 These systems exhibited ΦΔ values almost independent of the solvent polarity in contrast to donor–acceptor BODIPY dyads featuring SOCT-ISC. Recently, Durka and co-workers reported spiro 9-borafluorene complexes undergoing SOCT-ISC while not exhibiting solvent polarity dependence of ΦΔ values.36
Although predicting the efficiency of SOCT for a given structure in different media is still challenging, recently, we performed the first quantitative structure–property relationship (QSPR) study of these systems and developed a method for predicting singlet oxygen generation quantum yields for heavy-atom-free BODIPYs in different solvents based on a combination of quantum mechanical molecular descriptors and topological descriptors.37 QSPR analysis showed that structures containing a higher number of electron donor/acceptor subunits, e.g., donor–acceptor–donor (D–A–D) and acceptor–donor–acceptor triads, in general exhibit less pronounced solvent dependence of triplet state formation compared to D–A dyads. Such triads have been actively studied as materials for photovoltaics38 and it has been demonstrated that intramolecular charge transfer (ICT) in these systems is faster than in D–A dyads.39 Such triads also show weaker dependence of charge transfer on solvent polarity and exhibit efficient ICT even in non-polar solvents. Inspired by these observations, herein we investigated the possibility to attenuate the solvent polarity-dependence of triplet formation through SOCT-ISC in BODIPY by introducing two electron donating groups. For this study, we designed new triads by functionalizing the BODIPY core with anthracene or pyrene groups in positions 2 and 6 to facilitate the electron transfer. These newly synthesized triads were characterized using different spectroscopic techniques, demonstrating efficient charge transfer and triplet states formation. The BODIPY-anthracene triad (ABA) which showed higher triplet state yield was further studied as a photosensitizer in a two-component photoinitiating system. Here, the second component, N-phenylglycine, acts as a co-initiator reacting with excited states of ABA and forming free radicals. This system was applied by us for polymerization of methacrylate monomers in the volume of a photopolymerizable glass material under 532 nm laser patterning. The obtained high diffraction efficiency (up to 87%) of the resulting grating proved the excellent capability of ABA to initiate photopolymerization. Moreover, ABA showed comparable performance with respect to a reference diiodo-BODIPY photosensitizer (I2BDP) under the same conditions.
Results and discussion
Synthesis of BODIPY triads
The synthesis of target BODIPY triads is shown in Scheme 1. The synthesis started from 9-anthracenecarboxaldehyde and 2-pyrenylcarboxaldehyde which were reacted with 1-nitropropane to give the corresponding nitroalkenes 1a–b (step 1). These compounds were subjected to Barton–Zard reaction with ethyl isocyanacetate (step 2), delivering pyrrolecarboxylates 2a–b. The obtained products were cleanly transformed into pyrroles 3a–bvia the reduction with LiAlH4 (step 3). Pyrroles were reacted with benzaldehyde and the resulting intermediate dipyrromethanes were oxidized without isolation, followed by treatment with BF3·Et2O in the presence of N,N-diisopropylethylamine (step 4) to give compounds ABA and PBP. BODIPY products were isolated by silica gel column chromatography in good yields. Isolated products were fully characterized through 1H and 13C NMR spectroscopy and high-resolution mass spectrometry (HRMS, Fig. S7–S10, ESI†).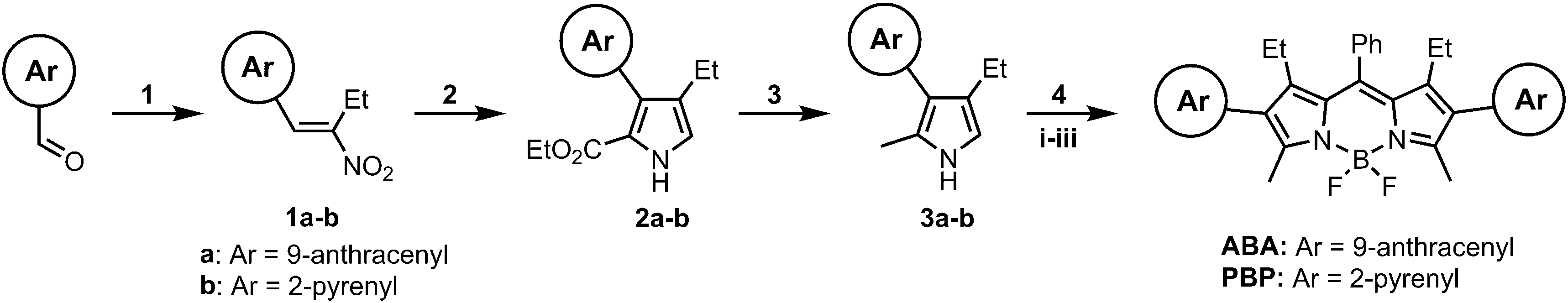 | ||
| Scheme 1 Synthesis of target compounds. Reagents and conditions: (1) 1-nitropropane (neat), (NH4)2CO3, reflux, 4–8 h, 59% yield for 1a and 63% yield for 1b; (2) CNCH2CO2Et, DBU, THF, room temperature (r.t.), 18 h, 78% yield for 2a and 66% yield for 2b; (3) LiAlH4, THF, reflux, 4 h, 99% yield for 3a and 93% yield for 3b; (4) (i) PhCHO, TFA, DCM, r.t., 18 h; (ii) DDQ, r.t., 1 h; (iii) BF3·Et2O, DIPEA, DCM, r.t., 2 h, 36% yield for ABA, 23% yield for PBP. | ||
Steady-state optical properties of the triads
The absorption and fluorescence emission properties of the prepared BODIPYs are presented in Fig. 1 and summarized in Table 1. For both triads the absorption spectra show transitions associated with the two subunits, indicating weak coupling of the chromophores in the ground state. The experimental and DFT-simulated spectra (Fig. S14 and S15, ESI†) are similar, with the main band corresponding to S0 → S1 transition in BODIPY. As shown in Fig. S16 (ESI†), triads have well-separated HOMO/LUMO distributions, indicating feasibility of the electron transfer from anthracene/pyrene subunits to the BODIPY in the excited state. Upon changing the solvent polarity, the shape and maxima of the absorption undergo little change. At the same time, the emission properties are greatly affected by solvent polarity. The emission spectrum of ABA in toluene (Fig. 1) shows a fluorescence band centred at 575 nm, characterized by a Stokes shift of 46 nm and an emission quantum yield (Φem) of 0.513. Unlike previously reported BODIPY-anthracene and pyrene dyads,40ABA and PBP do not display emission bands corresponding to the transition from the CT state which may be due to the negligible quantum yield of such emission. Increase in solvent polarity leads to a red shift of the emission and drop in quantum yields for both triads. In polar acetonitrile (ACN) the emission of ABA shows a Stokes shift of 125 nm and quantum yield of 0.025. Likewise, the excited state lifetime in ACN is much shorter (1.13 ns) than that in toluene (3.65 ns). This observation was reproduced in other solvents, showing a clear correlation between polarity, red shift of the emission, and its quantum yield. Such emission quenching in polar media is the signature of intramolecular electron transfer.41 Similar behaviour was observed for triad PBP, except for its slightly stronger emission in polar solvents compared to ABA. This indicates that although ICT takes place in dyad PBP, it is less efficient compared to the triad based on anthracene. This is consistent with higher oxidation potential of pyrene which makes it less efficient at quenching BODIPY fluorescence than anthracene.42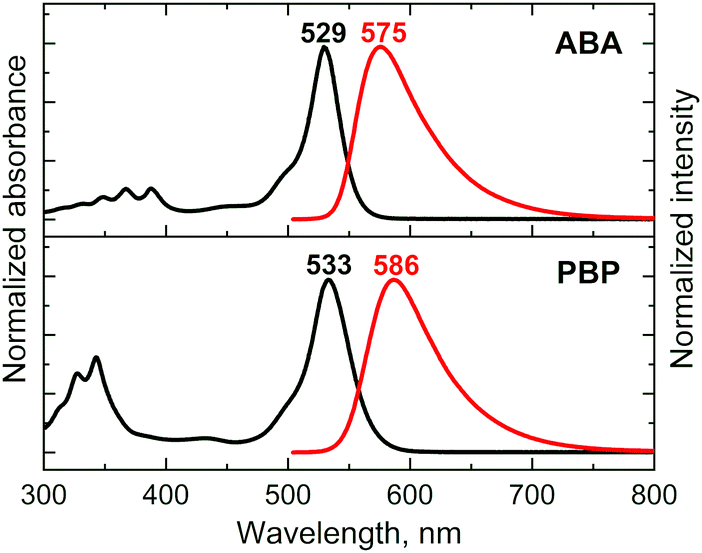 | ||
| Fig. 1 Absorption and emission spectra of ABA and PBP in toluene (λexc = 490 nm). | ||
| Compound | Solvent | λ abs (nm) | λ em (nm) | Φ em | Φ Δ |
|---|---|---|---|---|---|
| a Absorption maxima corresponding to the BODIPY chromophore. b The emission spectra were collected with excitation at the vibrational shoulder of the BODIPY absorption (490 nm). c Measured using Rhodamine 6G (Φem = 0.95 in ethanol) as a reference. d Measured using 2,6-diiodo-8-phenyl-1,3,5,7-tetramethylBODIPY (ΦΔ = 0.85 in toluene) as a reference. e DCM was not used due to possible photolysis of the sensor in this solvent, leading to overestimation of ΦΔ values.43 HEX – hexane, TOL – toluene, THF – tetrahydrofuran, DCM – dichloromethane, EtOH – ethanol, ACN – acetonitrile. | |||||
|
HEX | 528 | 561 | 0.617 | 0.22 |
| TOL | 529 | 575 | 0.513 | 0.289 | |
| THF | 528 | 580 | 0.291 | 0.307 | |
| DCM | 529 | 587 | 0.207 | n.d.e | |
| EtOH | 527 | 591 | 0.094 | 0.299 | |
| ACN | 525 | 640 | 0.025 | 0.14 | |
|
HEX | 531 | 573 | 0.676 | 0.18 |
| TOL | 533 | 586 | 0.634 | 0.203 | |
| THF | 530 | 586 | 0.445 | 0.185 | |
| DCM | 531 | 589 | 0.352 | n.d.e | |
| EtOH | 530 | 589 | 0.191 | 0.19 | |
| ACN | 527 | 602 | 0.078 | 0.09 | |
To rationalize the effect of anthracenyl and pyrenyl substituents on the geometry and electronic structure of the triads, density functional theory (DFT) calculations were performed (see ESI† for computational details). Calculated ground state geometries of ABA and PBP are presented in Fig. 2. ABA possesses Cs symmetry since the dihedral angle between the anthracene subunit and the BODIPY core (d(C4–C1–C25–C28), ψ1) equals 108°, whereas for the second anthracene residue the dihedral angle (d(C10–C11–C31–C34), ψ2) is equal to −73°. The PBP triad is strictly symmetrical (Cs point group) since both dihedral angles between the mean BODIPY plane and pyrene subunits are 65.5°. Similar molecular geometries were obtained in toluene and ACN: the root mean square deviation (RMSD) of Cartesian coordinates (toluene vs acetonitrile) equals 0.01 Å. The optimized geometry of ABA in S1 state shows rotation along C–C bond between anthracene subunits and BODIPY core: ψ1 and ψ2 were calculated to be 118° and −63° (Fig. S18, ESI†), respectively. Similarly, the S1 state geometry of PBP shows rotation with ψ1 and ψ2 between pyrene and the BODIPY planes of 54.5° (Fig. S20, ESI†). Hence, the dihedral angles of S1 state differ by 10° and 13.3° for ABA and PBP, respectively, as compared to the ground state geometries.
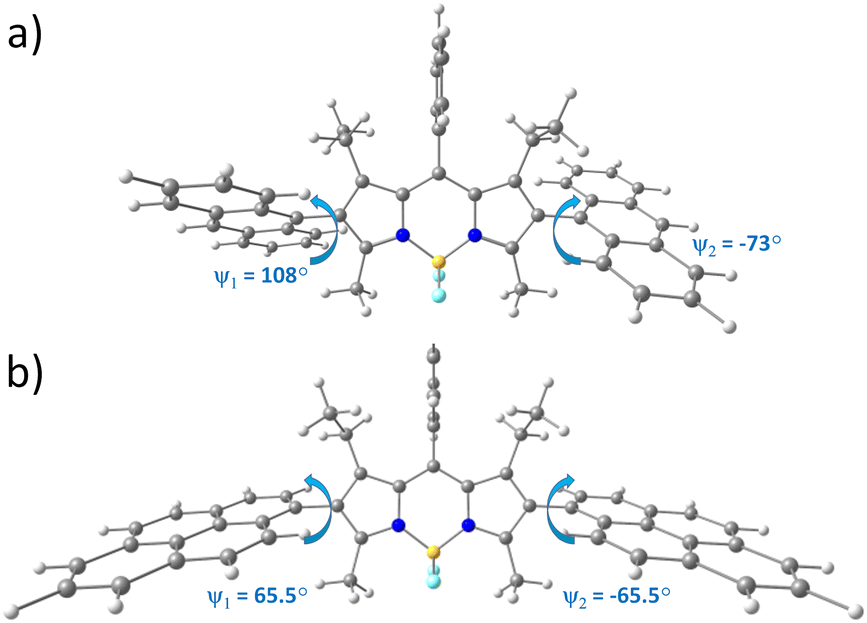 | ||
| Fig. 2 Optimized ground state geometries of (a) ABA and (b) PBP. | ||
The observed Stokes shifts for ABA and PBP are much larger compared to BODIPYs bearing aryl groups in the meso position (typically, <15 nm).44 Large Stokes shifts observed for both triads can be rationalized by the scheme presented in Fig. 3. In ABA, geometry relaxation following photoexcitation leads to a significant change of the frontier molecular orbital energies. Thus, the LUMO is stabilized by 0.1 eV at the S1 state geometry as compared to the S0 structure. At the same time, the HOMO is destabilized by 0.15 eV for the S1 state geometry as compared to the S0 state. As a result, the HOMO–LUMO energy gap is greatly decreased with S1 geometry relaxation. In PBP, geometry relaxation following photoexcitation leads to LUMO stabilization by 0.11 eV. The HOMO is destabilized by 0.14 eV for the S1 state geometry as compared to the S0 state (Fig. S22, ESI†). These results corroborate with previous reports on geometry relaxation-induced large Stokes shifts (up to 100 nm) in 2,6-bisthienyl BODIPY derivatives.45
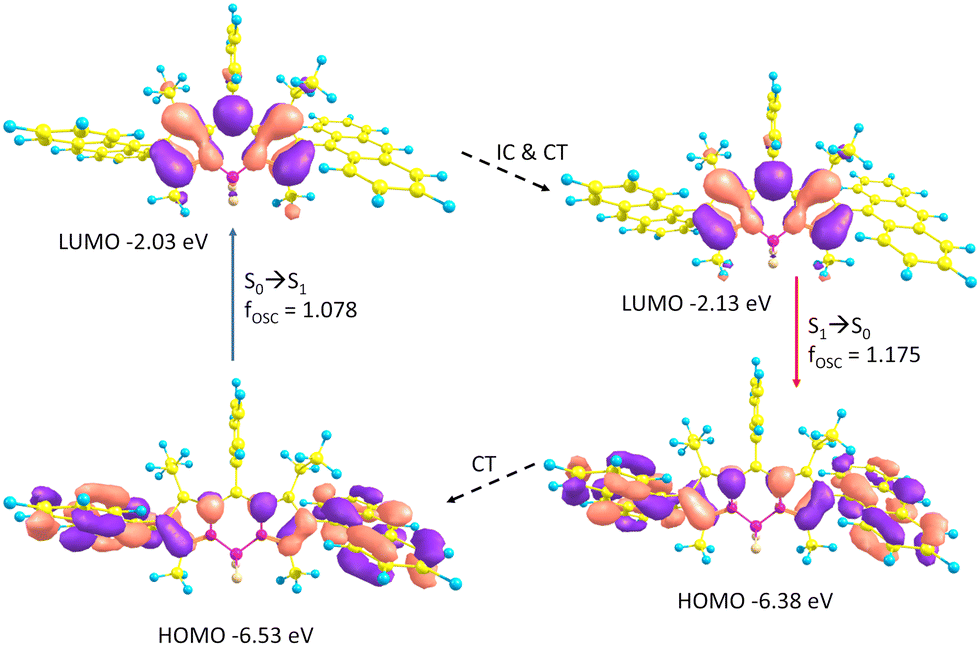 | ||
| Fig. 3 Rationalization of the large Stokes shift of ABA in ACN: the geometry relaxation upon photoexcitation and molecular orbitals involved in the vertical absorption and emission. The vertical excitations were calculated based on the optimized ground state geometry (S0), the emission was calculated based on the optimized geometry of the excited state (S1). | ||
To probe the efficiency of triplet state formation in compounds ABA and PBP, singlet oxygen sensitization was evaluated using the chemical trapping method with 1,9-dimethylanthracene (DMA). Irradiation of air-saturated solutions of the triads with 514 nm laser (20 mW cm−2) did not lead to any changes in the absorption spectra (Fig. S12 and S13, ESI†), proving sufficient photostability of these compounds and the absence of singlet oxygen addition which was previously observed by us for BODIPYs bearing anthracene moiety in the meso position.46 Irradiation of DMA solutions containing each BODIPY triad, resulted in formation of a corresponding endoperoxide and linear decrease of DMA absorption (Fig. S23a, ESI†), allowing to obtain ΦΔ value from comparison with reference photosensitizer, 2,6-diiodo-8-phenyl-1,3,5,7-tetramethylBODIPY (I2BDP, Fig. S23b, ESI†). As shown in Table 1, the resulting ΦΔ values only slightly vary depending on the solvent. The most efficient 1O2 sensitization was observed for ABA in toluene, tetrahydrofuran and ethanol (ΦΔ = 0.289–0.307). This correlates with rapid charge transfer and triplet state generation observed in the TA experiments (vide infra). Dyad PBP showed ΦΔ values of 0.185–0.2 in these solvents. In acetonitrile, ABA and PBP showed reduced ΦΔ values of 0.14 and 0.09, respectively. Such effect was observed for other BODIPY donor–acceptor systems undergoing SOCT-ISC in acetonitrile.47 As was shown in previous works, the efficiency of triplet state formation strongly depends on the energy gap between the CT state and the ground state. Highly polar solvents strongly stabilize the CT state through dipole–dipole interactions. This leads to a reduced CT-S0 energy gap which promotes recombination into the ground state and thus reduces ΦT. Acetonitrile is significantly more polar (εr = 37.5) compared to THF (εr = 7.6) and EtOH (εr = 24.5) and it can be expected that CT state of dyads in this solvent is strongly stabilized.
Reference BODIPY-anthracene and pyrene dyads bearing donor subunits in the meso-position of the BODIPY scaffold showed much more pronounced dependence of ΦΔ values on solvent polarity (Table S1, ESI†). For 8-anthryl-1,3,5,7-tetramethylBODIPY, ΦΔ values reduced by a factor of 10 upon changing the solvent from polar ethanol to non-polar toluene. Lower ΦΔ values for ABA and PBP compared to reference dyads can be rationalized taking into account different molecular geometries of these systems. meso-Substituted dyads are rigidly constrained in the orthogonal geometry due to the steric effect of methyl groups in positions 3 and 5.40 This provides high yields of triplet state formation via SOCT-ISC as orthogonal mutual arrangement compensates the change of spin magnetic momentum by a large change in the orbital magnetic momentum during the recombination.48 For anthracene/pyrene groups introduced in positions 2 or 6 the rotational barrier is lower allowing a deviation from orthogonality as is evidenced by computed S0, S1 and T1 excited state geometries of ABA and PBP (Fig. S18–S21, ESI†) and thus leading to reduced triplet state yields.
Femto- and nanosecond pump-probe transient absorption spectroscopy of the triads
To reveal the photoexcited state dynamics in the new triads, transient absorption (TA) spectroscopy was employed. Fig. 4 shows the picosecond–nanosecond (ps–ns) and nanosecond–microsecond (ns–μs) TA spectra and kinetics (inset) of selected spectral regions of ABA (a and c) and PBP (b and d) dissolved in toluene after photoexcitation at 505 nm (ps–ns TA) and 532 nm (ns–μs TA), respectively. The negative ΔT/T signals represent photoinduced absorption (PA), while positive ΔT/T signals represent photo-bleaching (PB). In Fig. 4a, the band centered at 520 nm (PB of BODIPY) showed no temporal evolution. On the other hand, the PA regions centered at ∼420 nm and ∼800 nm showed a spectral evolution with time, indicating the formation of charge-transfer (CT) states. The reason for the absence of a temporal evolution of the PB band is that both photoexcited singlet and CT states exhibit the same photobleaching, and the population of CT states compensates the decay of singlet states. The kinetics of the band at 550–570 nm exhibited a clear decay until 10 ps, and thereafter no further changes were observed until 500 ps (inset Fig. 4a). In contrast, the kinetics of the 770–870 nm band exhibited a clear rise due to the generation of CT states. An exponential fit to the kinetics yielded an inverse rate constant of 3.4 ps (singlet exciton decay) and 4.5 ps (CT state generation) for the bands at 550–570 nm and 770–870 nm, respectively. The ps–ns TA spectra of ABA in acetonitrile showed different spectral shape and evolution (Fig. S24, ESI†) compared those of ABA in toluene. Unlike in the case of toluene, the PB band of ABA in acetonitrile showed a clear decay after 50 ps. This is supported by the fast decay of the CT states (band centered at 630 nm). Table S2 (ESI†) summarizes the inverse generation rates of CT states in both toluene and acetonitrile solution.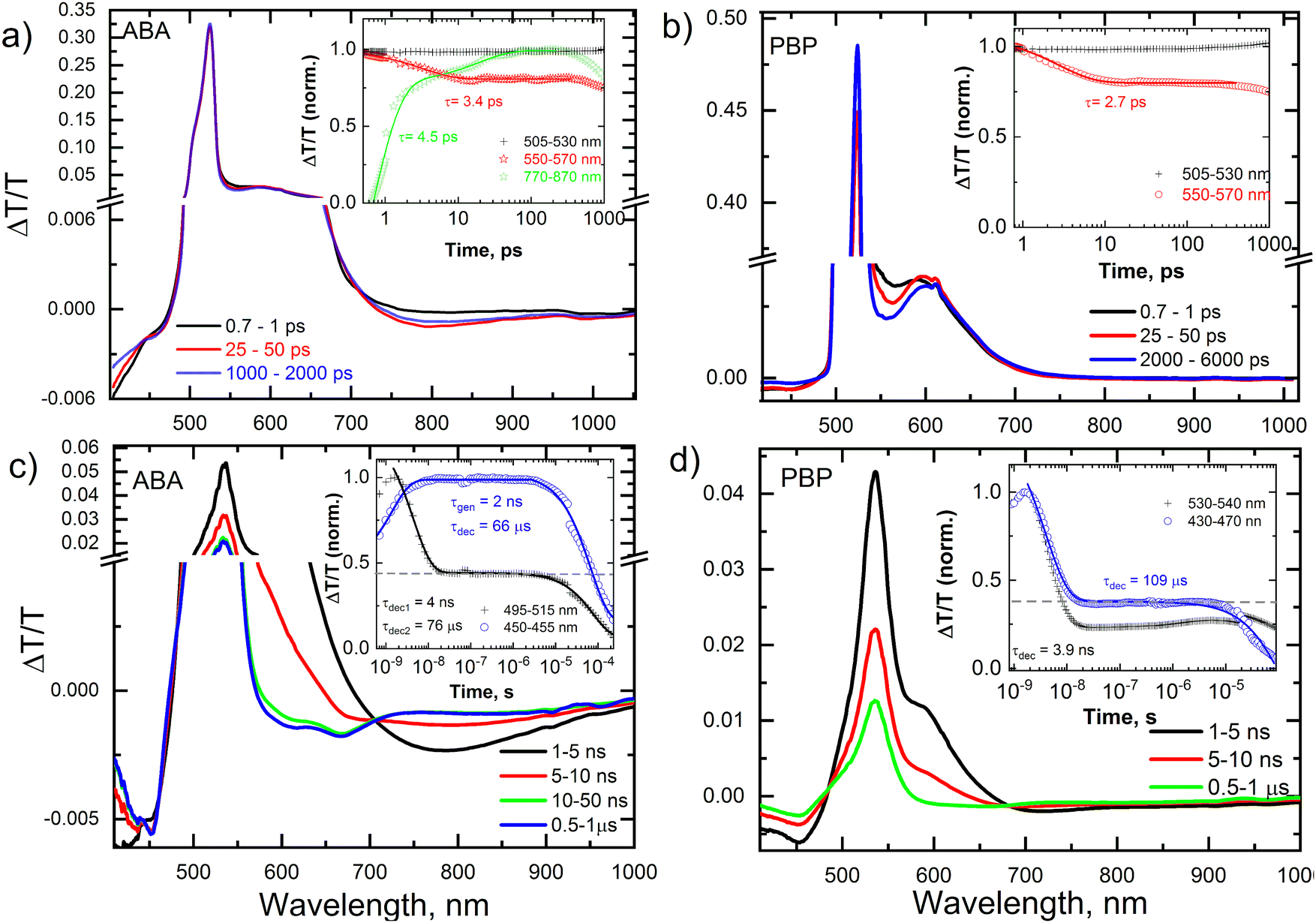 | ||
| Fig. 4 ps–ns TA spectra with kinetics (inset) of selected spectral region of ABA and PBP in toluene (a and b) after excitation with 505 nm laser pulses under inert atmosphere. ns–μs TA spectra with kinetics (inset) of selected spectral region of ABA and PBP in toluene (c and d) after excitation with 532 nm laser pulses under inert atmosphere. | ||
Fig. 4b shows the ps–ns TA spectra and associated kinetics of PBP in toluene. As in the case of ABA in toluene, the PB of PBP in toluene showed no spectral evolution, while the 550–570 nm band showed a fast decay, indicating the formation of CT states. An exponential fit to the kinetics shown in the inset of Fig. 4b yielded an inverse rate constant of 2.7 ps for the band at 550–570 nm. This indicates that the CT state formation is slightly faster in PBP in toluene compared to ABA in toluene. The spectral shape and evolution are different for PBP in acetonitrile compared to PBP in toluene solution (Fig. S24, ESI†). The inverse rate constants are provided in Table S2 (ESI†). In acetonitrile, both ABA and PBP showed early (after 50 ps), and faster decay of the CT states compared to their toluene counterparts. To further explore the CT state decay and generation and decay of triplet states, we performed also nanosecond–microsecond (ns–μs) TA spectroscopy.
Fig. 4c and d show the nanosecond–microsecond (ns–μs) TA spectra and kinetics (inset) of ABA and PBP, respectively, in toluene. Unlike the ps–ns TA spectra, the ns–μs TA spectra show the emergence of a very long-lived (several microseconds) excited state. Both ABA and PBP showed a clear difference in the spectral and kinetics evolution of the excited state. In ABA, the CT state peaked around 800 nm and decayed within ∼10 ns, causing the emergence of two new long-lived bands at 600 nm and 670 nm. Based on the previous reports,40,46 we assigned these bands to the BODIPY triplet state and confirmed this assignment by TA measurements in the presence of air (Fig. S25, ESI†). The ΔT/T values (amplitude of the signal) of the TA spectra measured in inert atmosphere (N2) and in air are significantly different, indicating that the triplets were quenched by oxygen present in air. An exponential fit to the kinetics shown in the inset of Fig. 4c yielded an inverse rate constant of 2 ns for triplet generation and 66 μs for the triplet decay. We obtained an estimated triplet yield of 44%, assuming both triplet and CT states exhibit the same absorption cross-section (see the horizontal dashed line in the inset) as was reported in a previous work.49 Furthermore, the triplet yield was independently estimated by using MCR-ALS (multivariate curve resolution – alternate least square) and by using the relative actinometry method50 with Rose Bengal in methanol as a reference and λpump = 532 nm, all yielding comparable values for PBP in toluene, shown in Table S2 (ESI†). Fig. 4d shows the ns–μs TA spectra and kinetics (inset) of PBP in toluene. Unlike in ABA, we did not observe a distinct spectral signature of triplets (since the CT state and triplet state photoinduced spectra overlap), yet the kinetics showed the presence of long-lived states, which we assigned to triplets. An exponential fit to the kinetics shown in the inset of Fig. 4d yielded a lifetime of 3.9 ns for the CT states (leading to triplet generation) and 109 μs for the triplet decay, and we obtained an estimated triplet yield of 36%. A similar analysis was done for ABA and PBP in acetonitrile and we obtained an estimated triplet yield of 23% for ABA and 13% for PBP in acetonitrile (Fig. S28, ESI†), respectively. Details are summarized in Table S2 (ESI†).
Photopolymerizable glass preparation and holographic recording
Due to its higher triplet state yield, ABA was chosen as a photosensitizer component for free-radical polymerization in a holographic recording material based on a photopolymerizable glass (PG), a hybrid organic–inorganic sol–gel material. Previously reported PG51 was sensitized by incorporation of Irgacure 784 as a photoinitiator with maximum absorption in the range of 400–480 nm. This makes the material less sensitive at 532 nm, the second harmonic of single frequency Nd:YAG lasers typically used for holographic recording. For full utilisation of the potential of these readily available light sources a sensitizer with optimum absorption near 532 nm wavelength is needed. In this work, the PG composition was modified by introducing a two-component photoinitiating system containing ABA and N-phenylglycine (NPG). NPG was demonstrated as very effective co-initiator for visible-light photoinduced polymerization of methacrylate monomers using various sensitizers, e.g. quinones,52 aromatic hydrocarbons,53 xanthene dyes54 and was applied in materials for holographic recording.55,56 The described mechanism of the sensitized photodecomposition of NPG involves an intermolecular electron transfer from NPG molecule to triplet excited state of the photosensitizer (3PS*) leading to corresponding radical anion (PS˙−) and NPG˙+ radical cation.57 The latter is readily deprotonated and decomposes to give anilinomethyl radical, PhNHCH2˙, that initiates the polymerization through an addition reaction onto the monomer double bond (Scheme S1, ESI†).58PG was synthesized by employing a four-step sol–gel process using two hybrid precursors: 3-trimethoxysilylpropyl methacrylate and a zirconium complex obtained by chelation of zirconium(IV) propoxide with methacrylic acid (see ESI† for synthetic details). After obtaining a homogeneous sol, ABA (0.025% mol with respect to the total amount of monomers) and NPG (1.4% mol) were added and the resulting composition was cross-linked by (3-aminopropyl)triethoxysilane to yield a fast-drying and water-resistant hybrid photopolymerizable glass (PG-ABA).
In order to evaluate the performance of ABA, a reference PG containing 2,6-diiodo-BODIPY (PG-I2BDP) and NPG was prepared using the method described above. I2BDP is a common triplet photosensitizer with triplet state yield near unity and excellent photostability, which account for its broad use in photocatalysis.59 Notably, it was shown to enable photopolymerization of acrylate monomers with high rates under low light intensities.60
Spectra of both PG-ABA and PG-I2BDP showed a wide absorption band with the maximum absorbance in the range of 500–550 nm (Fig. 5a) which confirmed the suitability of these materials for holographic recording with 532 nm laser. For the recording, dry layers with 220–260 μm thickness were obtained by a drop-casting method followed by temperature curing as described in the ESI.† Dry layers of PG-ABA and PG-I2BDP showed low light scattering as seen from a clear image of the pattern on the optical table observed through the layer in Fig. 5b. This allowed reducing losses due to scattering of light during holographic recording and achieving high diffraction efficiency of the grating.
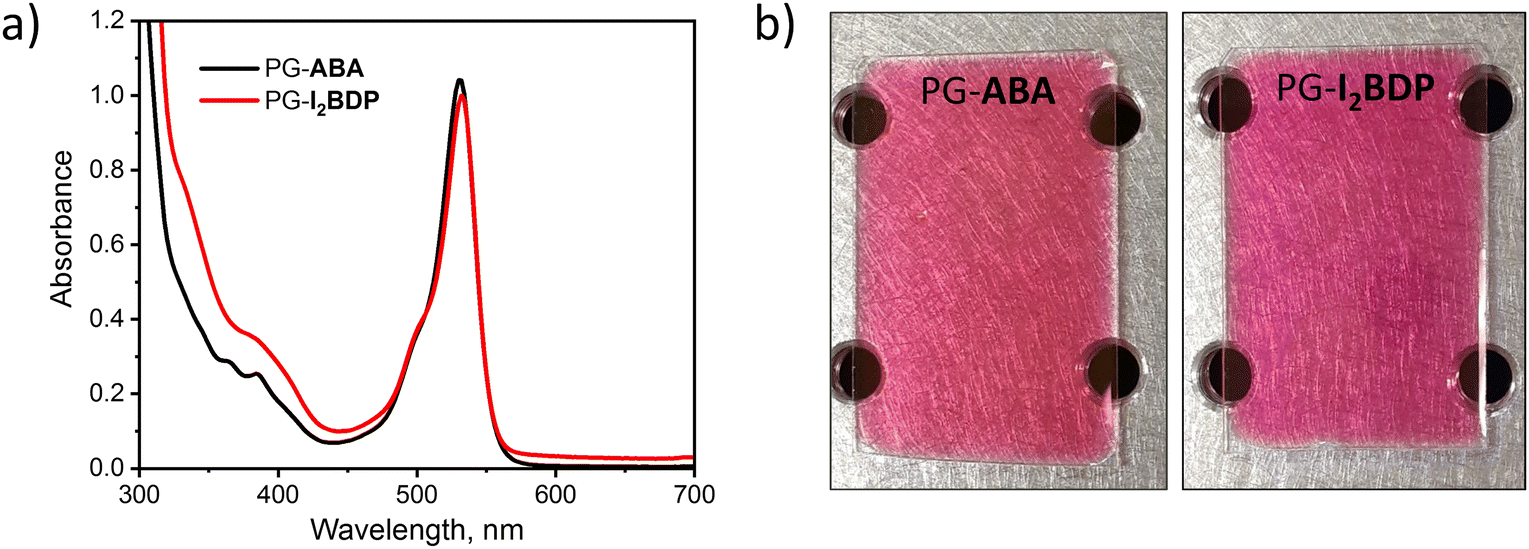 | ||
| Fig. 5 (a) UV-Vis absorption spectra of PG-ABA and PG-I2BDP sol dissolved in isopropanol. (b) Photographs of PG-ABA and PG-I2BDP layers with 260 μm thickness. | ||
Holographic recording of volume phase transmission gratings was carried out using the set-up presented in Fig. S29 (ESI†). As was proposed in our previous work,51 during holographic patterning of PG layers, photopolymerization is triggered in illuminated areas. Under these conditions, ABA is expected to undergo intermolecular charge transfer and form triplet excited states of BODIPY via SOCT-ISC, which further react with NPG to produce free radicals. Thus formed radicals react with methacrylate groups initiating polymerization and leading to the formation of rigidly structured nanoparticle-based domains in illuminated areas. As result of holographic recording, a periodic distribution of areas with different density is achieved, leading to the periodic distribution of the refractive index and thus the holographic grating formation (Fig. 6).
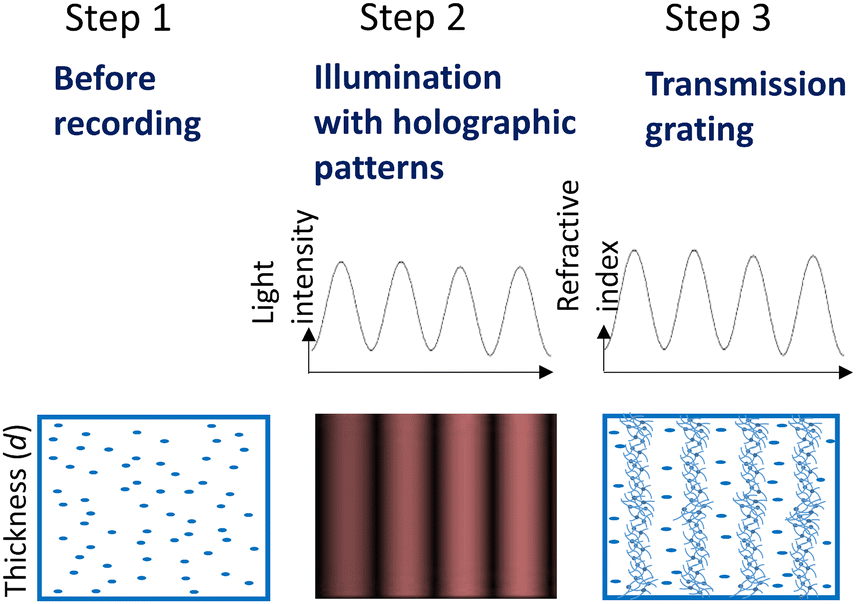 | ||
| Fig. 6 Grating formation in PG under holographic exposure. Step 1 shows the uniform distribution of monomers in PG layer before recording. Step 2 illustrates the sinusoidal profile of the light intensity produced by two coherent laser beams during recording. Step 3 displays spatially-varying refractive index within the PG layer due to density changes induced by polymerization of photoreactive components. | ||
To characterize the process of grating formation, diffraction efficiency growth curves were monitored in real time using a 633 nm laser during holographic recording (see Fig. S29 (ESI†) for details on the experimental set up). Obtained diffraction efficiency growth curves showed the successful formation of highly efficient gratings in PG-ABA and PG-I2BDP (Fig. 7a). The resulting photosensitivity was estimated from the linear part of the growth curves using the method described in the ESI.† As seen from Fig. 7a, the linear part of the curves for PG-ABA and PG-I2BDP has identical slopes, which denotes equal exposure sensitivity of the two compositions, found to be 6.1 × 10−5 mJ−1 cm2 (Fig. S30, ESI†). A shelf-life study confirmed that the recorded structures are stable and unchanged diffraction efficiency is observed over a period of 6 months (Fig. S31, ESI†).
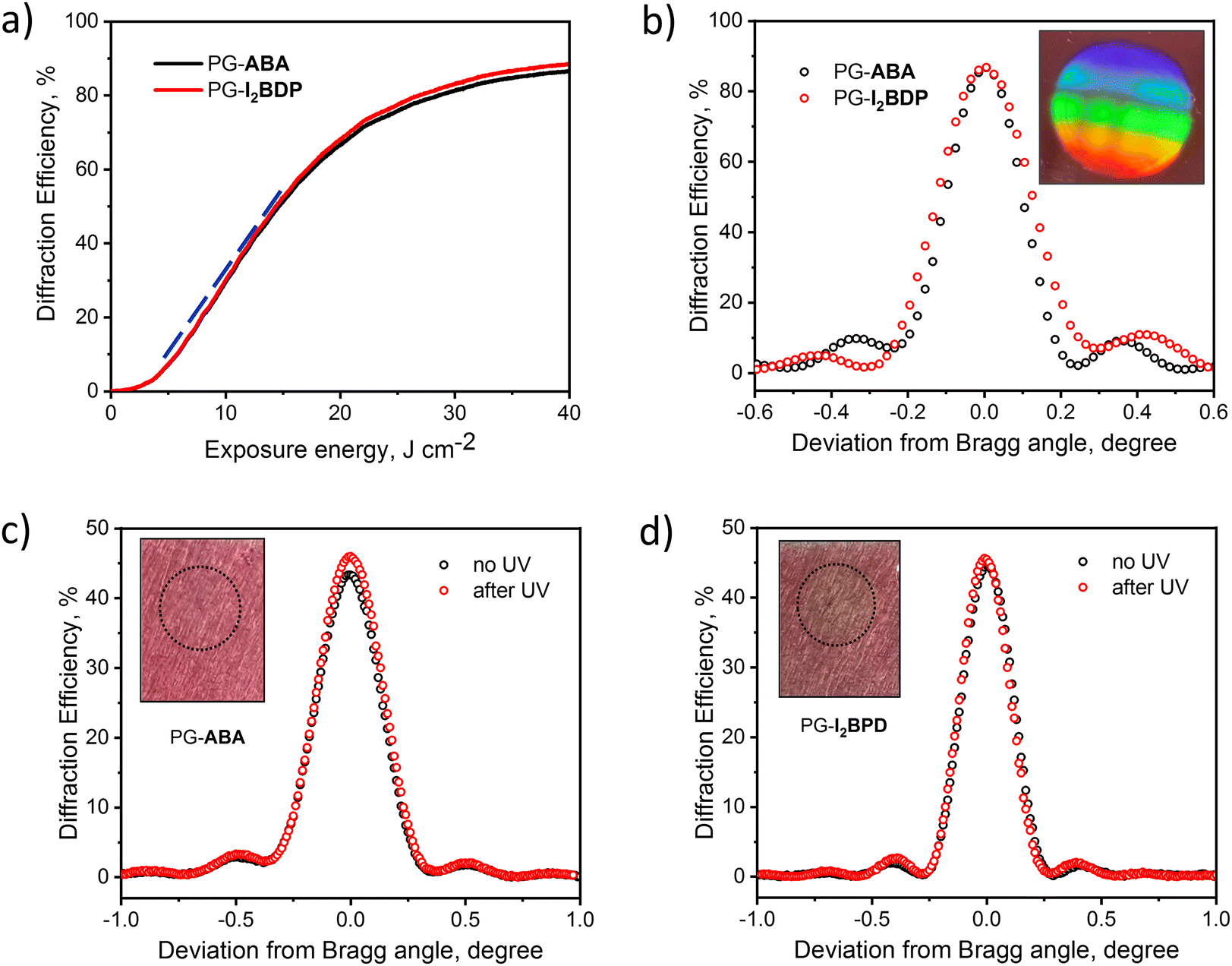 | ||
| Fig. 7 (a) Holographic recording of volume phase transmission gratings in PG-ABA and PG-I2BDP layers. Real-time diffraction efficiency growth curves showing the formation of transmission gratings with the spatial frequency of about 800 lines mm−1. The blue dashed line prepresents the curve's slope. (b) Angular selectivity curves of the transmission gratings recorded. Inset: Image of the light pattern created by a typical transmission grating recorded in PG-ABA and PG-I2BDP, under illumination with a white light source. (c and d) Experimental data on UV-curing. Angular selectivity curves of the transmission gratings recorded in PG-ABA and PG-I2BDP show unchanged diffraction efficiency before and after UV-curing using exposure energy of 22 J cm−2. Inset: The photograph of the layer after UV-curing indicates no layer cracking and confirms its stability to UV-exposure at current experimental conditions. | ||
A partial photobleaching of the used dyes was observed during the holographic recording as is seen from the UV-vis spectral changes of the studied PGs (Fig. S32, ESI†). This is in line with previous report on photobleaching of xanthene dyes initiated by NPG.57 It should be noted that bleaching of dyes in PG during holographic recording is beneficial for the development of holographic optical elements. Photobleaching of dyes under holographic exposure allows fabricating holographic optical elements with a certain degree of transparency that can be controlled by the recording light intensity and exposure time. Such holographic optical elements with controlled transparency are required for a number of applications including quickly advancing technologies such as 3D displays.61
Diffraction efficiency up to 87% was achieved using an exposure energy of 35 J cm−2 (Fig. 7b). This value is higher than for holographic photopolymers in which the diffusion of mobile species such as monomers, dyes and short polymer chains plays a dominant role in the grating formation.62 However, the total exposure energy required for PG-ABA and PG-I2BDP is in the same range as for previously reported photopolymer-filled glass in which the diffusion of mobile components through the nano-sized pores of the glass is hindered.63 Thus, relatively high value of the total exposure energy possibly indicates a slow diffusion of any mobile components in PG.
Refractive index modulation is another crucial parameter of holographic recording materials characterising their performance. In this work, the recorded transmission gratings are considered as volume gratings since the Klein-Cook Q-parameter calculated using eqn (S4) (ESI†) is 317 for 260 μm thick layers. Thus, the Kogelnik's coupled-wave theory64 can be applied. The refractive index modulation estimated by eqn (S5) (ESI†) was found to be dependent on the layer thickness for both materials, PG-ABA and PG-I2BDP. The values varied from 0.9 × 10−3 to 1.1 × 10−3 for layers with the thickness ranging from 260 to 220 μm, respectively (Fig. S33, ESI†). This value is close to one previously reported for gratings with similar thickness and spatial frequency recorded in a photopolymerizable glass sensitized with Irgacure 784.51 Thus, successful holographic recording of transmission gratings confirms comparable efficiency of both ABA-NPG and I2BDP-NPG photoinitiating systems for polymerisation of methacrylates in PG.
Among the various aspects of the holographic recording material, stability under UV-exposure is one of the key parameters required for the sustained performance of holographic optical elements and their successful implementations. In order to investigate the effect of UV-exposure on the grating stability, diffraction efficiency before and after UV-curing was monitored for PG-ABA and PG-I2BDP samples (Fig. 7c and d). For this experiment, transmission gratings with the diffraction efficiency of about 45% were recorded by using the total exposure energy of 22 J cm−2. After recording, corresponding samples were exposed to UV-A light by means of Dymax UV-curing System (ECE Series) using the same total exposure energy as used for recording (22 J cm−2). As seen from Fig. 7c and d, a slightly increased diffraction efficiency was observed for PG-ABA and PG-I2BDP. Moreover, no layer deterioration (cracking or breaking) was observed after UV-curing.
Conclusions
In summary, we prepared two novel donor–acceptor–donor systems comprised of BODIPY as an electron acceptor and anthracenes (ABA) or pyrenes (PBP) as donor subunits. These triads undergo efficient intramolecular charge transfer and form corresponding CT states in both polar and non-polar solvents. Femtosecond transient absorption spectra indicate that charge separation takes place on the timescale of a few picoseconds for both triads. Furthermore, recombination of CT states leads to formation of long-living BODIPY triplet excited states via SOCT-ISC mechanism without heavy atoms. Nanosecond transient absorption spectra show the formation of triplet states with 44% and 36% for ABA and PBP, respectively, in toluene. Notably, unlike previously investigated donor–acceptor systems, new triads showed only moderate solvent dependence of triplet state formation as is evidenced by singlet oxygen quantum yield values in different solvents.The two-component photoinitiating system containing ABA as a photosensitizer and NPG as a co-initiator was proposed for free-radical polymerization under 532 nm excitation. Feasibility of ABA-NPG photoinitiating system for the free-radical polymerization of methacrylates was demonstrated by holographic recording in a photopolymerizable hybrid sol–gel. Volume phase transmission gratings with the diffraction efficiency up to 87% were obtained in PG-ABA layers with 220–260 μm thickness confirming the efficiency of the ABA-NPG photoinitiating system. UV-resistance and sustained performance of holographic optical elements recorded on PG-ABA were proved by stable diffraction efficiency of transmission gratings under UV-curing. These results confirm stability of the diffraction grating under UV-exposure and provide further perspectives for utilisation of PG sensitized with heavy-atom-free BODIPYs as a holographic recording material with improved UV-resistance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. F. acknowledges TU Dublin Research Scholarship programme for the financial support of this work, Dr Alla Lemeune (ENS de Lyon) for HRMS measurements and Fred Odunjo (CREST, TU Dublin) for EDX measurements. A. A. B. acknowledges the Strategic Academic Leadership Programme “Priority 2030” of the Kazan Federal University. This publication is based on work supported by the KAUST Office of Sponsored Research (OSR) under award no. OSR-CRG2018-3746 and OSR-CRG2019-4025. C. S. P. D. C. would like to acknowledge Luis Gutiérrez-Arzaluz for help getting started with the modified microscope used for TRPL measurements.References
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- X. Zhao, Y. Hou, L. Liu and J. Zhao, Energy Fuels, 2021, 35, 18942 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Tompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- (a) H. L. Wang, C. H. Du, Y. Pu, R. Adur, P. C. Hammel and F. Y. Yang, Phys. Rev. Lett., 2014, 112, 197201 CrossRef CAS PubMed; (b) C. Du, H. Wang, F. Y. Yang and P. C. Hammel, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 140407 CrossRef.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944 RSC.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207 CrossRef CAS PubMed.
- (a) M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10 RSC; (b) E. Bassan, A. Gualandi, P. Giorgio Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607 RSC.
- (a) J. H. Golden, L. Estergreen, T. Porter, A. C. Tadle, D. M. R. Sylvinson, J. W. Facendola, C. P. Kubiak, S. E. Bradforth and M. E. Thompson, ACS Appl. Energy Mater., 2018, 1, 1083 CrossRef CAS; (b) W. Huang, X. Zhang, B. Chen, H. Miao, C. O. Trindle, Y. Wang, Y. Luo and G. Zhang, Chem. Commun., 2019, 55, 67 RSC; (c) J.-X. Wang, L.-Y. Niu, P.-Z. Chen, Y.-Z. Chen, Q.-Z. Yang and R. Boulatov, Chem. Commun., 2019, 55, 7017 RSC.
- (a) C. Trinh, K. O. Kirlikovali, S. Das, M. E. Ener, H. B. Gray, P. I. Djurovich, S. Bradforth and M. E. Thompson, J. Phys. Chem. C, 2014, 118, 21834 CrossRef CAS PubMed; (b) S. Das, W. G. Thornbury, A. N. Bartynski, M. E. Thompson and S. E. Bradforth, J. Phys. Chem. Lett., 2018, 9, 3264 CrossRef CAS PubMed.
- Y. Tsuga, M. Katou, S. Kuwabara, T. Kanamori, S. Ogura, S. Okazaki, H. Ohtani and H. Yuasa, Chem. – Asian J., 2019, 14, 2067 CrossRef CAS PubMed.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171 CrossRef CAS.
- (a) N. Rehmat, A. Toffoletti, Z. Mahmood, X. Zhang, J. Zhao and A. Barbon, J. Mater. Chem. C, 2020, 8, 4701 RSC; (b) Y. Zhao, A. A. Sukhanov, R. Duan, A. Elmali, Y. Hou, J. Zhao, G. G. Gurzadyan, A. Karatay, V. K. Voronkova and C. Li, J. Phys. Chem. C, 2019, 123, 18270 CrossRef CAS; (c) Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329 RSC.
- F. Faroldi, B. Bardi, I. Tosi, S. Doria, J. Isopi, L. Baldini, M. Di Donato, M. Marcaccio, F. Sansone and F. Terenziani, J. Mater. Chem. C, 2021, 9, 10899 RSC.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Angew. Chem., Int. Ed., 2020, 59, 11591 CrossRef CAS PubMed.
- (a) B. G. McCarthy, R. M. Pearson, C.-H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088 CrossRef CAS PubMed; (b) C. H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348 CrossRef CAS PubMed; (c) X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411 CrossRef CAS PubMed.
- F.-K. Bruder, R. Hagen, T. Rölle, M.-S. Weiser and T. Fäcke, Angew. Chem., Int. Ed., 2011, 50, 4552 CrossRef CAS PubMed.
- J. Guo, M. R. Gleeson and J. T. Sheridan, Phys. Res. Int., 2012, 16, ID803439 Search PubMed.
- C. M. Bigler, P.-A. Blanche and K. Sarma, Appl. Opt., 2018, 57, 2007 CrossRef PubMed.
- J. Xiao, J. Liu, X. Duan, X. Shi and Z. Zhang, Opt. Express, 2020, 28, 31316 CrossRef PubMed.
- D. Chemisana, J. Atencia, J. Marín-Sáez and M.-V. Collados, Opt. Express, 2016, 24, A720 CrossRef PubMed.
- H. Akbari, I. Naydenova, H. Ahmed, S. McCormack and S. Martin, Sol. Energy, 2017, 155, 103 CrossRef CAS.
- K. Murphy, V. Toal, S. Martin and I. Naydenova, Opt. Express, 2018, 26, 8916 CrossRef CAS PubMed.
- Y. Tomita, T. Aoi, S. Hasegawa, F. Xia, Y. Wang and J. Oshima, Opt. Express, 2020, 28, 28366 CrossRef CAS PubMed.
- M. Sevilla, J. Marín-Sáez, D. Chemisana, M.-V. Collados and J. Atencia, Photonics, 2021, 8, 465 CrossRef CAS.
- A. V. Velasco, M. L. Calvo and P. Cheben, J. Appl. Phys., 2013, 113, 033101 CrossRef.
- N. Zhang, J. Liu, J. Han, X. Li, F. Yang, X. Wang, B. Hu and Y. Wang, Appl. Opt., 2015, 54, 3645 CrossRef.
- J. Han, J. Liu, X. Yao and Y. Wang, Opt. Express, 2015, 23, 3534 CrossRef CAS PubMed.
- R. Mallavia, A. Fimia, C. García and R. J. Sastre, J. Mod. Opt., 2001, 48, 941 CrossRef CAS.
- Q. Gong, S. Wang, M. Huang and F. Gan, Mater. Lett., 2005, 59, 2969 CrossRef CAS.
- Y. Qi, H. Li, J. Guo, M. R. Gleeson and J. Sheridan, Opt. Commun., 2014, 320, 114 CrossRef CAS.
- N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568 RSC.
- A. Harriman, L. J. Mallon, G. Ulrich and R. Ziessel, Chem. Phys. Chem., 2007, 8, 1207 CrossRef CAS PubMed.
- Z. Mahmood, M. Taddei, N. Rehmat, L. Bussotti, S. Doria, Q. Guan, S. Ji, J. Zhao, M. Di Donato, Y. Huo and Y. H. Xing, J. Phys. Chem. C, 2020, 124, 5944 CrossRef CAS.
- P. H. Marek-Urban, M. Urban, M. Wiklińska, K. Paplińska, K. Woźniak, A. Blacha-Grzechnik and K. Durka, J. Org. Chem., 2021, 86, 12714 CrossRef CAS PubMed.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC.
- (a) E. Vauthey, Chem. Phys. Chem., 2012, 13, 2001 CrossRef CAS PubMed; (b) B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643 CrossRef CAS PubMed; (c) M. Soderberg, B. Dereka, A. Marrocchi, B. Carlotti and E. Vauthey, J. Phys. Chem. Lett., 2019, 10, 2944 CrossRef CAS PubMed; (d) X. Niu, Z. Kuang, M. Planells, Y. Guo, N. Robertson and A. Xia, Phys. Chem. Chem. Phys., 2020, 22, 15743 RSC.
- (a) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016 RSC; (b) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606 CrossRef CAS; (c) N. Kiseleva, D. Busko, B. S. Richards, M. A. Filatov and A. Turshatov, J. Phys. Chem. Lett., 2020, 11, 6560 CrossRef CAS PubMed.
- (a) J. Li, Y. Qian, L. Xie, Y. Yi, W. Li and W. Huang, J. Phys. Chem. C, 2015, 119, 2133 CrossRef CAS; (b) Y. Qian, M. Cai, X. Zhou, Z. Gao, X. Wang, Y. Zhao, X. Yan, W. Wei, L. Xie and W. Huang, J. Phys. Chem. C, 2012, 116, 12187 CrossRef CAS.
- E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124 CrossRef CAS.
- X.-F. Zhang and X. Li, J. Lumin., 2011, 131, 2263 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282 CrossRef CAS PubMed.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chemistry, 2019, 5, 138 CrossRef CAS.
- (a) Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194 CrossRef CAS PubMed; (b) Z. E. X. Dance, Q. Mi, D. W. McCamant, M. J. Ahrens, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2006, 110, 25163 CrossRef CAS PubMed; (c) D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136 CrossRef CAS PubMed.
- N. Kiseleva, M. A. Filatov, M. Oldenburg, D. Busko, M. Jakoby, I. A. Howard, B. S. Richards, M. O. Senge, S. M. Borisov and A. Turshatov, Chem. Commun., 2018, 54, 1607 RSC.
- J. S. de Melo, J. Pina, F. B. Dias and A. L. Maçanita, in Applied Photochemistry, ed. R. C. Evans, P. Douglas and H. D. Burrows, Springer, 2013, ch. 15, p. 533 Search PubMed.
- T. Mikulchyk, M. Oubaha, A. Kaworek, B. Duffy, M. Lunzer, A. Ovsianikov, S. E-Gul, I. Naydenova and D. Cody, Adv. Opt. Mater., 2022, 10, 2102089 CrossRef CAS.
- J. Jakubiak, X. Allonas, J. P. Fouassier, A. Sionkowska, E. Andrzejewska, L. A. Linden and J. F. Rabek, Polymer, 2003, 44, 5219 CrossRef CAS.
- S. Ikeda and S. Murata, J. Photochem. Photobiol., A, 2002, 149, 121 CrossRef CAS.
- J. Kabatc, Z. Kucybala, M. Pietrzak, F. Scigalski and J. Paczkowski, Polymer, 1999, 40, 735 CrossRef CAS.
- T. Mikulchyk, S. Martin and I. Naydenova, Appl. Opt., 2017, 56, 6348 CrossRef CAS PubMed.
- Q. Gong, S. Wang, M. Huang and F. Gan, Mater. Lett., 2005, 59, 2969 CrossRef CAS.
- G. Qiaoxia, H. Mingju and G. Fuxi, Dyes Pigm., 2006, 69, 204 CrossRef CAS.
- S. Ikeda, S. Murata, K. Ishii and H. Hamaguchi, Chem. Lett., 1999, 1009 CrossRef CAS.
- S. Lacombe and T. Pigot, Catal. Sci. Technol., 2016, 6, 1571 RSC.
- A. Stafford, D. Ahn, E. K. Raulerson, K.-Y. Chung, K. Sun, D. M. Cadena, E. M. Forrister, S. R. Yost, S. T. Roberts and Z. A. Page, J. Am. Chem. Soc., 2020, 142, 14733 CrossRef CAS PubMed.
- K. Hong, J. Yeom, C. Jang, O. Hong and B. Lee, Opt. Lett., 2014, 39, 127 CrossRef PubMed.
- T. Babeva, I. Naydenova, S. Martin and V. Toal, Opt. Express, 2008, 16, 8487 CrossRef PubMed.
- M. G. Schnoes, L. Dhar, M. L. Schilling, S. S. Patel and P. Wiltzius, Opt. Lett., 1999, 24, 658 CrossRef CAS PubMed.
- H. Kogelnik, Bell Syst. Tech. J., 1969, 48, 2909 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and analytical data for new compounds; optical spectra; computation details; sol–gel preparation and holographic recording procedure. See DOI: https://doi.org/10.1039/d2tc02263j |
| This journal is © The Royal Society of Chemistry 2022 |