
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile surface engineering of bio-waste derived amorphous carbon with SnO2 nanowires to enhance the efficacy of Li/Na storage†
S. Praveen
Kumar
,
Balla Rekha
Madhuri
,
Katchala
Nanaji
 ,
Srinivasan
Anandan
,
Tata Narasinga
Rao
and
Ramkrishna
Sahoo
*
,
Srinivasan
Anandan
,
Tata Narasinga
Rao
and
Ramkrishna
Sahoo
*
Centre for Nanomaterials, International Advanced Research Centre for Powder Metallurgy and New Materials (ARCI), Hyderabad-500005, Telangana, India. E-mail: ramkrishna.s@project.arci.res.in
First published on 10th March 2022
Abstract
Amorphous carbon is expected to be an excellent material for rechargeable battery anodes if the specific capacity can be enhanced by fabricating a composite with an appropriate material. To the contrary, SnO2 exhibits a very high specific capacity as a Li/Na-ion battery anode due to its conversion-alloying mechanism, but low cycling stability confines its practical applications. Hence, we demonstrate the strategy of synthesizing a SnO2 composite with non-activated carbon (NAC) derived from jute stick using facile wet chemical dispersion followed by annealing, which is a cost effective and scalable approach. The facile synthesis strategy proves to be beneficial for the uniform decoration of SnO2 nanowires on the top of the NAC, which helps to compensate the volume expansion during the electrochemical reaction. The C/SnO2 composite exhibits improved electrochemical performance compared to the NAC with a specific discharge/charge capacity of 1657/868 mA h g−1 at 25 mA g−1 specific current with high rate and stability as a Li-ion battery anode. Upon assembling the composite with C/LFP as a cathode, the full cell delivers the maximum energy density of 384 W h kg−1 with an average voltage of 2.3 V. It also demonstrates Na-ion storage behavior with a specific charge capacity of 217 mA h g−1 at 25 mA g−1 specific current. Hence, the facile, scalable synthesis route and excellent battery performance of the aforementioned composite may endorse this pathway for the development of low-cost rechargeable battery anodes.
Introduction
For the last few decades, an enormous surge in global energy demand has been observed and hence, advanced energy storage technology, especially the Li/Na-ion battery, has become extremely relevant in this regard. There is a constant pursuit of advanced anode materials for Li/Na-ion batteries, which can be rendered as extremely pivotal leading to the development of advanced energy storage technology.1–4 Though graphite and hard carbon are the commercial grade anode materials for Li and Na-ion batteries, respectively, other carbonaceous materials (such as soft carbon, amorphous carbon, porous carbon etc.) and transition metal oxides demonstrating conversion or conversion-alloying mechanisms are also promising materials as anodes.5–12 Tin oxide (e.g., SnO2) is one of such metal oxides that can readily participate in conversion reactions, followed by alloying with both Li and Na.13–21 Additionally, tin oxides are low-cost materials and they demonstrate a variety of properties including high theoretical capacity (both gravimetric and volumetric basis) and excellent safety. The typical electrochemical reactions of SnO2 with Li and Na-ions are as follows:| SnO2 + 4M+ + 4e− → Sn + 2M2O (M = Li+/Na+). |
| Sn + xM+ + xe− ↔ MxSn (O ≤ x ≤ 4.4 for Li & O ≤ x ≤ 3.75 for Na) |
Though a total ∼8.4 mole of Li participates in the electrochemical reaction, the theoretical capacity of SnO2 as a LIB anode is only 790 mA h g−1.13,14 This is primarily due to the loss of Li in the first irreversible cycle. During the first cycle, the irreversible conversion reaction initiates approximately at 1.5 V (vs. Li+/Li) accompanied by the reversible alloying reaction commencing below 0.5 V. The irreversible step in the first de-lithiation cycle sacrifices a significant amount of Li+ ions (∼ 4 moles) for the subsequent charge discharge cycles which costs a huge capacity loss of ∼712 mA h g−1 with an initial Coulombic efficiency (ICE) of 40–60%.2,13,14,22 The reversible alloying reaction driven by the in situ formed Sn in the first step actually supervises the electrochemical performance where the volume expansion is expected to be 200%.2,13,14,22 From the above discussion it is evident that apart from the several advantages of SnO2 as a Li/Na-ion battery anode, there are still a few critical issues and challenges that are particularly obligatory to be addressed and tackled with efficiency. To date several strategies have been adopted to compensate the strain due to the volume expansion–contraction of the SnO2 during the charge–discharge cycles and coarsening of Sn, which primarily includes the development of different nanostructures and different composites with carbonaceous materials.13,23–49 In the last few years, mostly solvo/hydrothermal processes have been exhaustively used to produce the desired nanostructures and composites; nevertheless, scaling up of the product using these processes is laborious.13,37,40,41,50 Other than this, Liu et al. synthesized 1D SnO2 nanorods adopting molten salt methods where SnCl4, NaCl and Na2CO3 were homogenously mixed and heated at 800 °C.51 The synthesized SnO2 exhibited high crystallinity with excellent phase purity where the authors claimed that high calcination temperature and the aging time acted as favorable factors for the formation of nanorods. Thermal evaporation is also a useful technique to synthesize SnO2 and its composites.51,52 Other than these, SnO2 has also been prepared by some other strategies, such as a sonochemical route, annealing of the precursor salts etc.43,46,50
Here, we have demonstrated the synthesis of a C/SnO2 composite using simple wet chemical dispersion of SnCl2 on jute stick-derived carbon followed by annealing at 350 °C in an air atmosphere. This process is one of the simplest and most scalable approaches to synthesize a C/SnO2 composite. The detailed synthesis of the carbon from jute sticks has already been reported.8 Here, the purpose of SnO2 decoration on the carbon surface is to increase the specific capacity of the non-activated carbon (NAC) without sacrificing the rate and cycling stability and thus low SnO2 content in the composite is desirable compared to the carbon content. The composite demonstrates a high specific charge capacity of 868 mA h g−1 at a specific current of 25 mA g−1 (437 mA h g−1 at 1 C, based on the discharge time). Comparably, the non-activated carbon (NAC) exhibits specific capacity of 320 mA h g−1 at the same current (213 mA h g−1 at 1 C, based on the discharge time) as a Li-ion battery anode. The composite maintains an excellent cycling stability of 70% of its initial value after 500 cycles. Na-ion battery performance has also been studied using the as-prepared composite, which delivers a reversible charge capacity of 217 mA h g−1 at 25 mA g−1 (C/9 rate).
Experimental section
Synthesis of amorphous carbon from jute powder
The synthesis of the porous carbon from the jute stick powder (bio waste) has already been reported.8 Briefly, 200 g of jute stick powder was preheated at 120 °C in air atmospheric conditions to remove the moisture content. This preheated jute powder was then carbonized in a tubular furnace at 900 °C for 2 h in a nitrogen atmosphere. The as-synthesized black powder was then washed thoroughly with DI water and acetone to remove all the impurities. The resulting black carbon powder was further ball-milled for 4 h using tungsten carbide balls. This non-activated carbon (NAC) was used for further studies and as one of the precursors for synthesizing the composite along with the SnO2 precursor. In this work, we have modified the synthesis process by avoiding the activation step with KOH and introduced the ball-milling part.Synthesis of the C/SnO2 composite
The C/SnO2 composite was prepared by using a simple wet chemical dispersion approach followed by annealing route. Initially, 40 mmol of SnCl2·5H2O hygroscopic salt was dissolved in DI water followed by the addition of 10 g of the as-prepared non-activated carbon (NAC) and stirred vigorously for a minimum of 6 h at ambient conditions. The resulting dispersion was then kept over a hot plate to evaporate the water content completely. Black flakes obtained after drying were ground to make an even textured black powder. The ground black powder was annealed at 350 °C for 4 h in an air atmosphere. After annealing, the powder was washed through filtration with DI water several times to remove the inorganic impurities. Finally, the filtered powder was dried at 70 °C and stored for further studies.Pure SnO2 was also synthesized using the same experimental route without using the NAC.
For comparison, another NAC and SnO2 composite were prepared where SnCl2 was mechanically mixed with NAC using a mortar and pastel. After that the mixture was treated exactly the same way as above and it is denoted as P-C/SnO2, where P stands for the physical mixture.
Material characterization
Details of the material characterization equipment have been mentioned in the ESI.†Electrochemical measurement
The as-prepared ball-milled non-activated carbon (NAC) and its composites with SnO2 (C/SnO2 & P-C/SnO2) were tested as a Li/Na-ion battery anode in half-cells using both a Swagelok cell and coin cell. Initially, a slurry was prepared with electro-active material (C/SnO2 and NAC), conductive carbon and polyvinylidene fluoride (PVDF) maintaining a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in N-methyl-2 pyrrolidene (NMP) solvent. The slurry was coated on the current collector (copper foil) using a doctor blade setup and dried for 16 h at 70 °C in a hot air oven. The dried foil was then subjected to a hot calendaring process in order to achieve compact pores and uniform thickness throughout the foil. The half-cells were then fabricated inside an argon-filled glove box using dried coated foil as the working electrode (diameter 12 mm; active material of 2–2.25 mg cm−2 for C/SnO2 composite and 2.75–3 mg cm−2 for NAC) and the Li/Na metal was used as the reference electrode and counter electrode for the Li/Na-ion battery whereas glass fiber was used as the separator. The whole electrochemical investigation was carried out using 1 M LiPF6 (in 1:1 EC:DEC) and 1 M NaClO4 (in PC) as the electrolyte for Li and Na-ion battery analysis, respectively.
:10:10 in N-methyl-2 pyrrolidene (NMP) solvent. The slurry was coated on the current collector (copper foil) using a doctor blade setup and dried for 16 h at 70 °C in a hot air oven. The dried foil was then subjected to a hot calendaring process in order to achieve compact pores and uniform thickness throughout the foil. The half-cells were then fabricated inside an argon-filled glove box using dried coated foil as the working electrode (diameter 12 mm; active material of 2–2.25 mg cm−2 for C/SnO2 composite and 2.75–3 mg cm−2 for NAC) and the Li/Na metal was used as the reference electrode and counter electrode for the Li/Na-ion battery whereas glass fiber was used as the separator. The whole electrochemical investigation was carried out using 1 M LiPF6 (in 1:1 EC:DEC) and 1 M NaClO4 (in PC) as the electrolyte for Li and Na-ion battery analysis, respectively.
Results and discussion
The C/SnO2 composite was synthesized via a wet-chemical dispersion followed by annealing route (Scheme 1). First, SnCl2·5H2O (40 mmol) powder was dispersed in de-ionized water and NAC was added followed by stirring where hydrolyzed Sn2+ ions were adhered uniformly on the surface of the NAC and the color of the dispersion instantly transformed from white to black. During the annealing, the surface adhered Sn2+ triggered nucleation and a uniform growth of SnO2 over the carbon surface could be observed. The facile synthesis route is a cost-effective and high-yield process. Here, chemical dispersion has been proven to be a crucial step for the synthesis of the composite, which has been discussed in the next section.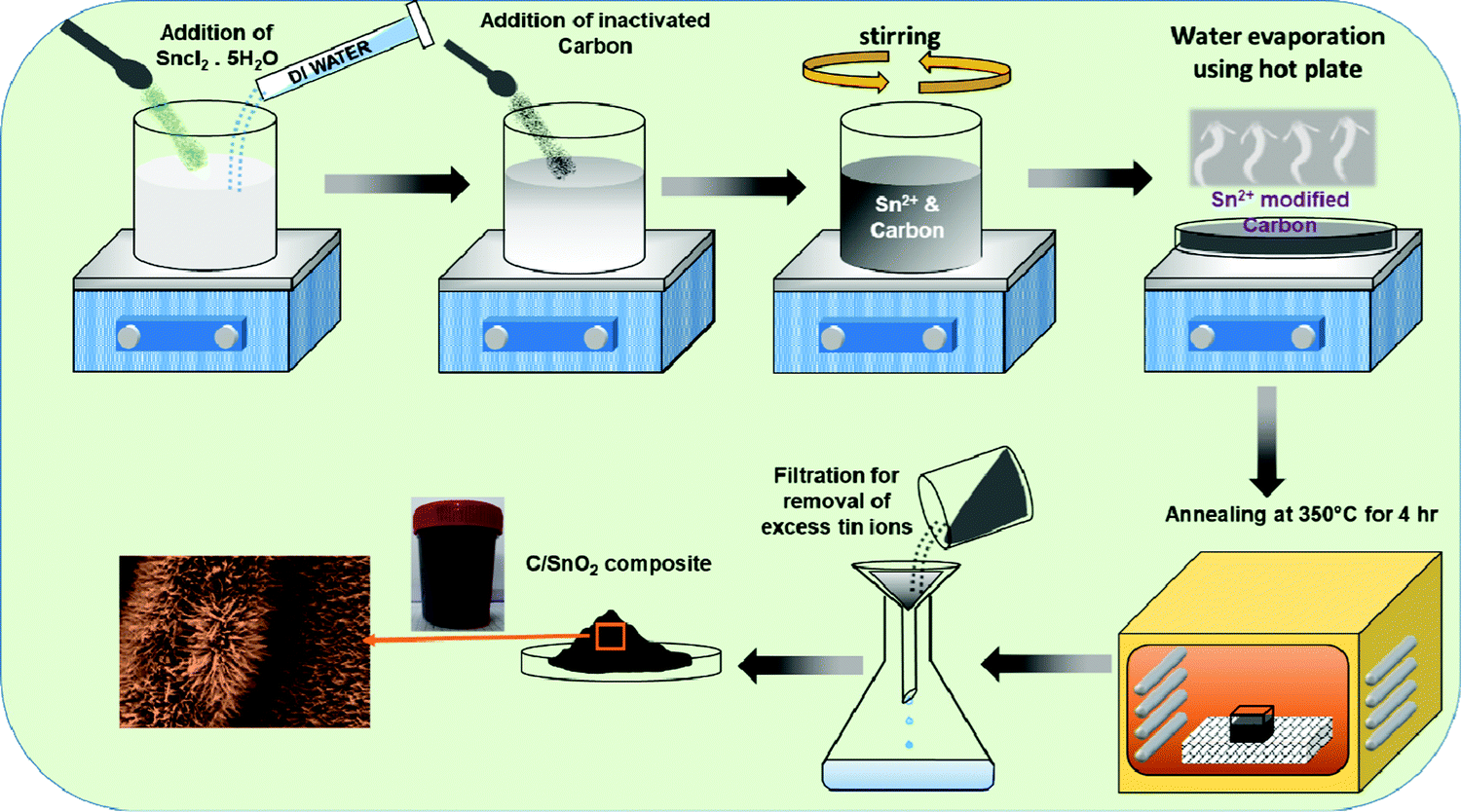 | ||
| Scheme 1 Schematic presentation of the synthesis of the C/SnO2 composite using the chemical dispersion method followed by annealing technique. | ||
Several physicochemical experiments have been carried out to characterize the as-synthesized materials in detail. The image collected from field emission scanning electron microscopy (FESEM) exhibits the flake like nature of the NAC (Fig. 1A). After the wet chemical dispersion and annealing process, the SnO2 nanowires were uniformly distributed all over the flat surface of the carbon (Fig. 1B and C). Here the chemical dispersion approach enacts a significant role towards the uniform growth of SnO2 on the carbon surface. Sn2+ ions were homogeneously adsorbed on the surface of the ball-milled non-activated carbon (NAC) during the stirring, resulting in the consistent growth of SnO2 upon annealing. The TEM image of the as-prepared composite also indicates the consistent distribution of the SnO2 nanowires over the carbon surface (Fig. 1D). HRTEM image analysis further confirms the presence of SnO2 phase, which is polycrystalline in nature (Fig. 1E and F). Elemental area mapping from the TEM image (Fig. 1G–I) demonstrates the spatial distribution of C, Sn and O elements confirming the homogeneous growth of SnO2 over the carbon surface. Such uniform decoration of nanostructures on the surface of carbonaceous materials is mostly prepared by a hydrothermal process, where vigorous reaction conditions control the nucleation of the metal ions.40,41,50 Here the simple wet chemical dispersion of the metal ions followed by annealing process has provided such a uniform growth of SnO2 nanowires on the carbon surface, which is not only a facile synthesis process but also scalable. In this work, approximately 15 g of the composite material was prepared in a single batch and it can be easily scaled up per batch depending on the necessity.
 | ||
| Fig. 1 FESEM images of (A) non-activated carbon (NAC) and (B and C) C/SnO2, (D) TEM image, (E) HRTEM image and (F) SAED pattern of C/SnO2. (F–I) Elemental area mapping of the C/SnO2 composite from the TEM image. | ||
In the context of comparison, a physical mixture of SnCl2 and NAC was annealed at the same temperature (350 °C) resulting in the random distribution of large SnO2 particles over the carbon surface as eventually confirmed from the FESEM image (Fig. S1, ESI†). Few reports are available in the literature, where researchers have used direct annealing of the Sn-salt to prepare SnO2.43,46,50,53–55 To the best of the authors’ knowledge, this work will be the first report where such a facile wet chemical dispersion and annealing of SnCl2 route has been employed to synthesize a controlled and uniform distribution of SnO2 nanowires on the carbon surface without using any surface modifier or stabilizer.
The X-ray diffraction (XRD) technique was implemented to identify the phase purity of the as-prepared materials as well as the crystal structure. The comparative XRD patterns of the pure carbon (NAC) (black line), pure SnO2 (red line) and C/SnO2 (blue line) composite are demonstrated in Fig. 2A. The characteristic broad peak at 23.2 and 43.7 for NAC refers to the amorphous character of the carbon (black line). The XRD patterns for the red line and blue line are indistinguishable where the patterns of the diffracted peaks are in good agreement with the tetragonal SnO2 having the space group P42/mnm (136) (ICDD PDF: 04-016-3047). The XRD pattern of the C/SnO2 does not illustrate any impurity except SnO2, which eventually suggests that a pure crystalline SnO2 phase has formed over the carbon flakes. Sn is the common impurity for SnO2 synthesis when the annealing of Sn-salt is done comparatively at elevated temperature (>550 °C).43 Here, the annealing temperature was purposely maintained at 350 °C to eliminate the probability of partial thermal reduction of SnO2 to Sn. Interestingly, the XRD does not exhibit any peak characteristic to the carbon materials in the composite, suggestive of the fact that the uniform dispersion of SnO2 materials could be achieved over the surface of the carbon. It is worth mentioning that peak broadening was observed in the case of the C/SnO2 composite manifesting the formation of the SnO2 nanostructure. The as-prepared composite and the carbon were further investigated using Raman spectroscopy and nitrogen adsorption–desorption isotherm analysis (detailed discussion has been made in the ESI,† Fig. S2A–E).
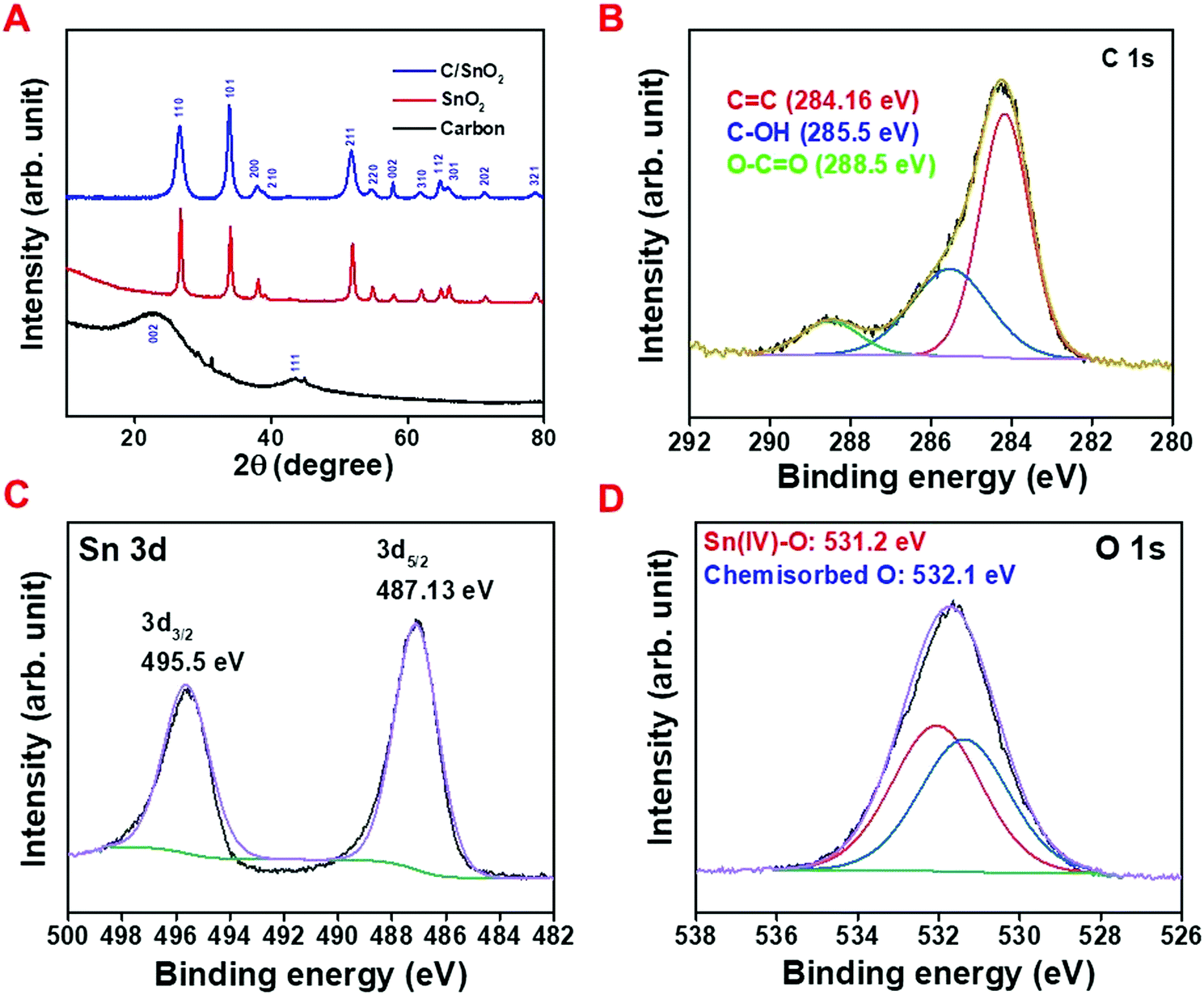 | ||
| Fig. 2 (A) Comparative X-ray diffraction pattern of NAC (black line), bare SnO2 (red line) and C/SnO2 (blue line), X-ray photo electron spectroscopy of the C/SnO2 composite; (B) C 1s, (C) Sn 3d and (D) O 1s. | ||
The X-ray photoelectron spectroscopic (XPS) technique was adopted to analyze the surface chemical analysis of the composite where the wide range spectrum exhibits the characteristic peaks of Sn 3d, Sn 3p, O 1s and C 1s (Fig. S2F, ESI†). The deconvoluted C 1s spectrum consists of three peaks where the peaks having binding energy values of 284.16 eV and 285.5 eV stand for the sp2 and sp3 carbon with a separation of 1.3 eV implying the amorphous character of the carbon and the peak at 288.5 eV corresponds to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group (Fig. 2B).8,56 The narrow range spectrum of Sn 3d exhibits two peaks at 487.13 V and 495.5 eV standing for the Sn 3d5/2 and Sn 3d3/2 of Sn(IV), respectively, confirming the SnO2 phase in the composite (Fig. 2C).19,57,58 The peaks at 531.2 eV and 532.19 eV in the deconvoluted O 1s spectrum correspond to the binding energy of Sn(IV)–O and chemisorbed oxygen (Fig. 2D).57,58 These results validate the formation of pure SnO2 on the surface of the amorphous carbon containing higher percentage of sp2 hybridization.
O functional group (Fig. 2B).8,56 The narrow range spectrum of Sn 3d exhibits two peaks at 487.13 V and 495.5 eV standing for the Sn 3d5/2 and Sn 3d3/2 of Sn(IV), respectively, confirming the SnO2 phase in the composite (Fig. 2C).19,57,58 The peaks at 531.2 eV and 532.19 eV in the deconvoluted O 1s spectrum correspond to the binding energy of Sn(IV)–O and chemisorbed oxygen (Fig. 2D).57,58 These results validate the formation of pure SnO2 on the surface of the amorphous carbon containing higher percentage of sp2 hybridization.
In order to explore the Li storage behavior of the NAC, galvanostatic charge–discharge (CD) studies were conducted at the working potential of 0.01 to 3 V (vs. Li+/Li) (Fig. 3A). The calculated specific discharge and charge capacity values are 657 mA h g−1 and 318 mA h g−1 having an initial Coulombic efficiency (ICE) of 48% at 25 mA g−1 specific current and it maintains the specific capacity of 121 mA h g−1 at 1000 mA g−1 (Fig. 3A and B).
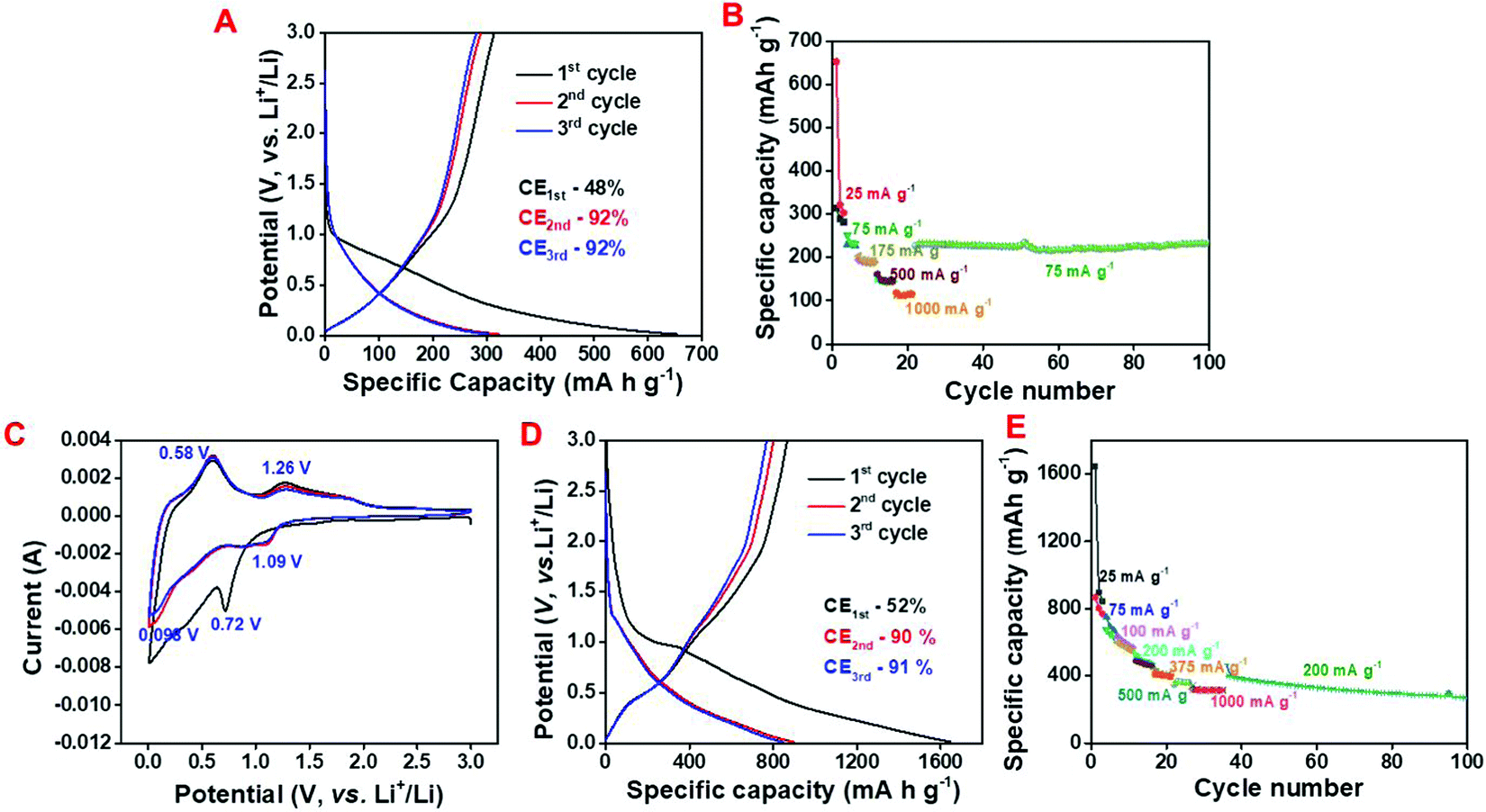 | ||
| Fig. 3 Half-cell electrochemical study; (A) galvanostatic charge–discharge curve and (B) rate capability curve of NAC, (C) cyclic voltammetry (CV) curve, (D) galvanostatic charge–discharge curve (CD) and (E) rate capability curve of C/SnO2. | ||
Moreover, the NAC maintains a specific capacity of 229 mA h g−1 for 100 charge–discharge cycles when the current returns to 75 mA g−1 suggesting the high rate capability of NAC as a LIB anode. In spite of the excellent rate performance of the NAC as a LIB anode, the relatively low specific capacity inhibits its actual potential towards the fabrication of a commercial grade anode. To investigate the electrochemical mechanism and performance of the C/SnO2 composite, cyclic voltammetry (CV) and CD were executed in the same working potential like NAC (Fig. 3C and D). Fig. 3C demonstrates the typical CV curve of the C/SnO2 at 1 mV s−1 where the sharp reduction peak at 0.72 V refers to the conversion of SnO2 to Sn and SEI formation is associated to the reduction of electrolyte. Upon further reduction a small hump at 0.98 V designates the alloying reaction of Sn and Li ions to form LiSnx and the oxidation peak at 0.58 V represents the corresponding de-alloying reaction.14,23,59 The reduction peak corresponding to the conversion of SnO2 to Sn is shifted to 1.09 V in the succeeding cycles and the oxidation peak at 1.26 V refers to the corresponding partial de-conversion of Sn and Li2O to SnO2 (Fig. 3C).14,23 The nature of the CD curve also supports the claim where a small plateau near 0.9 V in the cathodic curve of the C/SnO2 composite suggests the typical conversion reaction whereas the plateau behavior below 0.5 V stands for the alloying reaction (Fig. 3D). The C/SnO2 composite exhibits 1st cycle specific discharge and charge capacity values of 1657 mA h g−1 and 868 mA h g−1, respectively, having CE of 52% at 25 mA g−1 specific current where the low CE in the first cycle is due to the irreversibility of the Li2O/Sn to SnO2 and formation of SEI by decomposing the electrolyte.14,23 It is noteworthy that the C/SnO2 holds high surface area that can contribute to the low ICE due to the additional consumption of electrolyte. Successively in the 2nd and 3rd cycles at the same current the composite exhibits reversible discharge/charge capacity values of 907/817 and 856/790 mA h g−1 having CE of 90% and 92%, respectively, suggesting the formation of a stable SEI, which restricts the further decomposition of the electrolyte.2,14,23 In order to explore the rate performance of the as-prepared C/SnO2 composite, the specific capacity values have been calculated at successively increased specific currents where the composite maintains specific capacity of 316 mA h g−1 even at 1000 mA g−1 (Fig. 3E). Remarkably, the C/SnO2 maintains the specific capacity of 400 mA h g−1 when the specific current returns to 200 mA g−1 after 35 cycles and retains the capacity of 66% up to 100 cycles. This result promotes the resilient behavior of C/SnO2 towards different discharge currents, which can be beneficial for practical applications where high power is demanded. To date, several C/SnO2 composites have been explored to study the Li-ion storage behavior and the comparative analysis advocates that the as-prepared C/SnO2 composite by the low-cost synthesis process does not sacrifice the battery performance (Table S1, ESI†). Intending to establish the advantages of the wet chemical dispersion of SnCl2 with NAC, Li-ion battery behavior of the P-C/SnO2 composite (P stands for physical process) prepared by mechanical grinding of SnCl2 and carbon has also been investigated (details have been mentioned in the ESI,† Fig. S3A and B). This result clearly advocates the structural robustness of the C/SnO2 towards the electrochemical performance synthesized by the wet chemical dispersion technique; however the P-C/SnO2 composite prepared by the mechanical grinding process exhibits inferior rate and stability.
Apparently, SnO2 follows the conversion-based mechanism (diffusive) whereas amorphous carbon stores charge through the adsorption of Li-ions (capacitive). Thus, it is essential to evaluate the storage mechanism of the C/SnO2 composite as a Li-ion battery anode. The percentage capacitive/diffusive contribution of the C/SnO2 in each scan rate and the volumetric capacitive/diffusive contribution in the CV curve for a particular scan rate was evaluated using Dunn's equation;60
| i(V) = K1V + K2V1/2 |
The calculation was done by considering the current (i) and potential (V) from the CV curve of the C/SnO2 at different scan rates where it can be observed that with increase in scan rate from 1 mV s−1 to 5 mV s−1, the current of the different redox peaks increases (Fig. 4A).
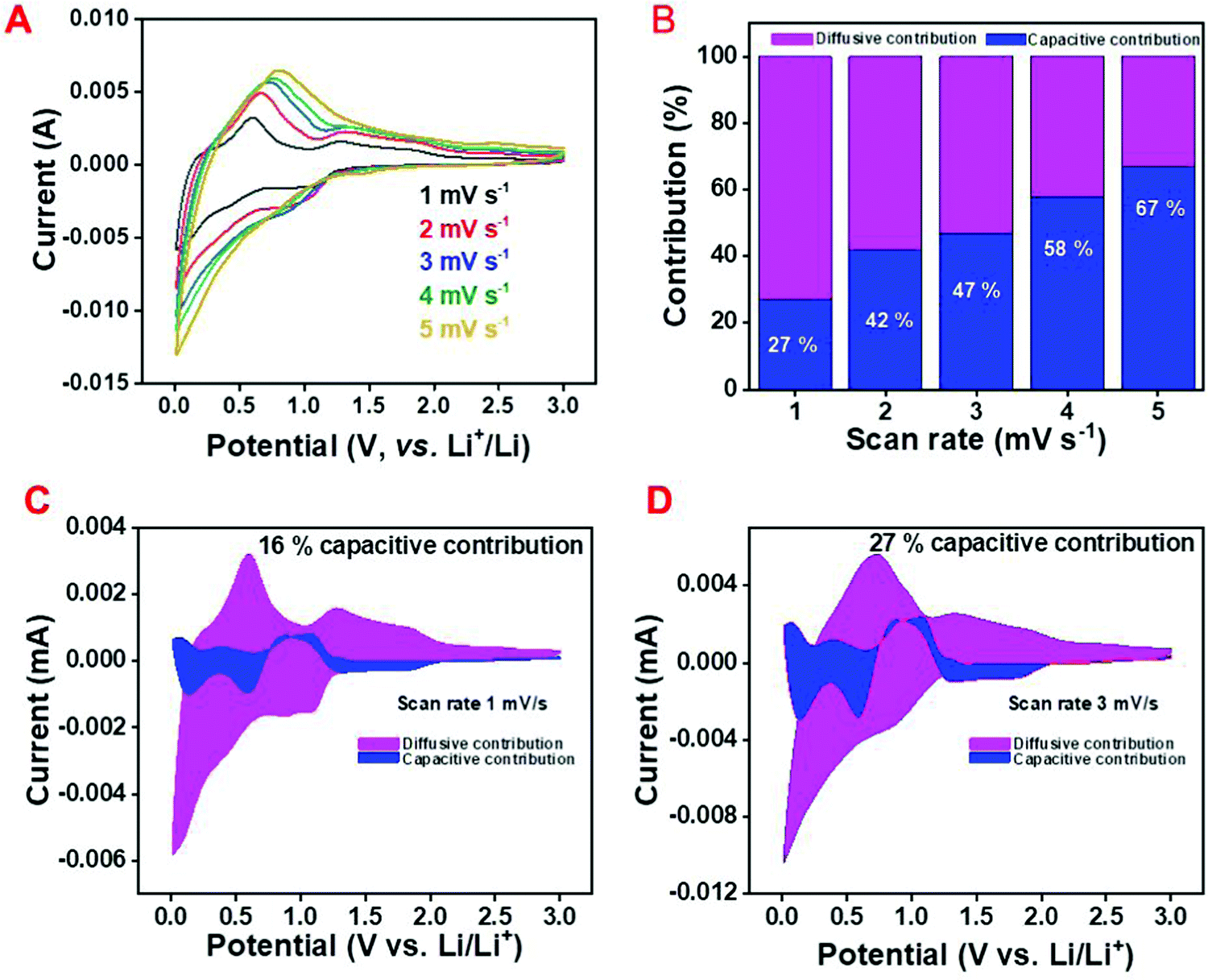 | ||
| Fig. 4 Kinetics of the Li-ion in the C/SnO2 composite; (A) cyclic voltammetry curve, (B) capacitive and diffusive contribution curve at different scan rates for a fixed potential (at 0.58 V), and capacitive and diffusive contribution in a cyclic voltammetry curve for a fixed scan rate, (C) 1 mV s−1 and (D) 3 mV s−1. | ||
Here the current (i) defines the potential for a particular peak and the change in potential for the assigned peak with the increase in scan rate has not been considered.60Fig. 4B demonstrates the capacitive contribution to be only 27% at 1 mV s−1 scan rate, which increases to 67% at a scan rate of 5 mV s−1 where the calculations have been carried out considering the oxidation peak of the de-alloying reaction (at 0.58 V). Similarly, the capacitance contribution of the total charge (considering the whole CV curve) increases from 16% to 27% with increase in the scan rate from 1 mV s−1 to 3 mV s−1 (Fig. 4C and D). From the above discussion it is conclusive that the C/SnO2 composite is majorly dominated by the diffusion-controlled reaction with a small contribution of the surface capacitive reaction.
Fig. 5A and B exhibit the cyclic stability performance of the as-prepared NAC and C/SnO2 at 200 mA g−1 and 375 mA h g−1 specific currents, respectively. It can be perceived that the NAC maintains ∼100% capacity retention even after 1000 cycles, whereas C/SnO2 retains ∼68% of its initial capacity after 1000 cycles. The high stability of activated carbon is mainly due to the adsorption-based mechanism. In contrast, the C/SnO2 composite maintains 67% of its initial capacity after 1000 charge–discharge cycles. Here, it is observed that initially the C/SnO2 exhibits a specific capacity of 410 mA h g−1 and the value decreases remarkably to 220 mA h g−1 within the first 150 cycles. After that, interestingly the specific capacity increases after 300 cycles, which reaches up to 278 mA h g−1 after ∼500 cycles and is maintained up to 1000 cycles (Fig. 5B). The initial decrease in the specific capacity may be attributed to the gradual expansion in volume of the SnO2 nanostructures due to the conversion and alloying reaction, which deteriorates the structure of the composite. The sudden increase in the specific capacity after 300 cycles could be due to the structural reconstruction of the material, which provides more active sites and hence electrolyte penetration occurs more profoundly.60–63
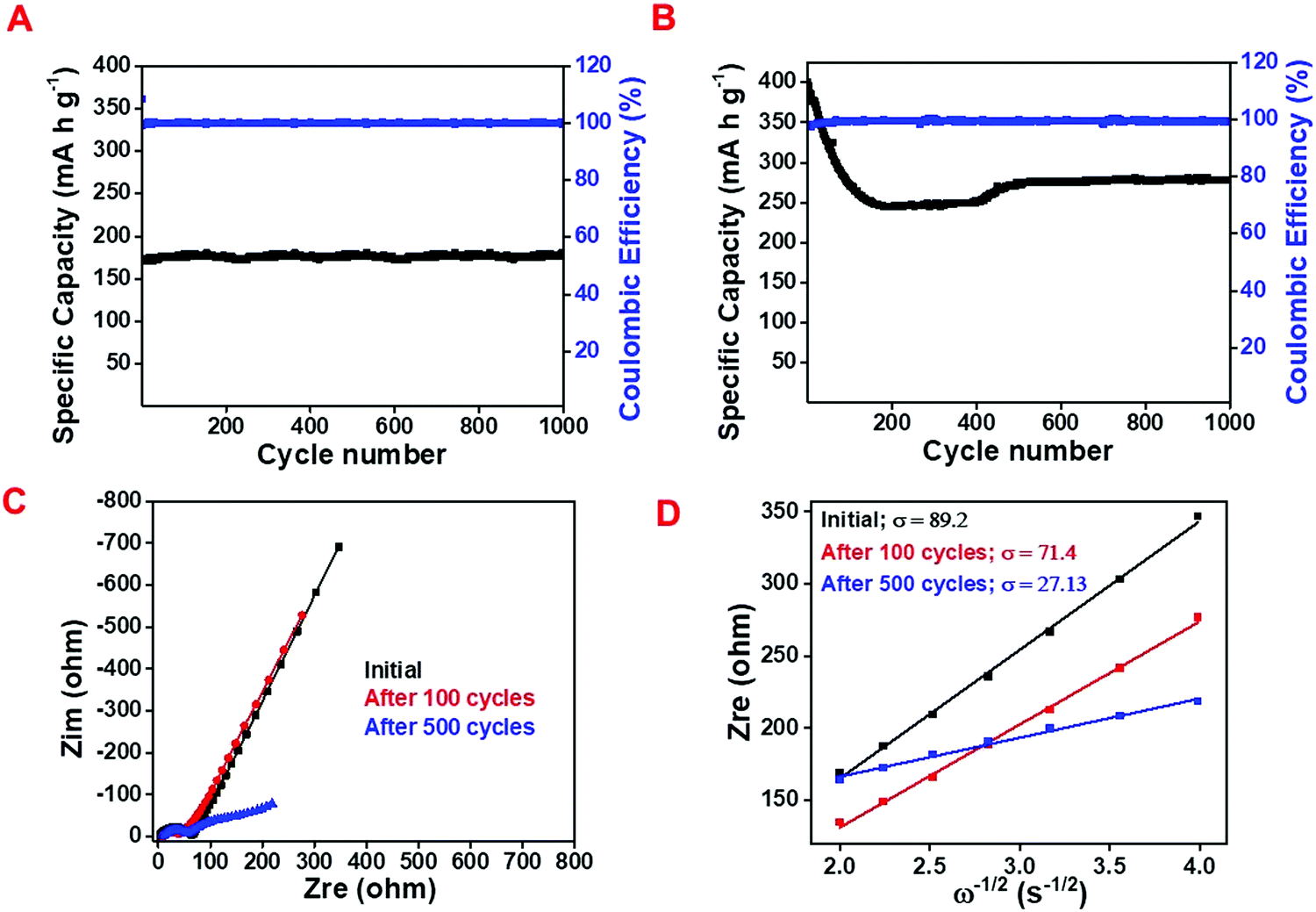 | ||
| Fig. 5 Cyclic stability curve for (A) NAC at 200 mA g−1 and (B) C/SnO2 at 375 mA h g−1, (C) electrochemical impedance spectra (EIS) of C/SnO2 at different stages of the cycles, (D) real impedance (Zrel) vs. angular frequency (ω) plot where the slot refers to the Warburg impedance coefficient (σ). | ||
From the above results it is apparent that surface engineering of the NAC with SnO2 nanowires increases the specific capacity without significantly dropping the rate capability and stability, which is the sole objective of this work. The synergistic effect between carbon and SnO2 nanowires and the uniform distribution of SnO2 nanowires over the surface of NAC provide structural and mechanical stability to the composite, which is reflected during the rate capability and stability results. The wet chemical dispersion results in the uniform adhesion of Sn2+ on the surface of the NAC. Upon calcination, this directs uniform 1D growth of SnO2 on the top of the carbon without any agglomeration. As a matter of fact, the C/SnO2 composite maintains the ability to withstand severe volume expansion due to the formation of LiSnX. In the case of the composite prepared by the mechanical grinding of SnCl2 salt with NAC followed by annealing, the agglomerated bulk morphology of the SnO2 on the surface of NAC is observed, which directly affects the battery performance of the composite. It has already been mentioned that in the first 50 cycles the mechanical grinding-based composite (P-C/SnO2) exhibits a sharp specific capacity fall to 22% of its initial value owing to the structural instability of the bulk SnO2 during the lithiation and de-lithiation of Li-ions in the subsequent cycles.
So as to analyze the electrochemical performance of the C/SnO2 as a Li-ion battery anode more distinctly, electrochemical impedance spectra (EIS) were studied at different stages of charge–discharge cycles (Fig. 5C). The calculated impedance parameters (Table S2, ESI†) suggest that internal resistance (RS) associated with the solid electrode and electrode–electrolyte interface increases from 2.9 Ohm to 6.7 Ohm upon 100 charge–discharge cycles and further increases to 7.4 Ohm after 500 charge–discharge cycles. This increase in RS value can be due to the weakening in the contact in between the current collector and the electrode in successive cycles. In contrast to the RS, the semicircle part in the EIS curve attributable to the charge transfer resistance (RCT) increases significantly from 62.1 Ohm to 32.6 Ohm after 100 cycles, indicating the increase in kinetics due to the activation of the redox sites in the subsequent cycles, which in turn facilitates further Li-ion storage. Initially the electrode is not wet enough by the liquid electrolyte and thus offers less redox active sites towards the electrolyte while with increase in the charge–discharge cycles, the wettability of the electrode increases as well as the redox active sites, which can be observed form the stability curve (Fig. 5B). The slope region in the EIS defines the impedance due to the diffusion and the calculated Warburg coefficient (σ) signifies the drop in diffusion resistance values from 89.2 Ohm s−1/2 to 71.47 Ohm s−1/2 after the 100 charge–discharge cycles. This observation suggests the decrease in diffusion resistance with the increase in cycles as confirmed from the calculated diffusion coefficient (Fig. 5D). It has been observed that the Diffusion coefficient calculated from the EIS curve increases after 100 cycles (2.12 × 10−14 cm2 s−1) as compared to its initial value (1.36 × 10−14 cm2 s−1); this in turn, indicates the concurrent increment of the diffusion of Li-ions with the increment in the number of cycles. The DLi+ value significantly increases after 500 cycles supporting the simultaneous increase in specific capacity with the increase in charge–discharge cycles after 100 cycles.
Intending to examine the real application, a full cell was fabricated using carbon coated LiFePO4 (C/LFP) as the cathode and C/SnO2 as the anode. The C/LFP was synthesized by a reported method.64 The cathodic performance of the C/LFP was investigated in the potential window of 2 to 4 V (vs. Li+/Li) having a specific capacity of 156 mA h g−1 and 137 mA h g−1 at C/10 and 1C (Fig. 6A and Fig. S4, ESI†). It is worth mentioning that both the cathode and anode were assembled in the full cell fabrication without any pretreatment, such as pre-lithiation or pre-cycling. In most of the studies, the authors have usually performed pre-cycling of the cathode and anode or pre-lithiation of the anode to avoid complications due to SEI formation.65Fig. 6A demonstrates the charge–discharge curves in half-cells where C/LFP and C/SnO2 exhibit specific capacity values of 137 and 437 mA h g−1 at 1C. Thus, theoretically the mass ratio was expected to be ∼1:3. (C/SnO2:C/LFP), but practically a 1:4 ratio of the same was used to compensate for the low ICE of C/SnO2. The full cell study was carried out within 0.8 to 3.5 V working voltage range where the lower cut-off voltage (0.8 V) accounts for the conversion and alloying reaction of the C/SnO2 (Fig. 6B). The calculated specific capacity values of the full cell are 167, 131, 100, 92 and 77 mA h g−1 at the specific current of 60 (∼C/2.5), 120 (∼1C), 275 (∼3C), 350 (∼4C) and 575 mA g−1 (∼8C) endorsing the excellent rate performance (Fig. 6C). Moreover, the cell exhibits a specific capacity of 90 mA h g−1 when the specific current returns to 120 mA g−1 after 20 cycles and retains 45% capacity after 50 cycles. The full cell exhibits an average voltage of 2.3 V and a maximum energy density of 384 W h kg−1 considering the active weight of the anode. In a few cases, researchers restricted the lower cut-off voltage to 2 V to increase the rate and stability by avoiding the conversion reaction, whereas in the present case the C/LFP//C/SnO2 exhibits good rate capability and stability even though the lower cut-off voltage is 0.8 V.
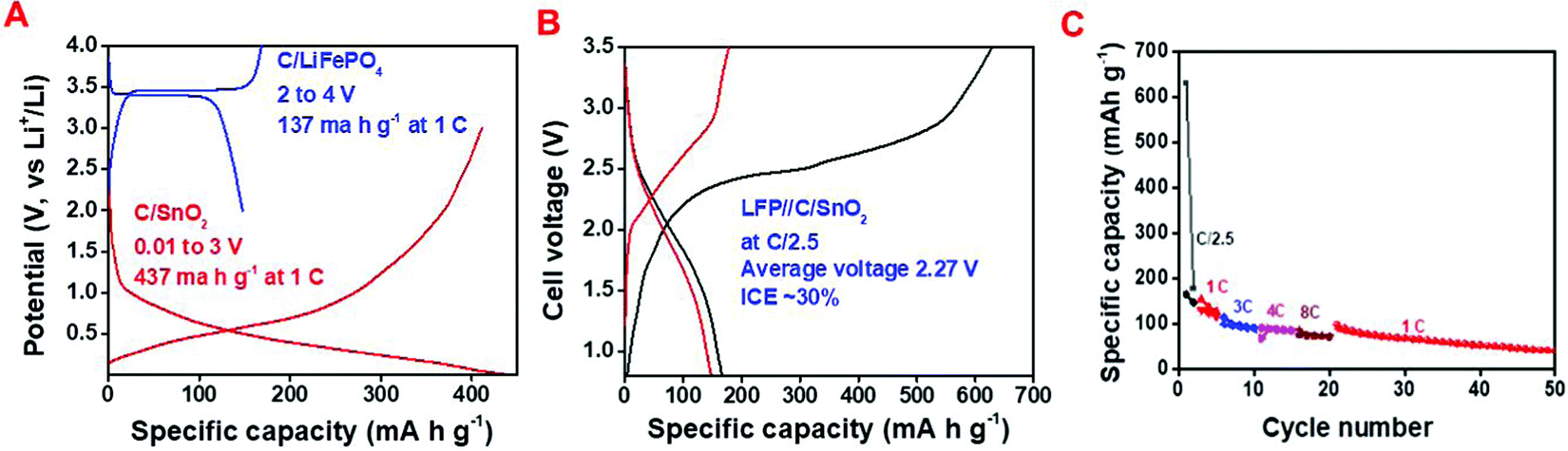 | ||
| Fig. 6 Full cell analysis; C/LFP as the cathode and C/SnO2 as the anode; (A) charge–discharge profile of C/LFP and C/SnO2 at 1C in the half cell, (B) charge–discharge curve at C/2.5 and (C) rate capability curve of the full cell. | ||
The Na-ion storage behavior has also been explored for both the samples and a comparative study has been mentioned here. The CD profile suggests that NAC delivers discharge and charge capacity values of 420 mA h g−1 and 177 mA h g−1 in the 1st cycle with an ICE of 42% at 25 mA h g−1 specific current following the adsorption–desorption mechanism whereas at 1000 mA g−1 it maintains the specific capacity of 87 mA g−1 (Fig. 7A and B). As observed from the galvanostatic charge–discharge profile, C/SnO2 exhibits a 1st cycle discharge and charge capacity of 953 and 220 mA h g−1 at 25 mA g−1 current having the ICE of 22% and the specific capacity value decreases to 91 mA h g−1 at 500 mA g−1 (Fig. 7C and D).
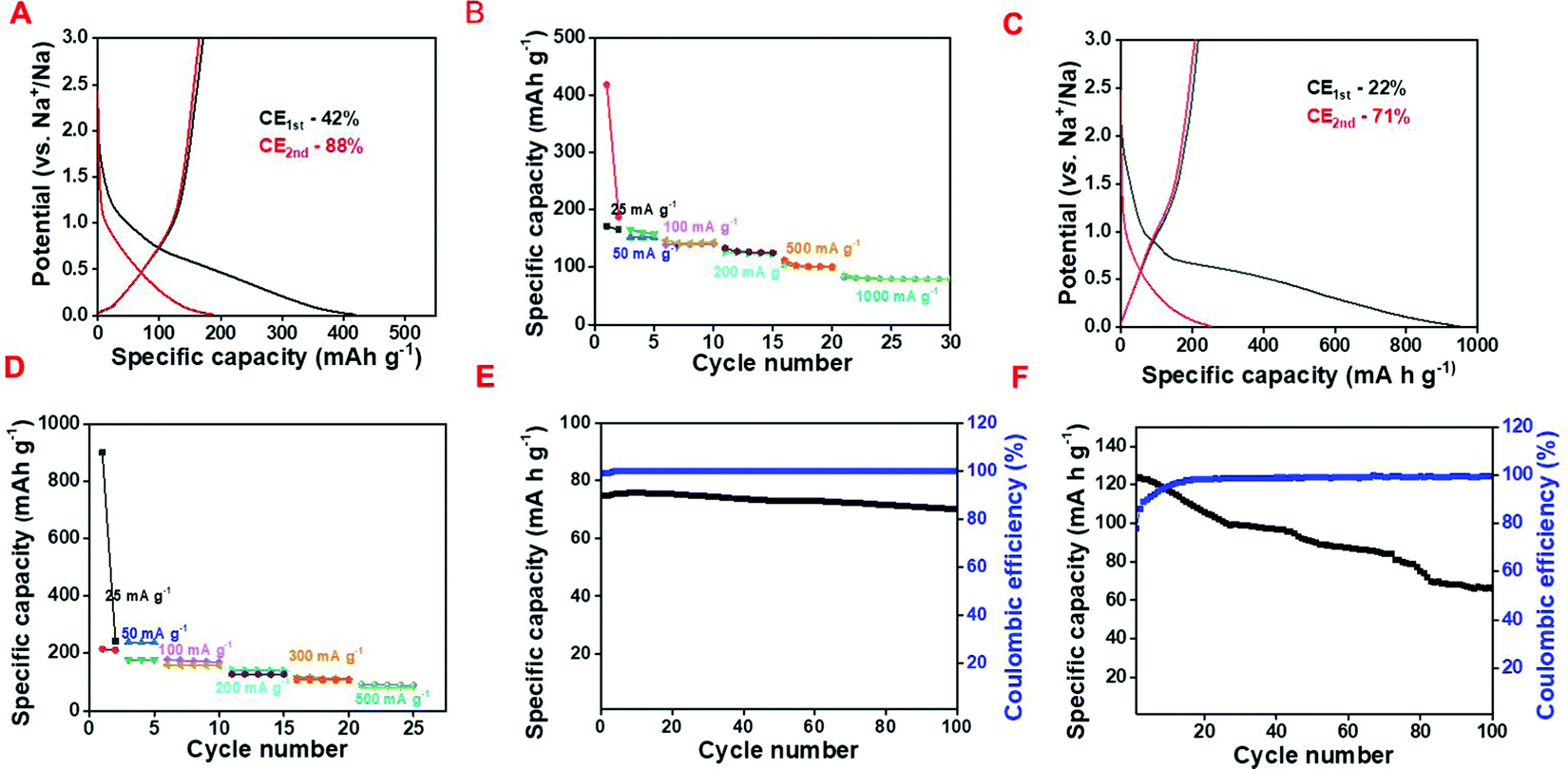 | ||
| Fig. 7 Half-cell electrochemical study as a Na-ion battery; (A) galvanostatic charge–discharge curve and (B) rate capability curve of NAC, (C) galvanostatic charge–discharge curve and (D) rate capability curve of C/SnO2, cycling stability curve of (E) NAC at 1000 mA g−1 and (F) C/SnO2 at 200 mA g−1. | ||
Such a low ICE is generated predominantly due to the irreversible conversion reaction and the high electrolyte consumption during the formation of the SEI. Even in the second cycle the CE is lower, which may be due to the unstable SEI formation in the first cycle. As a matter of fact, less Na-ions are participating in the oxidation process, which results in the low specific capacity value as well. In order to scrutinize the cycling stability, a charge–discharge experiment was performed up to 100 cycles for both NAC and C/SnO2 and it can be seen that NAC maintains almost 94% of its initial capacity whereas C/SnO2 maintains 70% at a specific current of 1000 mA g−1 and 200 mA g−1, respectively (Fig. 7E and F). The above results substantiate that large volume expansion–contraction of SnO2 during the charge–discharge cycles perturbs the mechanical integrity of the composite resulting in crack formation within the SEI, which hampers the stability as well as the rate capability of the C/SnO2 as a Na-ion battery. In order to comprehensively realize the battery performance of the C/SnO2 composite more distinctly, EIS was performed before and after the 100 cycles and different impedance parameters were calculated (details have been mentioned in the ESI,† Fig. S5).
From the above discussion it is conspicuous that the non-activated carbon (NAC) exhibits excellent Li/Na-ion battery performance in terms of rate capability and cycling stability, but the specific capacity values are not satisfactory. Thus, conversion-alloying based SnO2 has been introduced on the surface of the NAC to enhance the specific capacity without sacrificing the rate and cycling stability significantly. As a Li-ion battery anode, the C/SnO2 composite proves to be an excellent anode material, where the specific capacity value increases significantly with excellent rate and cycling stability and the composite demonstrates a specific capacity of 431 mA h g−1 at 1C which is even higher than the commercial graphite. On the other hand, in the case of the Na-ion battery, although C/SnO2 exhibits improved specific capacity value compared to the NAC, the larger size of the Na-ion hampers the formation of a stable SEI affecting the rate capability and cycling stability. The plausible reaction mechanism of the as-prepared C–SnO2 as a Li/Na-ion battery anode has been demonstrated in Scheme 2. It is noteworthy that the Na-ion storage study of SnO2 demands special attention, which is the possible direction for our future studies.
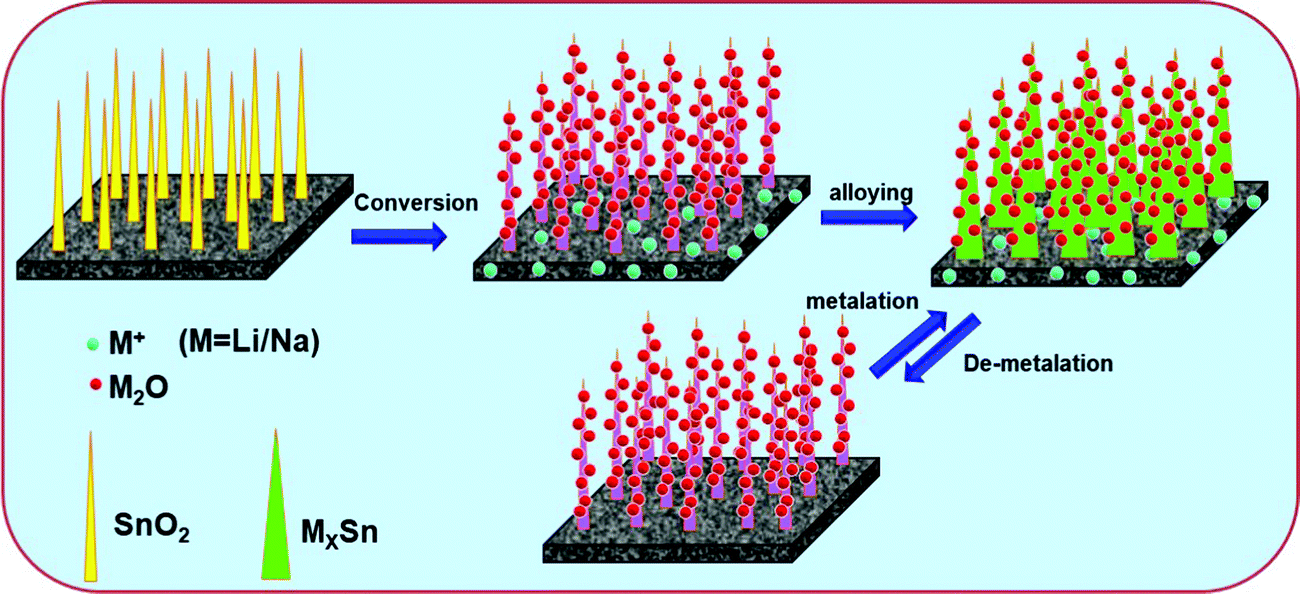 | ||
| Scheme 2 Schematic presentation of the Li/Na-ion insertion and de-insertion reaction mechanism of the C/SnO2 composite. | ||
Conclusion
Here, a C/SnO2 composite has been reported starting from jute-derived non-activated carbon (NAC) using a wet chemical dispersion followed by annealing approach. The synthesis process is facile, cost effective and easily scalable, and is considered as the most desirable parameter for industrial purposes. The detailed characterizations of the as-synthesized materials have been achieved using different physicochemical techniques and it has been concluded that the pure and crystalline SnO2 nanowires are uniformly distributed over the carbon flakes. The as-prepared C/SnO2 has been tested as both a LIB and NIB and it has been found that the composition with SnO2 enhances the specific capacity of NAC significantly. The C/SnO2 composite exhibits specific capacity values of 868 mA h g−1 and 217 mA h g−1 at 25 mA g−1 as a LIB and NIB anode, respectively, and those of NAC are 320 mA h g−1 and 177 mA h g−1 at the same current. The full cell assembly of the C/SnO2 with C/LFP provides 2.3 V average cell voltage having an energy density of 384 Wh kg−1, which validates the viability of the C/SnO2 in the real cell. Here, it can be concluded that the surface engineering of jute-derived non-activated carbon with SnO2 nanowires renders the composite as a better anode material for the LIB/NIB anode and the facile synthesis approach can be extended to synthesize low cost metal oxide based anode materials for rechargeable batteries.Author contributions
P. Kumar and B. R. Madhuri have contributed in conducting the experiments for synthesis and electrochemical characterizations of the materials and in writing the manuscript. K. Nanji and S. Anandan have contributed through scientific inputs in materials synthesis and characterization. T. N. Rao has contributed through scientific inputs and reviewing of the manuscript. R. Sahoo has conceptualized and supervised the work, conducted the experiments for battery performance, data interpretation, acquisition of funding, writing, reviewing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Bijoy Kumar Das, ARCI for the Raman analysis of the samples. The work has been financially supported by Department of Science and Technology, Govt. of India (DST INSPIRE Faculty award DST/INSPIRE/04/2018/001825).References
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- Y. Xia, S. Han, Y. Zhu, Y. Liang and M. Gu, Energy Storage Mater., 2019, 18, 125–132 CrossRef.
- Q. Shi, J. Zhou, S. Ullah, X. Yang, K. Tokarska, B. Trzebicka, H. Q. Ta and M. H. Rümmeli, Energy Storage Mater., 2021, 34, 735–754 CrossRef.
- A. Bauer, J. Song, S. Vail, W. Pan, J. Barker and Y. Lu, Adv. Energy Mater., 2018, 8, 1702869 CrossRef.
- Y. Li, Y. Lu, P. Adelhelm, M. M. Titirici and Y. S. Hu, Chem. Soc. Rev., 2019, 48, 4655–4687 RSC.
- D. Saurel, B. Orayech, B. Xiao, D. Carriazo, X. Li and T. Rojo, Adv. Energy Mater., 2018, 8, 1703268 CrossRef.
- S. J. Kang, K. H. Kim, J. Kang, D. Y. Kim, S. A. Chae, N. Saito and S. Y. Choi, J. Mater. Chem. A, 2019, 7, 16149–16160 RSC.
- K. Nanaji, T. N. Rao, U. V. Varadaraju and S. Anandan, Int. J. Energy Res., 2020, 44, 2289–2297 CrossRef CAS.
- D. Puthusseri, M. Wahid and S. Ogale, ACS Omega, 2018, 3, 4591–4601 CrossRef CAS PubMed.
- Y. Ma, Y. Ma, G. Giuli, T. Diemant, R. J. Behm, D. Geiger, U. Kaiser, U. Ulissi, S. Passerini and D. Bresser, Sustainable Energy Fuels, 2018, 2, 2601–2608 RSC.
- Y. Zhang, Q. Zhou, J. Zhu, Q. Yan, S. X. Dou and W. Sun, Adv. Funct. Mater., 2017, 27, 1702317 CrossRef.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- J. S. Chen and X. W. (David) Lou, Small, 2013, 9, 1877–1893 CrossRef CAS PubMed.
- Y. Wang, Z. X. Huang, Y. Shi, J. I. Wong, M. Ding and H. Y. Yang, Sci. Rep., 2015, 5, 9164 CrossRef PubMed.
- Y. Li, Y. Zhao, C. Ma, J. Shi and Y. Zhao, J. Alloys Compd., 2020, 821, 153506 CrossRef CAS.
- F. Li, G. Luo, W. Chen, Y. Chen, Y. Fang, M. Zheng and X. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 36949–36959 CrossRef CAS PubMed.
- X. Lu, Q. Mao, Q. Xiong, S. Pan, X. Duan and Z. Ji, J. Solid State Chem., 2019, 277, 556–563 CrossRef CAS.
- B. Dursun, E. Topac, R. Alibeyli, A. Ata, O. Ozturk and R. Demir-Cakan, J. Alloys Compd., 2017, 728, 1305–1314 CrossRef CAS.
- H. Bian, X. Xiao, S. Zeng, M. F. Yuen, Z. Li, W. Kang, D. Y. W. Yu, Z. Xu, J. Lu and Y. Y. Li, J. Mater. Chem. A, 2017, 5, 2243–2250 RSC.
- Z. Huang, H. Hou, G. Zou, J. Chen, Y. Zhang, H. Liao, S. Li and X. Ji, Electrochim. Acta, 2016, 214, 156–164 CrossRef CAS.
- Y. C. Lu, C. Ma, J. Alvarado, T. Kidera, N. Dimov, Y. S. Meng and S. Okada, J. Power Sources, 2015, 284, 287–295 CrossRef CAS.
- S. Zhao, C. D. Sewell, R. Liu, S. Jia, Z. Wang, Y. He, K. Yuan, H. Jin, S. Wang, X. Liu and Z. Lin, Adv. Energy Mater., 2020, 10, 1902657 CrossRef CAS.
- R. Hu, Y. Ouyang, T. Liang, H. Wang, J. Liu, J. Chen, C. Yang, L. Yang and M. Zhu, Adv. Mater., 2017, 29, 1605006 CrossRef PubMed.
- H. Ying and W. Q. Han, Adv. Sci., 2017, 4, 1700298 CrossRef PubMed.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- Y. Feng, K. Wu, J. Ke, H. Dong, X. Huang, C. Bai, D. Xiong and M. He, J. Power Sources, 2020, 467, 228357 CrossRef CAS.
- R. Hu, D. Chen, G. Waller, Y. Ouyang, Y. Chen, B. Zhao, B. Rainwater, C. Yang, M. Zhu and M. Liu, Energy Environ. Sci., 2016, 9, 595–603 RSC.
- G. Kilibarda, D. V. Szabó, S. Schlabach, V. Winkler, M. Bruns and T. Hanemann, J. Power Sources, 2013, 233, 139–147 CrossRef CAS.
- J. Wang, F. Fang, T. Yuan, J. Yang, L. Chen, C. Yao, S. Zheng and D. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 3544–3553 CrossRef CAS PubMed.
- J. Li, Y. Zhao, N. Wang and L. Guan, Chem. Commun., 2011, 47, 5238–5240 RSC.
- R. S. Kalubarme, B. B. Kale and S. W. Gosavi, Nanotechnology, 2011, 23, 035402 Search PubMed.
- Y. Zhang, J. Xiao, Q. Lv, L. Wang, X. Dong, M. Asif, J. Ren, W. He, Y. Sun, F. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 38201–38210 CrossRef CAS PubMed.
- J. Liu, Y. Li, X. Huang, R. Ding, Y. Hu, J. Jiang and L. Liao, J. Mater. Chem., 2009, 19, 1859–1864 RSC.
- F. Tian, Y. Cheng, Y. Zhang, Q. Zhao, Q. Shi, Y. Zhang, C. Zhou, S. Yang and X. Song, Mater. Lett., 2021, 284, 129019 CrossRef CAS.
- L. Y. Jiang, X. L. Wu, Y. G. Guo and L. J. Wan, J. Phys. Chem. C, 2009, 113, 14213–14219 CrossRef CAS.
- H. Bin Wu, J. S. Chen, X. W. (David) Lou and H. H. Hng, J. Phys. Chem. C, 2011, 115, 24605–24610 CrossRef.
- W. Wang, P. Li, Y. Fu and X. Ma, J. Power Sources, 2013, 238, 464–468 CrossRef CAS.
- J. Liu, W. Li and A. Manthiram, Chem. Commun., 2010, 46, 1437–1439 RSC.
- Y. Li, Q. Meng, J. Ma, C. Zhu, J. Cui, Z. Chen, Z. Guo, T. Zhang, S. Zhu and D. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 11146–11154 CrossRef CAS PubMed.
- M. Wang, S. Li, Y. Zhang and J. Huang, Chem. – Eur. J., 2015, 21, 16195–16202 CrossRef CAS PubMed.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868–2874 CrossRef CAS.
- R. Hu, H. Zhang, J. Liu, D. Chen, L. Yang, M. Zhu and M. Liu, J. Mater. Chem. A, 2015, 3, 15097–15107 RSC.
- H. Wang, G. Jiang, X. Tan, J. Liao, X. Yang, R. Yuan and Y. Chai, Inorg. Chem. Commun., 2018, 95, 67–72 CrossRef CAS.
- Q. Wang, J. Xu, G. Shen, Y. Guo, X. Zhao, Y. Xia, H. Sun, P. Hou, W. Xie and X. Xu, Electrochim. Acta, 2019, 297, 879–887 CrossRef CAS.
- X. Ao, J. Jiang, Y. Ruan, Z. Li, Y. Zhang, J. Sun and C. Wang, J. Power Sources, 2017, 359, 340–348 CrossRef CAS.
- Y. Hong, W. Mao, Q. Hu, S. Chang, D. Li, J. Zhang, G. Liu and G. Ai, J. Power Sources, 2019, 428, 44–52 CrossRef CAS.
- B. Cao, Z. Liu, C. Xu, J. Huang, H. Fang and Y. Chen, J. Power Sources, 2019, 414, 233–241 CrossRef CAS.
- L. Zu, Q. Su, F. Zhu, B. Chen, H. Lu, C. Peng, T. He, G. Du, P. He, K. Chen, S. Yang, J. Yang and H. Peng, Adv. Mater., 2017, 29, 1701494 CrossRef PubMed.
- D. A. Shnawah, M. F. M. Sabri, I. A. Badruddin, S. B. M. Said, M. B. A. Bashir, N. M. Sharif and M. H. Elsheikh, J. Alloys Compd., 2015, 622, 184–188 CrossRef CAS.
- Y. Li, S. Zhu, Q. Liu, J. Gu, Z. Guo, Z. Chen, C. Feng, D. Zhang and W. J. Moon, J. Mater. Chem., 2012, 22, 2766–2773 RSC.
- Y. Liu, C. Zheng, W. Wang, C. Yin and G. Wang, Adv. Mater., 2001, 13, 1883–1887 CrossRef CAS.
- Z. R. Dai, J. L. Gole, J. D. Stout and Z. L. Wang, J. Phys. Chem. B, 2002, 106, 1274–1279 CrossRef CAS.
- D. H. Youn, A. Heller and C. B. Mullins, Chem. Mater., 2016, 28, 1343–1347 CrossRef CAS.
- A. Kumar and H. M. Jena, Results Phys., 2016, 6, 651–658 CrossRef.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- B. Lesiak, L. Kövér, J. Tóth, J. Zemek, P. Jiricek, A. Kromka and N. Rangam, Appl. Surf. Sci., 2018, 452, 223–231 CrossRef CAS.
- M. Kwoka, L. Ottaviano, M. Passacantando, G. Czempik, S. Santucci and J. Szuber, Appl. Surf. Sci., 2008, 254, 8089–8092 CrossRef CAS.
- M. U. Yousaf, E. Pervaiz, S. Minallah, M. J. Afzal, L. Honghong and M. Yang, Results Phys., 2019, 14, 102455 CrossRef.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS.
- R. Sahoo, T. H. Lee, D. T. Pham, T. H. T. Luu and Y. H. Lee, ACS Nano, 2019, 13, 10776–10786 CrossRef CAS PubMed.
- H. H. Fan, H. H. Li, K. C. Huang, C. Y. Fan, X. Y. Zhang, X. L. Wu and J. P. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 10708–10716 CrossRef CAS PubMed.
- H. Xue, D. Y. W. Yu, J. Qing, X. Yang, J. Xu, Z. Li, M. Sun, W. Kang, Y. Tang and C. S. Lee, J. Mater. Chem. A, 2015, 3, 7945–7949 RSC.
- Y. Chu, L. Guo, B. Xi, Z. Feng, F. Wu, Y. Lin, J. Liu, D. Sun, J. Feng, Y. Qian and S. Xiong, Adv. Mater., 2018, 30, 1704244 CrossRef PubMed.
- P. M. Pratheeksha, E. H. Mohan, B. V. Sarada, M. Ramakrishna, K. Hembram, P. V. V. Srinivas, P. J. Daniel, T. N. Rao and S. Anandan, Phys. Chem. Chem. Phys., 2017, 19, 175–188 RSC.
- J. Song, P. Yan, L. Luo, X. Qi, X. Rong, J. Zheng, B. Xiao, S. Feng, C. Wang, Y. S. Hu, Y. Lin, V. L. Sprenkle and X. Li, Nano Energy, 2017, 40, 504–511 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ya00021g |
| This journal is © The Royal Society of Chemistry 2022 |